
Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Phage Isolation
2.2. Single Plaque Purification
2.3. Large-Scale Amplification of SSBI34
2.4. Phage Stability and One-Step Growth Curve
2.5. Host Range
2.6. Bacterial Growth Reduction Assay
2.7. Transmission Electron Microscopy
2.8. Genome Sequencing and Bioinformatic Analysis
2.9. Phylogenetic Analysis of SSBI34
2.10. Statistical Analysis
3. Results
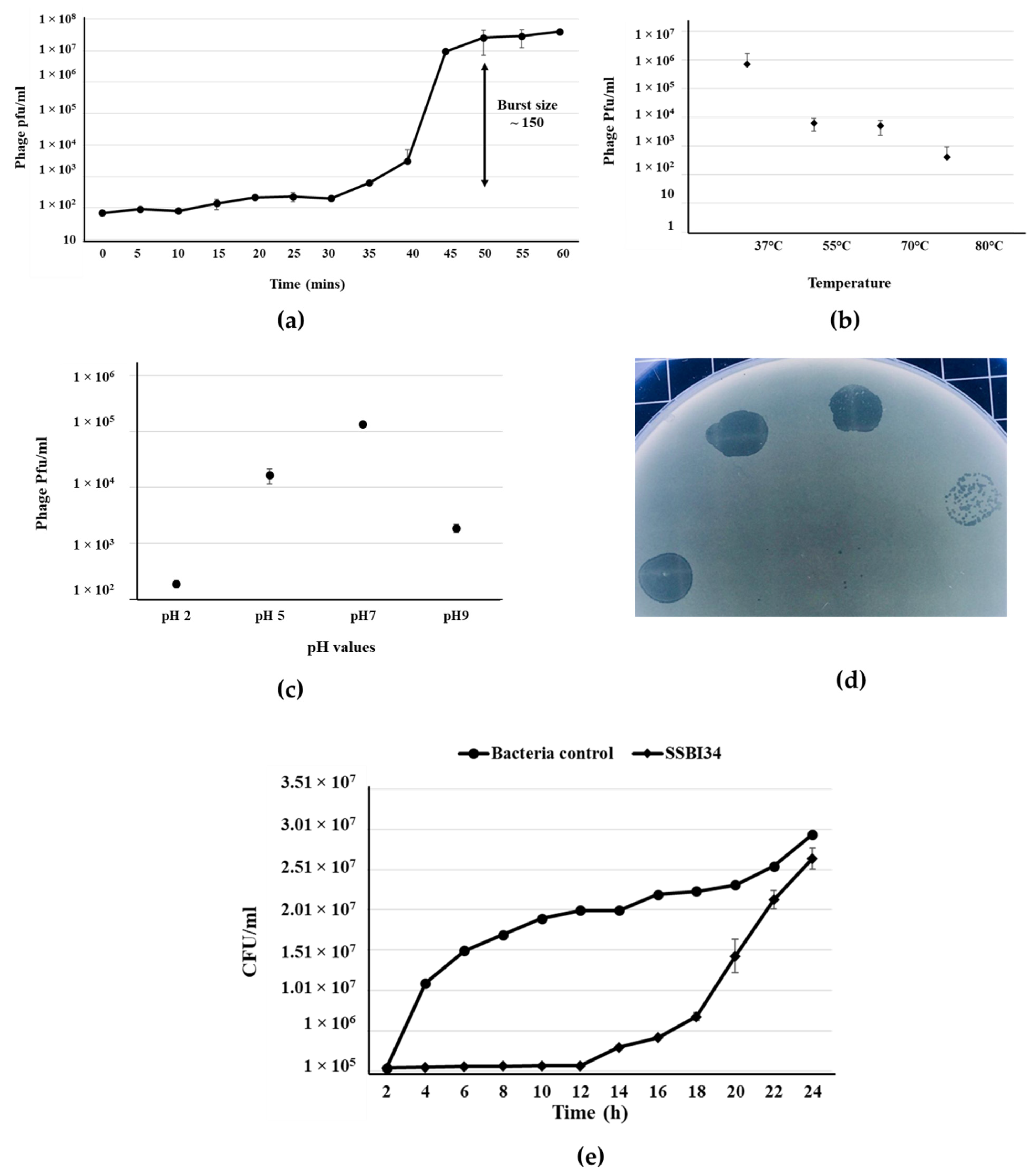
3.1. Phage Isolation and Characterization
3.2. Genome Characterization of Salmonella-Phage-SSBI34
3.2.1. Structural ORF Analysis of SSBI34
3.2.2. ORFs Involved in RNA Synthesis and Modification
3.2.3. DNA Replication, Modification, and Metabolism
3.2.4. Cell Wall Hydrolases
3.2.5. Phylogenetic Analysis of SSBI34
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liyanage, G.Y.; Pathmalal, M. Risk of Prophylactic Antibiotics in Livestock and Poultry Farms—A Growing Problem for Human and Animal Health. Pharm. J. Sri Lanka 2017, 7, 13–22. [Google Scholar] [CrossRef]
- Agyare, C.; Boamah, V.E.; Zumbi, C.N.; Osei, F.B. Antibiotic use in poultry production and its effects on bacterial resistance. Antimicrob Resist Glob. Threat 2018, 33–50. [Google Scholar]
- Kumar, A.; Patyal, A.; Panda, A.K. Sub-therapeutic use of antibiotics in animal feed and their potential impact on environmental and human health: A comprehensive review. J. Anim. Feed Sci. Technol. 2018, 6, 25. [Google Scholar]
- Angulo, F.J.; Johnson, K.R.; Tauxe, R.V.; Cohen, M.L. Origins and consequences of antimicrobial-resistant nontyphoidal Salmonella: Implications for the use of fluoroquinolones in food animals. Microb. Drug Resist. 2000, 6, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Salazar, E.; Gudiño, M.E.; Sevillano, G.; Zurita, J.; Guerrero-López, R.; Jaramillo, K.; Calero-Cáceres, W. Antibiotic resistance of Salmonella strains from layer poultry farms in central Ecuador. J. Appl. Microbiol. 2020, 128, 1347–1354. [Google Scholar] [CrossRef]
- Frenzen, P.D.; Buzby, J.C.; Roberts, T. An Updated Estimate of the Economic Costs of Human Illness Due to Food Borne Salmonella in the United States. J. Food Prot. 2020, 83, 959–967. [Google Scholar] [CrossRef]
- Suijkerbuijk, A.W.M.; Bouwknegt, M.; Mangen, M.J.J.; de Wit, G.A.; van Pelt, W.; Bijkerk, P.; Friesema, I.H.M. The economic burden of a Salmonella Thompson outbreak caused by smoked salmon in the Netherlands, 2012–2013. Eur. J. Public Health 2016, 27, 325–330. [Google Scholar] [CrossRef][Green Version]
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Döpfer, D.; Fazil, A.; Fischer-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001921. [Google Scholar] [CrossRef]
- Havelaar, A.H.; Kirk, M.D.; Torgerson, P.R.; Gibb, H.J.; Hald, T.; Lake, R.J.; Praet, N.; Bellinger, D.C.; de Silva, N.R.; Gargouri, N.; et al. World Health Organization Global Estimates and Regional Comparisons of the Burden of Foodborne Disease in 2010. PLoS Med. 2015, 12, e1001923. [Google Scholar] [CrossRef]
- Xiang, Y.; Li, F.; Dong, N.; Tian, S.; Zhang, H.; Du, X.; Zhou, X.; Xu, X.; Yang, H.; Xie, J.; et al. Investigation of a Salmonellosis Outbreak Caused by Multidrug Resistant Salmonella Typhimurium in China. Front. Microbiol. 2020, 11, 801. [Google Scholar] [CrossRef]
- Folster, J.P.; Grass, J.E.; Bicknese, A.; Taylor, J.; Friedman, C.R.; Whichard, J.M. Characterization of resistance genes and plasmids from outbreaks and illness clusters caused by Salmonella resistant to ceftriaxone in the United States, 2011–2012. Microb. Drug Resist. 2017, 23, 188–193. [Google Scholar] [CrossRef]
- Kariuki, S.; Gordon, M.A.; Feasey, N.; Parry, C.M. Antimicrobial resistance and management of invasive Salmonella disease. Vaccine 2015, 33, C21–C29. [Google Scholar] [CrossRef]
- Rathore, M.H.; Bux, D.; Hasan, M. Multidrug-resistant Salmonella typhi in Pakistani children: Clinical features and treatment. South. Med. J. 1996, 89, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Soomro, A.H.; Khaskheli, M.; Bhutto, M.B.; Shah, G.; Memon, A.; Dewani, P. Prevalence and antimicrobial resistance of Salmonella serovars isolated from poultry meat in Hyderabad, Pakistan. Turk. J. Vet. Anim. Sci. 2011, 34, 455–460. [Google Scholar]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Adebayo, O.S.; Gabriel-Ajobiewe, R.; Taiwo, M.O.; Kayode, J.S. Phage therapy: A potential alternative in the treatment of multi-drug resistant bacterial infections. J. Microbiol. Exp. 2017, 5, 173. [Google Scholar]
- Higgins, J.P.; Higgins, S.E.; Guenther, K.L.; Huff, W.; Donoghue, A.M.; Donoghue, D.J.; Hargis, B.M. Use of a specific bacteriophage treatment to reduce Salmonella in poultry products. Poult. Sci. 2005, 84, 1141–1145. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.P.; Filho, R.L.A.; Higgins, S.E.; Wolfenden, A.D.; Tellez, G.; Hargis, B.M. Evaluation of Salmonella-Lytic Properties of Bacteriophages Isolated from Commercial Broiler Houses. Avian Dis. 2008, 52, 139–142. [Google Scholar] [CrossRef]
- Atterbury, R.J.; Van, M.A.P.B.; Ortiz, F.; Lovell, M.A.; Harris, J.A.; de Boer, A.; Wagenaar, J.A.; Allen, V.M.; Barrow, P.A. Bacteriophage therapy to reduce Salmonella colonization of broiler chickens. Appl. Environ. Microbiol. 2007, 73, 4543–4549. [Google Scholar] [CrossRef]
- Wagenaar, J.A.; Bergen, M.A.P.V.; Mueller, M.A.; Wassenaar, T.M.; Carlton, R.M. Phage therapy reduces Campylobacter jejuni colonization in broilers. Vet. Microbiol. 2005, 109, 275–283. [Google Scholar] [CrossRef]
- Carrillo, C.; Atterbury, R.J.; El-Shibiny, A.; Connerton, P.L.; Dillon, E.; Scott, A.; Connerton, I.F. Bacteriophage therapy to reduce Campylobacter jejuni colonization of broiler chickens. Appl. Environ. Microbiol. 2005, 71, 6554–6563. [Google Scholar] [CrossRef]
- Smith, H.W.; Huggins, M.B.; Shaw, K.M. The Control of Experimental Escherichia coli Diarrhoea in Calves by Means of Bacteriophages. Microbiology 2018, 133, 1111–1126. [Google Scholar] [CrossRef] [PubMed]
- Huff, W.E.; Huff, G.R.; Rath, N.C.; Balog, J.M.; Donoghue, A.M. Bacteriophage Treatment of a Severe Escherichia coli Respiratory Infection in Broiler Chickens. Avian Dis. 2003, 47, 1399–1405. [Google Scholar] [CrossRef] [PubMed]
- Budiati, T.; Rusul, G.; Wan-Abdullah, W.N.; Arip, Y.M.; Ahmad, R.; Thong, K.L. Prevalence, antibiotic resistance and plasmid profiling of Salmonella in catfish (Clarias gariepinus) and tilapia (Tilapia mossambica) obtained from wet markets and ponds in Malaysia. Aquaculture 2013, 372–375, 127–132. [Google Scholar] [CrossRef]
- Shanmugasamy, M.; Velayutham, T.; Rajeswar, J. Inv A gene specific PCR for detection of Salmonella from broilers. Vet. World 2011, 4, 562. [Google Scholar] [CrossRef]
- Biemer, J.J. Antimicrobial susceptibility testing by the Kirby-Bauer disc diffusion method. Ann. Clin. Lab. Sci. 1973, 3, 135–140. [Google Scholar]
- Frye, J.G.; Jackson, C.R. Genetic mechanisms of antimicrobial resistance identified in Salmonella enterica, Escherichia coli, and Enteroccocus spp. isolated from U.S. food animals. Front. Microbiol. 2013, 4, 135. [Google Scholar] [CrossRef]
- Ramachandran, A.; Shanthi, M.; Sekar, U. Detection of blaCTX-Mextended spectrum betalactamase producing salmonella enterica serotype typhi in a tertiary care centre. J. Clin. Diagn. Res. 2017, 11, DC21–DC24. [Google Scholar] [CrossRef]
- El-Tayeb, M.A.; Ibrahim, A.S.S.; Al-Salamah, A.A.; Almaary, K.S.; Elbadawi, Y.B. Prevalence, serotyping and antimicrobials resistance mechanism of Salmonella enterica isolated from clinical and environmental samples in Saudi Arabia. Braz. J. Microbiol. 2017, 48, 499–508. [Google Scholar] [CrossRef]
- Bao, H.; Zhang, H.; Wang, R. Isolation and characterization of bacteriophages of Salmonella enterica serovar Pullorum. Poult. Sci. 2011, 10, 2370–2377. [Google Scholar] [CrossRef]
- Cormier, J.; Janes, M. A double layer plaque assay using spread plate technique for enumeration of bacteriophage MS2. J. Virol. Methods 2014, 196, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; McDonald, R.J. Genomic structure of phage F22, a hybrid between serologically and morphologically unrelated Salmonella typhimurium bacteriophages P22 and Fels 2. Genet. Res. 1986, 48, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Capra, M.L.; Quiberoni, A.; Reinheimer, J. Phages of Lactobacillus casei/paracasei: Response to environmental factors and interaction with collection and commercial strains. J. Appl. Microbiol. 2006, 100, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Kropinski, A.M. Practical Advice on the One-Step Growth Curve. Methods Mol. Biol. 2018, 1681, 41–47. [Google Scholar] [CrossRef]
- Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of Bacteriophages Using the Small Drop Plaque Assay System. Bacteriophages Methods Mol. Biol. 2009, 501, 81–85. [Google Scholar] [CrossRef]
- O’Flynn, G.; Coffey, A.; Fitzgerald, G.F.; Ross, R.P. The newly isolated lytic bacteriophages st104a and st104b are highly virulent against Salmonella enterica. J. Appl. Microbiol. 2006, 101, 251–259. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree, a Graphical Viewer of Phylogenetic Trees and as a Program for Producing Publication-Ready Figures. In Molecular Evolution, Phylogenetics and Epidemiology; 2006; Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 4 January 2022).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Chevenet, F.; Brun, C.; Bañuls, A.-L.; Jacq, B.; Christen, R. TreeDyn: Towards dynamic graphics and annotations for analyses of trees. BMC Bioinform. 2006, 7, 439. [Google Scholar] [CrossRef]
- King, G.; Murray, N.E. Restriction alleviation and modification enhancement by the Rac prophage of Escherichia coli K-12. Mol. Microbiol. 1995, 16, 769–777. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Sullivan, M.B.; Knezevic, P.; van Zyl, L.; Sarkar, B.L.; Dutilh, B.E.; Alfenas-Zerbini, P.; Łobocka, M.; Tong, Y.; Brister, J.R. Taxonomy of prokaryotic viruses: 2018-2019 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2020, 165, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Brister, J.R. How to name and classify your phage: An informal guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- Shin, H.; Lee, J.-H.; Kim, Y.; Ryu, S. Complete genome sequence of Cronobacter sakazakii bacteriophage CR3. Am. Soc. Microbiol. 2012, 86, 6367–6368. [Google Scholar] [CrossRef][Green Version]
- Toribio, A.L.; Pickard, D.; Cerdeño-Tárraga, A.M.; Petty, N.K.; Thomson, N.; Salmond, G.; Dougan, G. Complete genome sequences of two Citrobacter rodentium bacteriophages, CR8 and CR44b. Genome Announc. 2014, 2, e00146-14. [Google Scholar] [CrossRef] [PubMed]
- Buttimer, C.; Lynch, C.; Hendrix, H.; Neve, H.; Noben, J.-P.; Lavigne, R.; Coffey, A. Isolation and characterization of Pectobacterium phage vB\_PatM\_CB7: New insights into the genus Certrevirus. Antibiotics 2020, 9, 352. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, W.I.; Kwon, Y.C.; Cha, K.E.; Kim, M.; Myung, H. A newly isolated bacteriophage, PBES 02, infecting Cronobacter sakazakii. J. Microbiol. Biotechnol. 2016, 26, 1629–1635. [Google Scholar] [CrossRef]
- Delesalle, V.A.; Tanke, N.T.; Vill, A.C.; Krukonis, G.P. Testing hypotheses for the presence of tRNA genes in mycobacteriophage genomes. Bacteriophage 2016, 6, e1219441. [Google Scholar] [CrossRef]
- Bailly-Bechet, M.; Vergassola, M.; Rocha, E. Causes for the intriguing presence of tRNAs in phages. Genome Res. 2007, 17, 1486–1495. [Google Scholar] [CrossRef]
- Kelly, D.; McAuliffe, O.; O’Mahony, J.; Coffey, A. Development of a broad-host-range phage cocktail for biocontrol. Bioeng. Bugs 2011, 2, 31–37. [Google Scholar] [CrossRef]
- Chaturongakul, S.; Ounjai, P. Phage–host interplay: Examples from tailed phages and Gram-negative bacterial pathogens. Front. Microbiol. 2014, 5, 442. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | Position | ORF | Map Region | Total AA | Putative Function | Query Coverage | Similarity | GB Ac. No. | Organism |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 45,076–46,560 | 80 | II | 494 AA | Phage terminase, large subunit, PBSX family TC | 99% | 85.63% | YP_006383016.1 | Cronobacter phage CR3 |
| 2 | 46,576–48,087 | 81 | II | 503 AA | Putative portal protein | 99% 99% | 77.51% 76.53% | YP_009014964.1 QQG33308.1 | Cronobacter phage CR9 Pectobacterium phage PcCB7 V |
| 3 | 48,156–48,733 | 82 | II | 191 AA | Putative prohead protease | 98% 98% | 77.37% 78.42% | QUL77265.1 YP_009014965.1 | Escherichia phage UPEC06 Cronobacter phage CR9 |
| 4 | 49,814–50,248 | 84 | II | 144 AA | putative head stabilization/decoration protein | 99% | 65.36% 68.06% 66.67% | QEG12074.1 QUL77267.1 YP_009014967.1 | Klebsiella phage vB_KaeM_KaOmega Escherichia phage UPEC06 Cronobacter phage CR9 |
| 5 | 50,272–51,264 | 85 | II | 330 AA | putative major capsid protein | 99% | 81.21% | YP_009014968.1 ATS9340 | Cronobacter phage CR9 Pectobacterium phage DU_PP_I |
| 6 | 51,998–55,276 | 89 | II | 1092 AA | Virion structural protein/ Putative tail fiber protein | 99% | 61.63% 60.54 | ARB11484.1 YP_009014970.1 | Pectobacterium phage vB_PatM_CB7 Cronobacter phage CR9 |
| 7 | 55,317–55,967 | 90 | II | 216 AA | Hypothetical protein (Putative structural protein tail fiber protein collagen triple helix) | 99% | 60.19% 53.46% | ATS9340 ARB11485.1 | Pectobacterium phage DU_PP_I Pectobacterium phage vB_PatM_CB7 |
| 8 | 55,969–56,316 | 92 | II | 115 AA | Hypothetical protein putative tail fiber protein | 92% | 57.01% | YP_009014972.1 | Cronobacter phage CR9 |
| 9 | 56,372–57,160 | 93 | II | 262 AA | hypothetical protein tail fiber like protein | 91% | 51.89% | YP_006383026.1 | Cronobacter phage CR3 |
| 10 | 57,747–58,277 | 94 | II | 176 AA | hypothetical protein CR8 head completion adaptor | 98% | 72.57% | YP_009042249.1 | Cronobacter phage CR8 |
| 11 | 58,340–58,801 | 95 | II | 153 AA | Putative RNA polymerase/virion morphogenesis protein | 99% | 75.82% 75.16% | ATS93412.1 YP_009014974.1 | Pectobacterium phage DU_PP_I Cronobacter phage CR9 |
| 12 | 58,816–59,256 | 96 | II | 146 AA | hypothetical protein Putative minor capsid protein | 99% 99% | 75.34% 74.66% | YP_007392682.1 DAM41403.1 | Pectobacterium phage phiTE Myoviridae sp. |
| 13 | 59,256–59,834 | 97 | II | 192 AA | hypothetical protein putative tail to head joining protein | 95% 90% | 64.13% 66.68% | YP_006383030.1 QUL77277.1 | Cronobacter phage CR3 Escherichia phage UPEC06 |
| 14 | 59,938–61,353 | 98 | II | 471 AA | Putative structural protein 1 (probable tail sheath protein) | 99% | 78.09% 77.87% | YP_006383031.1 YP_009042253.1 | Cronobacter phage CR3 Cronobacter phage CR8 |
| 15 | 61,357–61,839 | 99 | II | 160 AA | Hypothetical protein putative tail tube protein | 96% | 76.28% | YP_009014978.1 | Cronobacter phage CR9 |
| 16 | 61,916–62,389 | 100 | II | 157 AA | Tail assembly chaperon protein | 96% | 74.51% | YP_009042255.1 | Cronobacter phage CR8 |
| 17 | 62,707–65,178 | 101 | II | 823 AA | Tail tape measure domain (controls tail length) | 99% 84% | 55.58% 60.96% | QEG12088.1 ATS93421.1 | Klebsiella phage vB_KaeM_KaOmega Pectobacterium phage DU_PP_I |
| 18 | 65,246–60,127 | 102 | II | 293 AA | Putative tail tape measure protein | 98% | 61.17% | QUL77284.1 | Escherichia phage UPEC06 |
| 19 | 66,483–67,451 | 104 | II | 322 AA | Putative tail protein CR9 | 99% | 73.62% | YP_009014984.1 | Cronobacter phage CR9 |
| 20 | 67,461–68,108 | 105 | II | 215 AA | Putative base plate assembly protein | 97% | 66.23% | QEG12092.1 | Klebsiella phage vB_KaeM_KaOmega |
| 21 | 68,118–68,642 | 106 | II | 174 AA | Putative tail lysozyme/part of base plate wedge protein | 99% | 72.32% 71.19% | QEG12093.1 ARB11502.1 | Klebsiella phage vB_KaeM_KaOmega Pectobacterium phage vB_PatM_CB7 |
| 22 | 68,737–70,224 | 107 | II | 495 AA | Putative base plate assembly protein | 99% | 76.97% | YP_009188994.1 | Cronobacter phage PBES 02 |
| 23 | 70,236–70,883 | 108 | II | 215 AA | Putative base plate wedge protein | 97% 97% | 81.99% 78.20% | YP_009014988.1 QQG33332.1 | Cronobacter phage CR9 Pectobacterium phage PcCB7V |
| 24 | 70,897–72,417 | 109 | II | 506 AA | XXXCH domain containing protein (Putative tail fiber domain) | 99% | 50.90% 50.09% | QEG12096.1 YP_009042265.1 | Klebsiella phage vB_KaeM_KaOmega Cronobacter phage CR8 |
| 25 | 72,420–72,935 | 110 | II | 171 AA | Putative tail fiber assembly protein | 98% 98% | 59.76% 57.65% | QUL77293.1 QEG12097.1 | Escherichia phage UPEC06 Klebsiella phage vB_KaeM_KaOmega |
| 26 | 73,313–73,774 | 112 | II | 153 AA | Putative membrane protein | 99% | 57.52% 55.56% | QEG12099.1 YP_009042268.1 | Klebsiella phage vB_KaeM_KaOmega Cronobacter phage CR8 |
| 27 | 73,794–74,066 | 113 | II | 90 AA | Putative membrane protein | 91% | 73.49% | YP_009851591.1 | Erwinia phage pEp_SNUABM_01 |
| 28 | 74,059–77,580 | 114 | II | 1173 AA | Putative tail fiber protein 2 | 99% 99% | 32.98% 33.92% | QQG33338.1 YP_009189001.1 | Pectobacterium phage PcCB7V Cronobacter phage PBES |
| Phage | Total ORF | Genome Size (bp) | G + C Content (%) | Accession No. | Querry Coverage | Shared Proteins (%) ** |
|---|---|---|---|---|---|---|
| Identity (%) * | ||||||
| SE-Phage-SSBI34 | 234 | 141,095 | 44% | MZ520832 | 100 | 100% |
| 100 | ||||||
| Klebsiella phage vB_KaeM_KaOmega, | 317 | 149,489 | 50.5 | MN013077.1 | 7% | 8.11% |
| 77.94% | ||||||
| Cronobacter phage CR9 | 281 | 151,924 | 50.6 | JQ691611.1 | 5% | 17.52% |
| 74.78% | ||||||
| Pectobacterium phage DU_PP_I | 267 | 144,959 | 50.1 | MF979560.1 | 5% | 2.99% |
| 77.25% | ||||||
| Pectobacterium phage PcCB7V | 269 | 146,054 | 50.4% | MW367417.1 | 4% | 6% |
| 77.78% | ||||||
| Cronobacter phage PBES 02 | 270 | 149,732 | 50.7 | KT353109.1 | 4% | 5.55% |
| 76.56% | ||||||
| Cronobacter phage CR8, | 269 | 149,162 nt | 50.8 | KC954774.1 | 4% | 7.69% |
| 76.44% | ||||||
| Cronobacter phage CR3 | 265 | 149,273 | 50.9 | JQ691612.1 | 4% | 15% |
| 76.46% | ||||||
| Pectobacterium phage DU_PP_IV | 268 | 145,233 | 50.3 | MF979563.1 | 3% | 0 |
| 76.49% | ||||||
| Pectobacterium phage phiTE | 242 | 142,349 | 50.1 | JQ015307.1 | 2% | 6.83% |
| 76.23% | ||||||
| Acinetobacter phage ABPH49 | 252 | 149,960 | 50.8 | MH533020.1 | 1% | 0.85% |
| 72.87% | ||||||
| Escherichia phage UPEC06 | 318 | 143,140 | 41.2 | MW250786.1 | 1% | 8.54% |
| 76.50% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sattar, S.; Ullah, I.; Khanum, S.; Bailie, M.; Shamsi, B.; Ahmed, I.; Abbas Shah, T.; Javed, S.; Ghafoor, A.; Pervaiz, A.; et al. Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae. Viruses 2022, 14, 241. https://doi.org/10.3390/v14020241
Sattar S, Ullah I, Khanum S, Bailie M, Shamsi B, Ahmed I, Abbas Shah T, Javed S, Ghafoor A, Pervaiz A, et al. Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae. Viruses. 2022; 14(2):241. https://doi.org/10.3390/v14020241
Chicago/Turabian StyleSattar, Sadia, Inam Ullah, Sofia Khanum, Marc Bailie, Bushra Shamsi, Ibrar Ahmed, Tahir Abbas Shah, Sundus Javed, Aamir Ghafoor, Amna Pervaiz, and et al. 2022. "Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae" Viruses 14, no. 2: 241. https://doi.org/10.3390/v14020241
APA StyleSattar, S., Ullah, I., Khanum, S., Bailie, M., Shamsi, B., Ahmed, I., Abbas Shah, T., Javed, S., Ghafoor, A., Pervaiz, A., Sohail, F., Imdad, K., Tariq, A., Bostan, N., Ali, I., & Altermann, E. (2022). Genome Analysis and Therapeutic Evaluation of a Novel Lytic Bacteriophage of Salmonella Typhimurium: Suggestive of a New Genus in the Subfamily Vequintavirinae. Viruses, 14(2), 241. https://doi.org/10.3390/v14020241