

Monitoring the Establishment of VOC Gamma in Minas Gerais, Brazil: A Retrospective Epidemiological and Genomic Surveillance Study
 ,
,  , , , , , , , , , , ,
, , , , , , , , , , ,  , , , , and add
Show full author list
, , , , and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
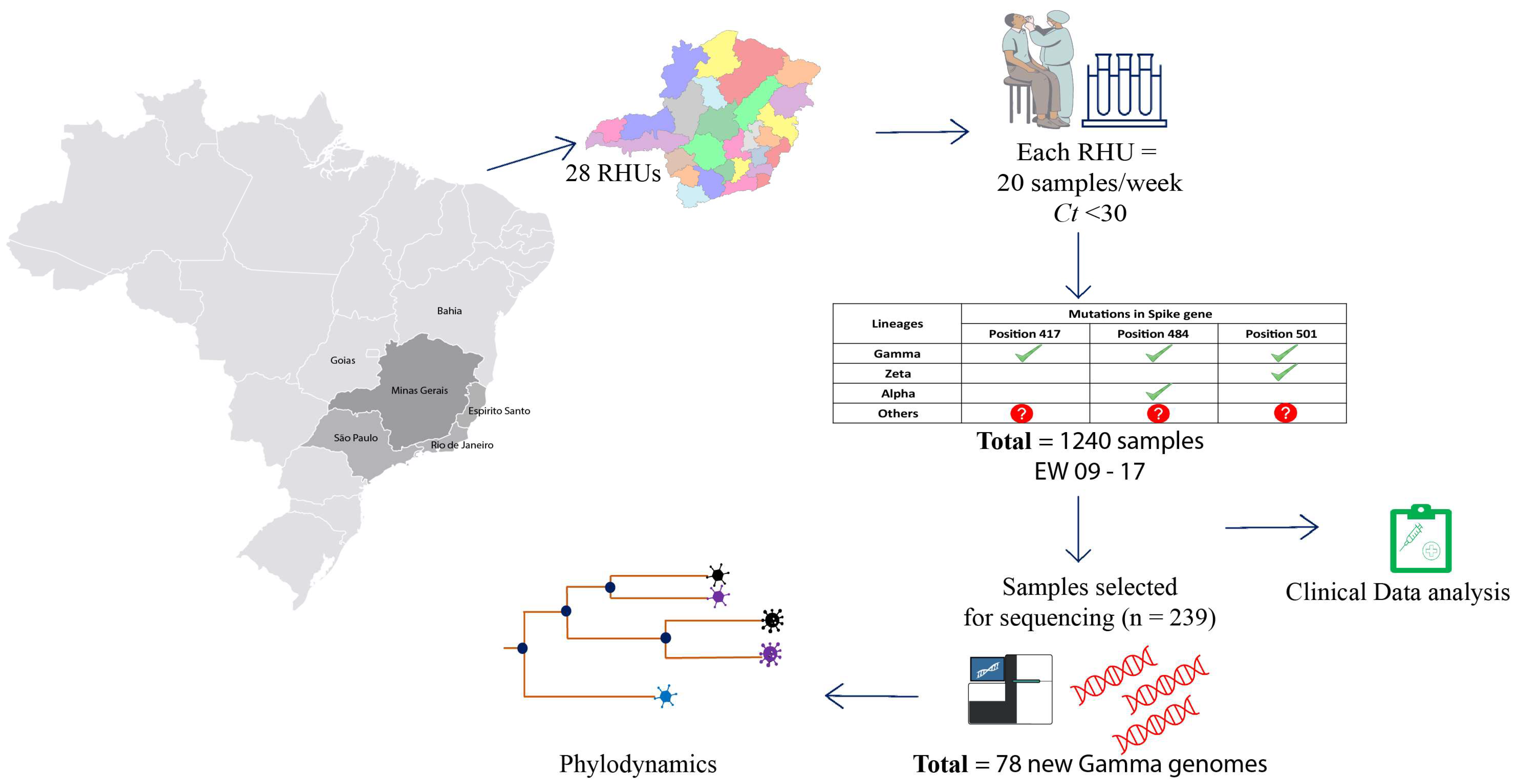
2.1. Study Design
2.2. Epidemiological Data and Clinical Outcome Analysis
2.3. cDNA Synthesis and rhAmp Genotyping Assay
2.4. Library Preparation and Genome Sequencing
2.5. SARS-CoV-2 Genome Assembly and Lineage Classification
2.6. Phylogenetic and Phylodynamic Analysis of VOCs in Minas Gerais
3. Results
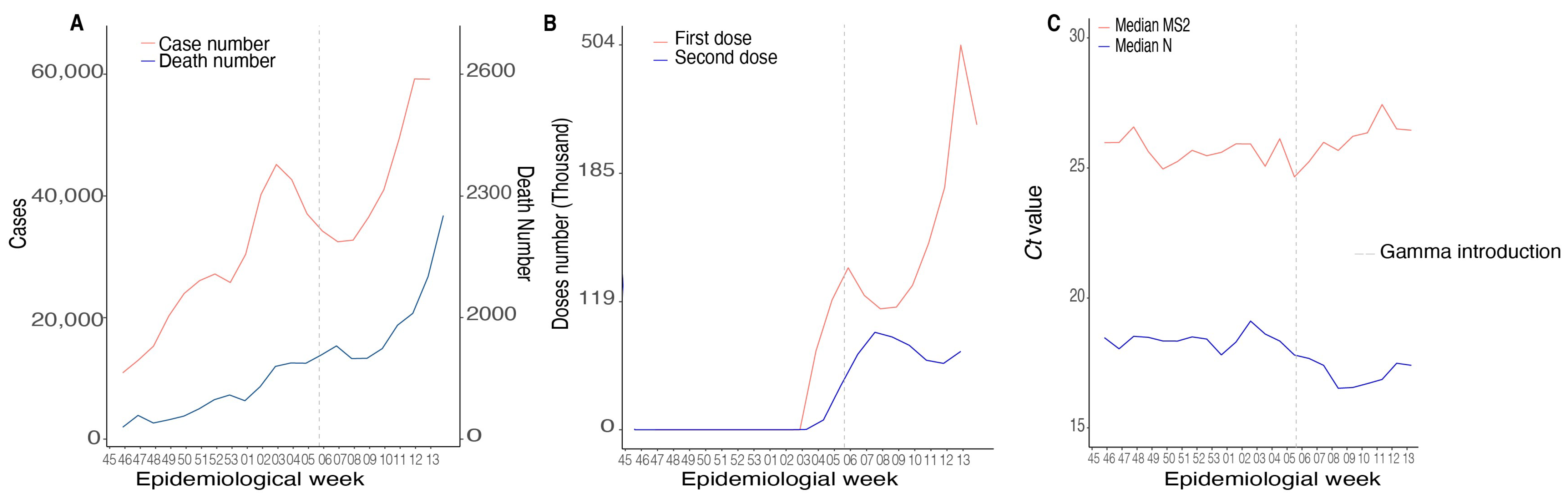
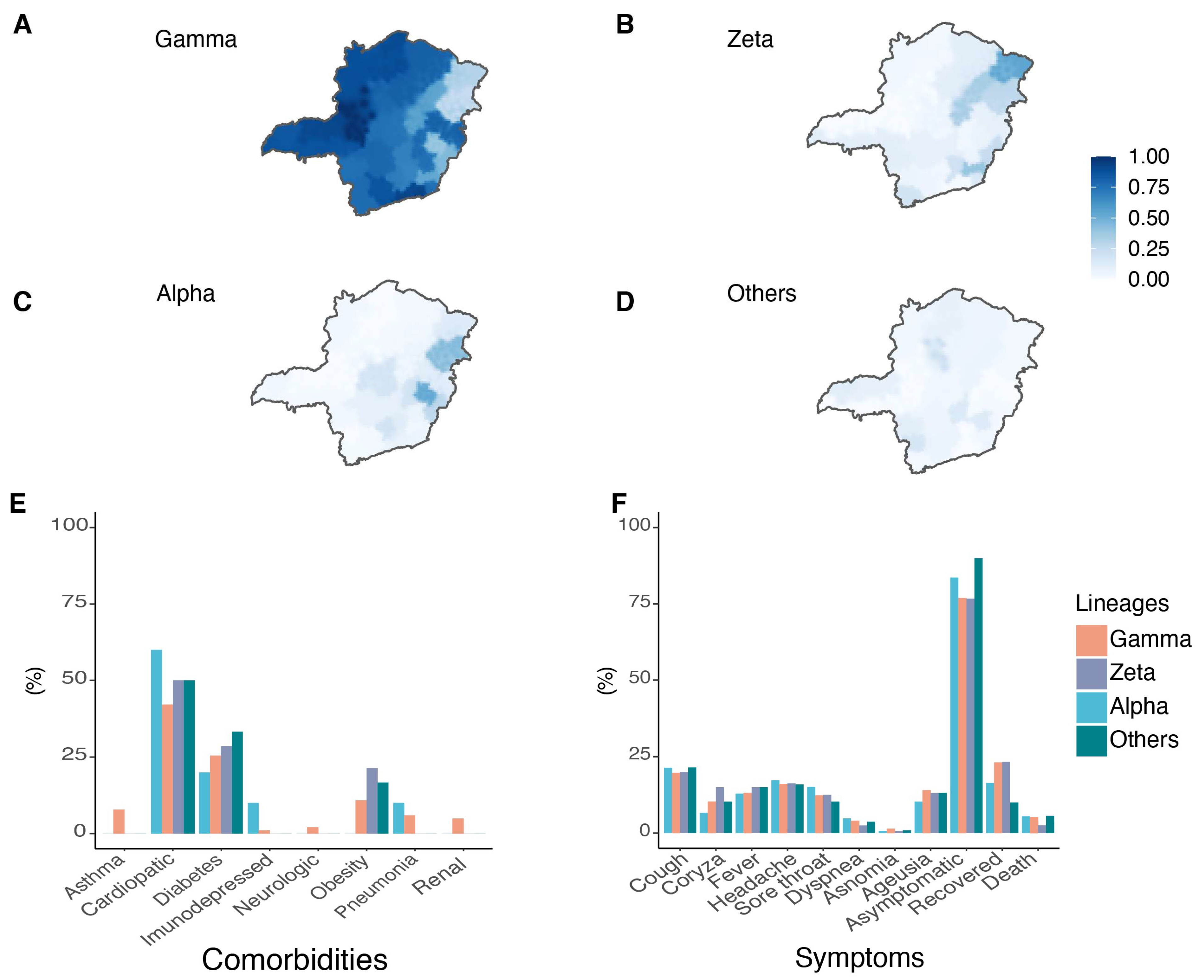
3.1. Epidemiological Data and Clinical Aspects of SARS-CoV-2 in MG State
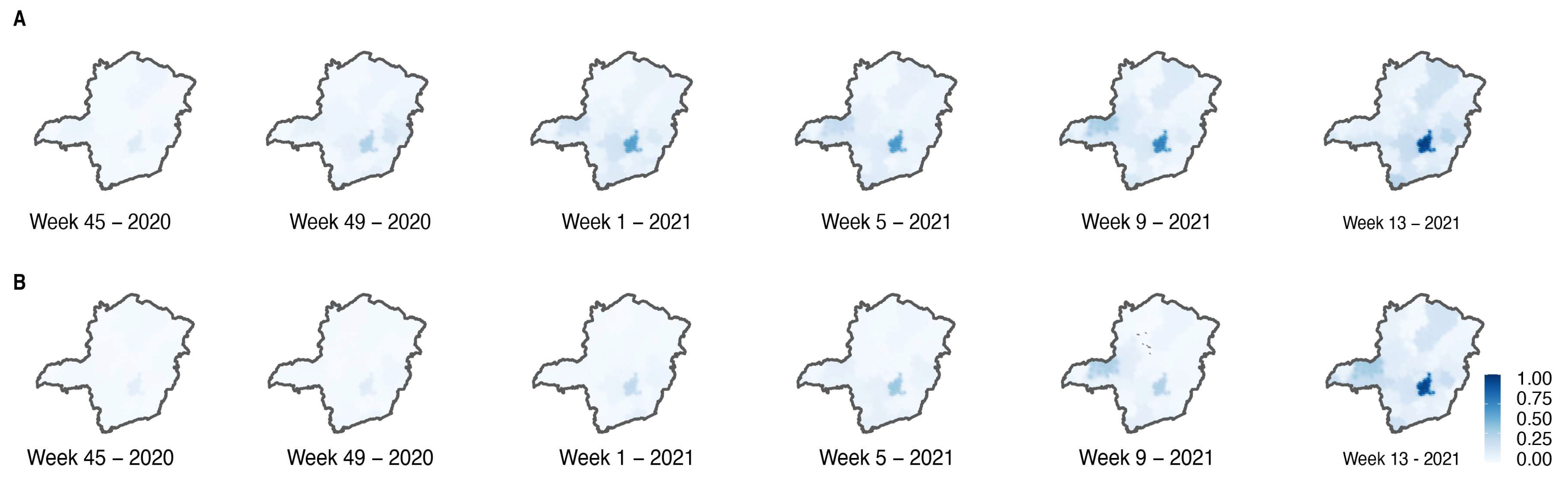
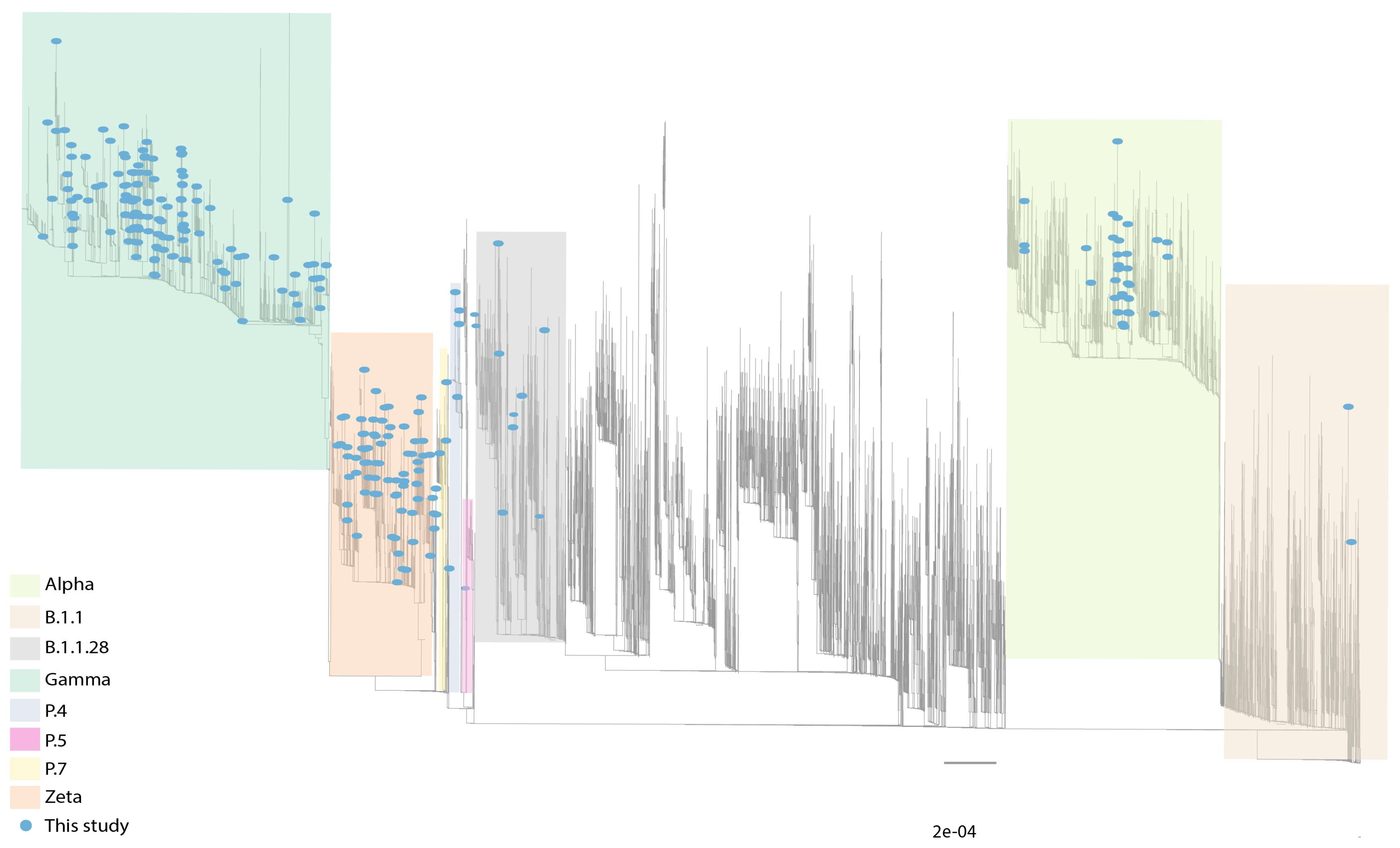
3.2. Circulation of SARS-CoV-2 Variants in MG State
3.3. Sequencing Results
3.4. Circulation of Other SARS-CoV-2 Lineages in MG
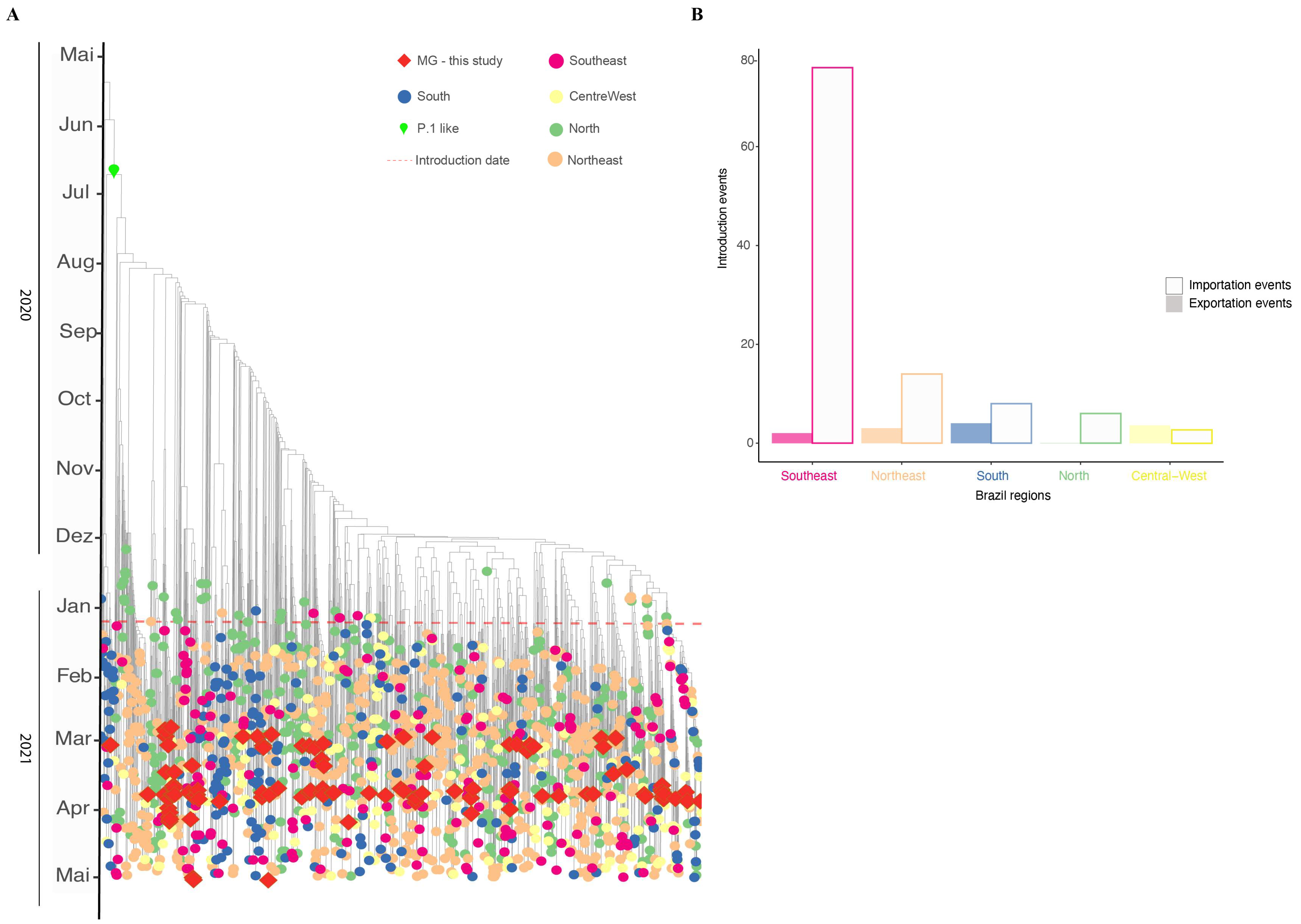
3.5. Phylodynamics of VOC Gamma Introduction into MG
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. WHO COVID-19 Weekly Epidemiological Update; World Health Organization: Geneva, Switzerland, 2022; pp. 1–23. [Google Scholar]
- Brizzi, A.; Whittaker, C.; Servo, L.M.S.; Hawryluk, I.; Prete, C.A.J.; de Souza, W.M.; Aguiar, R.S.; Araujo, L.J.T.; Bastos, L.S.; Blenkinsop, A.; et al. Spatial and Temporal Fluctuations in COVID-19 Fatality Rates in Brazilian Hospitals. Nat. Med. 2022, 28, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Lai, S.; Gao, G.F.; Shi, W. The Emergence, Genomic Diversity and Global Spread of SARS-CoV-2. Nature 2021, 600, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Gidari, A.; Sabbatini, S.; Bastianelli, S.; Pierucci, S.; Busti, C.; Monari, C.; Luciani Pasqua, B.; Dragoni, F.; Schiaroli, E.; Zazzi, M.; et al. Cross-Neutralization of SARS-CoV-2 B.1.1.7 and P.1 Variants in Vaccinated, Convalescent and P.1 Infected. J. Infect. 2021, 83, 467–472. [Google Scholar] [CrossRef]
- Naveca, F.G.; Claro, I.; Giovanetti, M.; de Jesus, J.G.; Xavier, J.; Iani, F.C.d.M.; do Nascimento, V.A.; de Souza, V.C.; Silveira, P.P.; Lourenço, J.; et al. Genomic, Epidemiological and Digital Surveillance of Chikungunya Virus in the Brazilian Amazon. PLoS Negl. Trop. Dis. 2019, 13, e0007065. [Google Scholar] [CrossRef] [PubMed]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. SARS-CoV-2 Spike Mutations, L452R, T478K, E484Q and P681R, in the Second Wave of COVID-19 in Maharashtra, India. Microorganisms 2021, 9, 1542. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.d.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Volz, E.; Mishra, S.; Chand, M.; Barrett, J.C.; Johnson, R.; Geidelberg, L.; Hinsley, W.R.; Laydon, D.J.; Dabrera, G.; O’Toole, Á.; et al. Assessing Transmissibility of SARS-CoV-2 Lineage B.1.1.7 in England. Nature 2021, 593, 266–269. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 Variant: Unique Features and Their Impact on Pre-Existing Antibodies. J. Autoimmun. 2022, 126, 102779. [Google Scholar] [CrossRef]
- Claro, I.M.; da Silva Sales, F.C.; Ramundo, M.S.; Candido, D.S.; Silva, C.A.M.; de Jesus, J.G.; Manuli, E.R.; de Oliveira, C.M.; Scarpelli, L.; Campana, G.; et al. Local Transmission of SARS-CoV-2 Lineage B.1.1.7, Brazil, December 2020. Emerg. Infect. Dis. 2021, 27, 970–972. [Google Scholar] [CrossRef]
- Slavov, S.N.; Giovanetti, M.; Dos Santos Bezerra, R.; Fonseca, V.; Santos, E.V.; Rodrigues, E.S.; Adelino, T.; Xavier, J.; Borges, J.S.; Evaristo, M.; et al. Molecular Surveillance of the On-Going SARS-COV-2 Epidemic in Ribeirao Preto City, Brazil. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 93, 104976. [Google Scholar] [CrossRef] [PubMed]
- Voloch, C.M.; da Silva Francisco, R.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.d.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic Characterization of a Novel SARS-CoV-2 Lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95, e00119-21. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.; Mojsiejczuk, L.; Acuña, D.; Alexay, S.; Amadio, A.; Aulicino, P.; Debat, H.; Fay, F.; Fernández, F.; Giri, A.A.; et al. Cost-Effective Method to Perform SARS-CoV-2 Variant Surveillance: Detection of Alpha, Gamma, Lambda, Delta, Epsilon, and Zeta in Argentina. Front. Med. 2021, 8, 755463. [Google Scholar] [CrossRef]
- Paulo, S.; Nanev, S.; Bezerra, S.; Strazza, E.; Vieira, E.; Serrano, J.; De, L.; Ribeiro, G.; Patan, L.; Ranieri, A.; et al. Genomic Monitoring of the SARS-CoV-2 B1.1.7 (WHO VOC Alpha) in The. Virus Res. 2022, 308, 198643. [Google Scholar] [CrossRef]
- Castro, M.C.; Kim, S.; Barberia, L.; Ribeiro, A.F.; Gurzenda, S.; Ribeiro, K.B.; Abbott, E.; Blossom, J.; Rache, B.; Singer, B.H. Spatiotemporal Pattern of COVID-19 Spread in Brazil. Science 2021, 372, 821–826. [Google Scholar] [CrossRef] [PubMed]
- OPAS. Alerta Epidemiológico COVID-19: Aumento de Hospitalizações e Mortes Entre Pacientes Com Menos de 60 Anos de Idade. 26 de abril de 2021; Organização Pan-Americana da Saúde: Washington, DC, USA, 2021. [Google Scholar]
- Brüssow, H.; Zuber, S. Can a Combination of Vaccination and Face Mask Wearing Contain the COVID-19 Pandemic? Microb. Biotechnol. 2022, 15, 721–737. [Google Scholar] [CrossRef] [PubMed]
- Gräf, T.; Bello, G.; Venas, T.M.M.; Pereira, E.C.; Paixão, A.C.D.; Appolinario, L.R.; Lopes, R.S.; Mendonça, A.C.D.F.; da Rocha, A.S.B.; Motta, F.C.; et al. Identification of a Novel SARS-CoV-2 P.1 Sub-Lineage in Brazil Provides New Insights about the Mechanisms of Emergence of Variants of Concern. Virus Evol. 2021, 7, veab091. [Google Scholar] [CrossRef]
- Lionel, A.; Wickham, H.; Henry, M.L. CRAN, 2022-09-24 05:40:02 UTC. Package ‘Rlang.’ 2022. Available online: https://cran.r-project.org/web/packages/rlang/index.html (accessed on 27 November 2022).
- Almeida, R.; Arruda, D.; Nascimento, I.; Santiago, B.; Silva, W.; Andrade, P.R.; Kauê, C.; Braga, V. CRAN, 2022-08-16 07:50:02 UTC. Package ‘Geobr’ R Topics Documented. 2020. Available online: https://cran.r-project.org/web/packages/geobr/index.html (accessed on 27 November 2022).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2022; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 27 November 2022).
- Jason, A.; Brunson, C.; Read, Q.D. Package ‘Ggalluvial’; 2020; ISBN 0387286950. Available online: https://cran.r-project.org/web/packages/ggalluvial/index.html (accessed on 27 November 2022).
- Sumner, M.; Cook, I.; Baston, D. Package ‘Sf.’ 2022. Available online: https://cran.r-project.org/web/packages/sf/sf.pdf (accessed on 27 November 2022).
- Romero, F.; Moreira, R.; Mirela, D.; Mariani, D.; Herlinger, A.L.; Schiffler, F.B.; Rossi, Á.D.; Leitão, I.D.C.; Miranda, S.; Augusto, M.; et al. Epidemiological Dynamics of SARS-CoV-2 VOC Gamma in Rio de Janeiro, Brazil. Virus Evol. 2021, 7, veab087. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Tavare, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Some Math. Quest. Biol./DNA Seq. Anal. Ed. Robert M. Miura 1986, 57–86. Available online: https://agris.fao.org/agris-search/search.do?recordID=US201301755037 (accessed on 27 November 2022).
- Yang, Z. Maximum Likelihood Phylogenetic Estimation from DNA Sequences with Variable Rates over Sites: Approximate Methods. J. Mol. Evol. 1994, 39, 306–314. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; Corado, A.D.L.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P. 1 emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- Silva, J.P.; De Lima, A.B.; Alvim, L.B.; Mendonça, C.P.T.B.; Fonseca, P.L.C.; Moreira, F.R.R.; Queiroz, D.C.; Ferreira, J.G.G.; Ferreira, A.C.S.; Souza, R.P.; et al. Delta Variant of SARS-CoV-2 Replacement in Brazil: A National Epidemiologic Surveillance Program. Viruses 2022, 14, 847. [Google Scholar] [CrossRef]
- Carrillo-Larco, R.M.; Altez-Fernandez, C. Anosmia and Dysgeusia in COVID-19: A Systematic Review. Wellcome Open Res. 2020, 5, 94. [Google Scholar] [CrossRef]
- Izcovich, A.; Ragusa, M.A.; Tortosa, F.; Lavena Marzio, M.A.; Agnoletti, C.; Bengolea, A.; Ceirano, A.; Espinosa, F.; Saavedra, E.; Sanguine, V.; et al. Prognostic Factors for Severity and Mortality in Patients Infected with COVID-19: A Systematic Review. PLoS ONE 2020, 15, e0241955. [Google Scholar] [CrossRef] [PubMed]
- Oran, D.P.; Topol, E.J. The Proportion of SARS-CoV-2 Infections That Are Asymptomatic: A Systematic Review. Ann. Intern. Med. 2021, 174, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-D.; Ding, M.; Dong, X.; Zhang, J.-J.; Kursat Azkur, A.; Azkur, D.; Gan, H.; Sun, Y.-L.; Fu, W.; Li, W.; et al. Risk Factors for Severe and Critically Ill COVID-19 Patients: A Review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chi, J.; Lv, W.; Wang, Y. Obesity and Diabetes as High-Risk Factors for Severe Coronavirus Disease 2019 (COVID-19). Diabetes Metab. Res. Rev. 2021, 37, e3377. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Bittar, C.; Possebon, F.S.; Ullmann, L.S.; Geraldini, D.B.; da Costa, V.G.; de Almeida, L.G.P.; da S Sanches, P.R.; Nascimento-Júnior, N.M.; Cilli, E.M.; Artico Banho, C.; et al. The Emergence of the New P.4 Lineage of SARS-CoV-2 With Spike L452R Mutation in Brazil. Front. Public Health 2021, 9, 745310. [Google Scholar] [CrossRef]
- Francisco Junior, R.d.S.; Lamarca, A.P.; de Almeida, L.G.P.; Cavalcante, L.; Machado, D.T.; Martins, Y.; Brustolini, O.; Gerber, A.L.; Guimarães, A.P.d.C.; Gonçalves, R.B.; et al. Turnover of SARS-CoV-2 Lineages Shaped the Pandemic and Enabled the Emergence of New Variants in the State of Rio de Janeiro, Brazil. Viruses 2021, 13, 2013. [Google Scholar] [CrossRef]
- Sant’Anna, F.H.; Muterle Varela, A.P.; Prichula, J.; Comerlato, J.; Comerlato, C.B.; Roglio, V.S.; Mendes Pereira, G.F.; Moreno, F.; Seixas, A.; Wendland, E.M. Emergence of the Novel SARS-CoV-2 Lineage VUI-NP13L and Massive Spread of P.2 in South Brazil. Emerg. Microbes Infect. 2021, 10, 1431–1440. [Google Scholar] [CrossRef]
- Mitchell, P.K.; Martins, M.; Reilly, T.; Caserta, L.C.; Anderson, R.R.; Cronk, B.D.; Murphy, J.; Goodrich, E.L.; Diel, D.G. SARS-CoV-2 B.1.1.7 Variant Infection in Malayan Tigers, Virginia, USA. Emerg. Infect. Dis. 2021, 27, 3171–3173. [Google Scholar] [CrossRef]
- Washington, N.L.; Gangavarapu, K.; Zeller, M.; Bolze, A.; Cirulli, E.T.; Schiabor Barrett, K.M.; Larsen, B.B.; Anderson, C.; White, S.; Cassens, T.; et al. Emergence and Rapid Transmission of SARS-CoV-2 B.1.1.7 in the United States. Cell 2021, 184, 2587–2594.e7. [Google Scholar] [CrossRef]
- Campos, K.R.; Sacchi, C.T.; Abbud, A.; Caterino-de-Araujo, A. SARS-CoV-2 variants in severely symptomatic and deceased persons who had been vaccinated against COVID-19 in São Paulo, Brazil. Rev. Panam Salud Publica 2021, 45, e126. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.L.C.; Moreira, F.R.R.; de Souza, R.M.; Guimarães, N.R.; Carvalho, N.O.; Adelino, T.E.R.; Alves, H.J.; Alvim, L.B.; Candido, D.S.; Coelho, H.P.; et al. Tracking the Turnover of SARS-CoV-2 VOCs Gamma to Delta in a Brazilian State (Minas Gerais) with a High-Vaccination Status. Virus Evol. 2022, 8, veac064. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, D.C.; Carobin, N.V.; de Araújo e Santos, L.C.G.; Fonseca, P.L.C.; Braga-Paz, I.L.; Dias, R.C.; Ferreira, J.G.G.; Freitas, T.R.; Menezes, D.; Nolasco, S.C.V.M.; et al. SARS-CoV-2 Omicron BA.1, BA.2, and XAG Identification during Routine Surveillance on a University Campus in Belo Horizonte, Brazil, 2022. Braz. J. Microbiol. 2022, 54, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Chenchula, S.; Karunakaran, P.; Sharma, S.; Chavan, M. Current Evidence on Efficacy of COVID-19 Booster Dose Vaccination against the Omicron Variant: A Systematic Review. J. Med. Virol. 2022, 94, 2969–2976. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alves, H.J.; de Araújo, J.L.F.; Fonseca, P.L.C.; Moreira, F.R.R.; Bonfim, D.M.; Queiroz, D.C.; Miguita, L.; de Souza, R.M.; Geddes, V.E.V.; Costa, W.C.; et al. Monitoring the Establishment of VOC Gamma in Minas Gerais, Brazil: A Retrospective Epidemiological and Genomic Surveillance Study. Viruses 2022, 14, 2747. https://doi.org/10.3390/v14122747
Alves HJ, de Araújo JLF, Fonseca PLC, Moreira FRR, Bonfim DM, Queiroz DC, Miguita L, de Souza RM, Geddes VEV, Costa WC, et al. Monitoring the Establishment of VOC Gamma in Minas Gerais, Brazil: A Retrospective Epidemiological and Genomic Surveillance Study. Viruses. 2022; 14(12):2747. https://doi.org/10.3390/v14122747
Chicago/Turabian StyleAlves, Hugo José, João Locke Ferreira de Araújo, Paula Luize Camargos Fonseca, Filipe Romero Rebello Moreira, Diego Menezes Bonfim, Daniel Costa Queiroz, Lucyene Miguita, Rafael Marques de Souza, Victor Emmanuel Viana Geddes, Walyson Coelho Costa, and et al. 2022. "Monitoring the Establishment of VOC Gamma in Minas Gerais, Brazil: A Retrospective Epidemiological and Genomic Surveillance Study" Viruses 14, no. 12: 2747. https://doi.org/10.3390/v14122747
APA StyleAlves, H. J., de Araújo, J. L. F., Fonseca, P. L. C., Moreira, F. R. R., Bonfim, D. M., Queiroz, D. C., Miguita, L., de Souza, R. M., Geddes, V. E. V., Costa, W. C., Oliveira, J. S. d., Medeiros, E. L. A., Souza, C. S. A. d., Saliba, J. W., Menezes, A. L., Oliveira, E. S. d., Adelino, T. E. R., Guimaraes, N. R., Ribeiro, A. A., ... Aguiar, R. S. (2022). Monitoring the Establishment of VOC Gamma in Minas Gerais, Brazil: A Retrospective Epidemiological and Genomic Surveillance Study. Viruses, 14(12), 2747. https://doi.org/10.3390/v14122747