
A Characterization and an Evolutionary and a Pathogenicity Analysis of Reassortment H3N2 Avian Influenza Virus in South China in 2019–2020



Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Virus Isolation
2.2. Whole-Genome Sequencing of AIV Isolates
2.3. Reference Strains Information
2.4. Phylogenetic Analyses
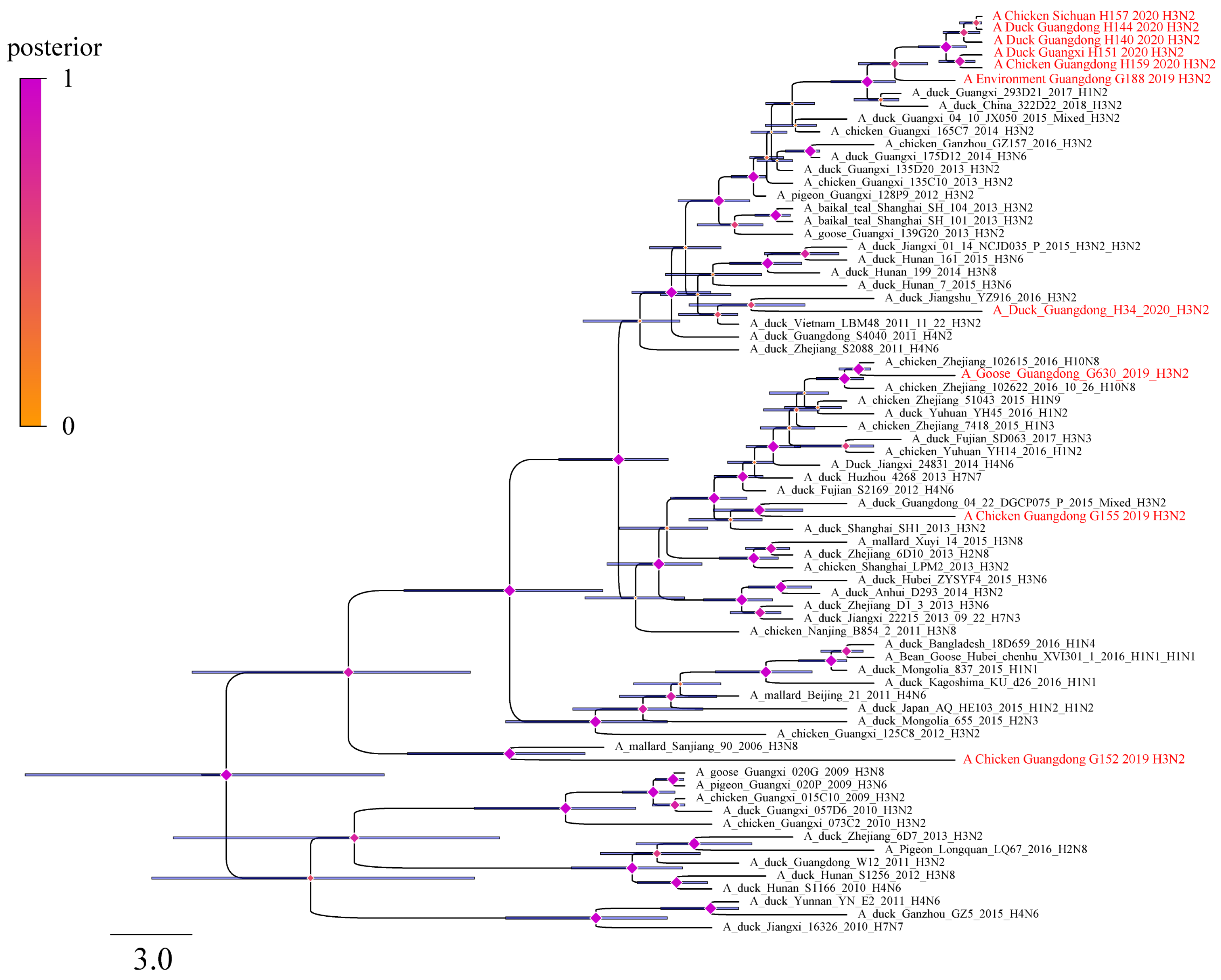
2.5. Estimating Substitution Rates
2.6. Amino Acid Analysis
2.7. Pathogenicity Test of BALB/c-Mice
3. Results
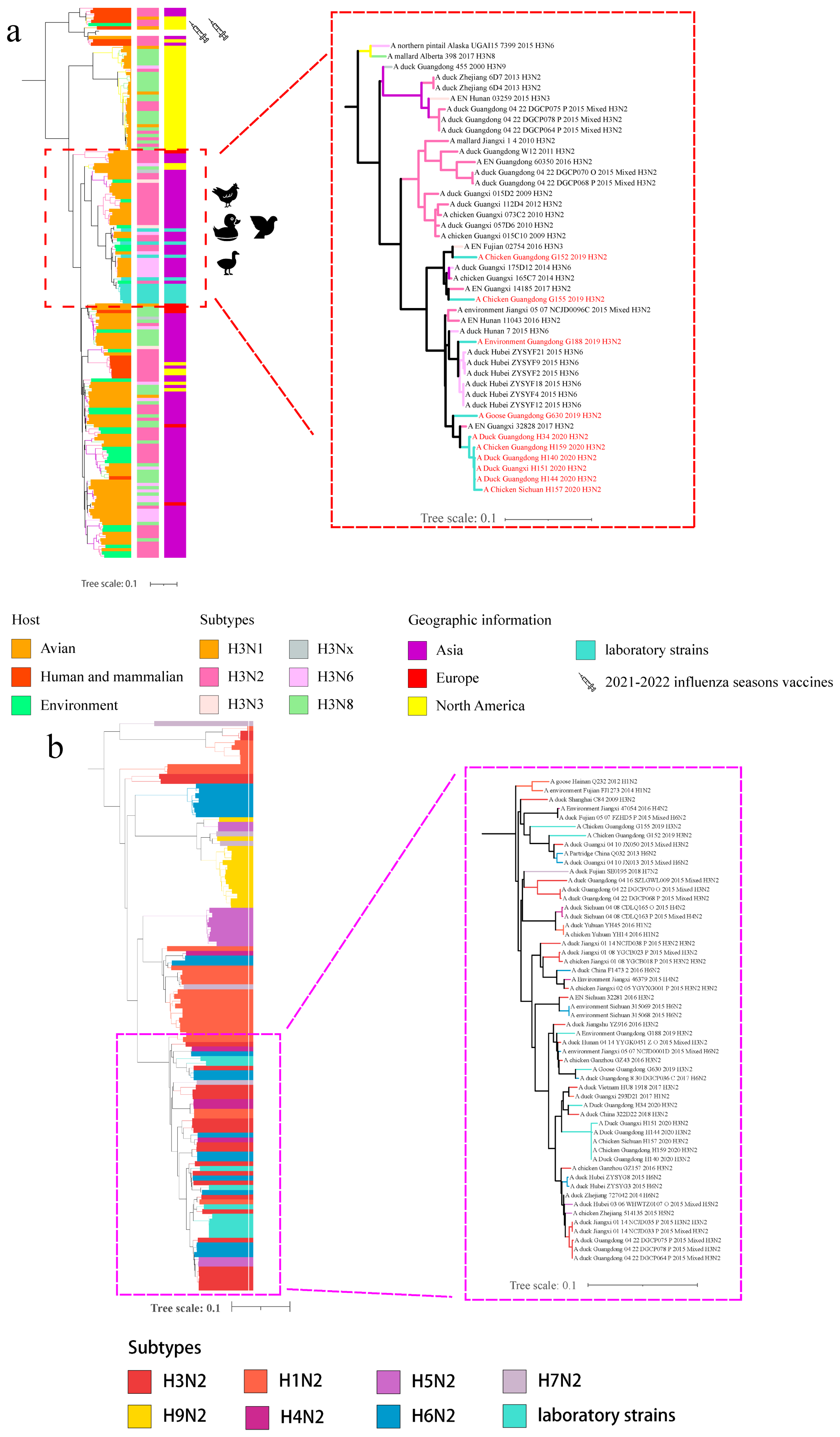
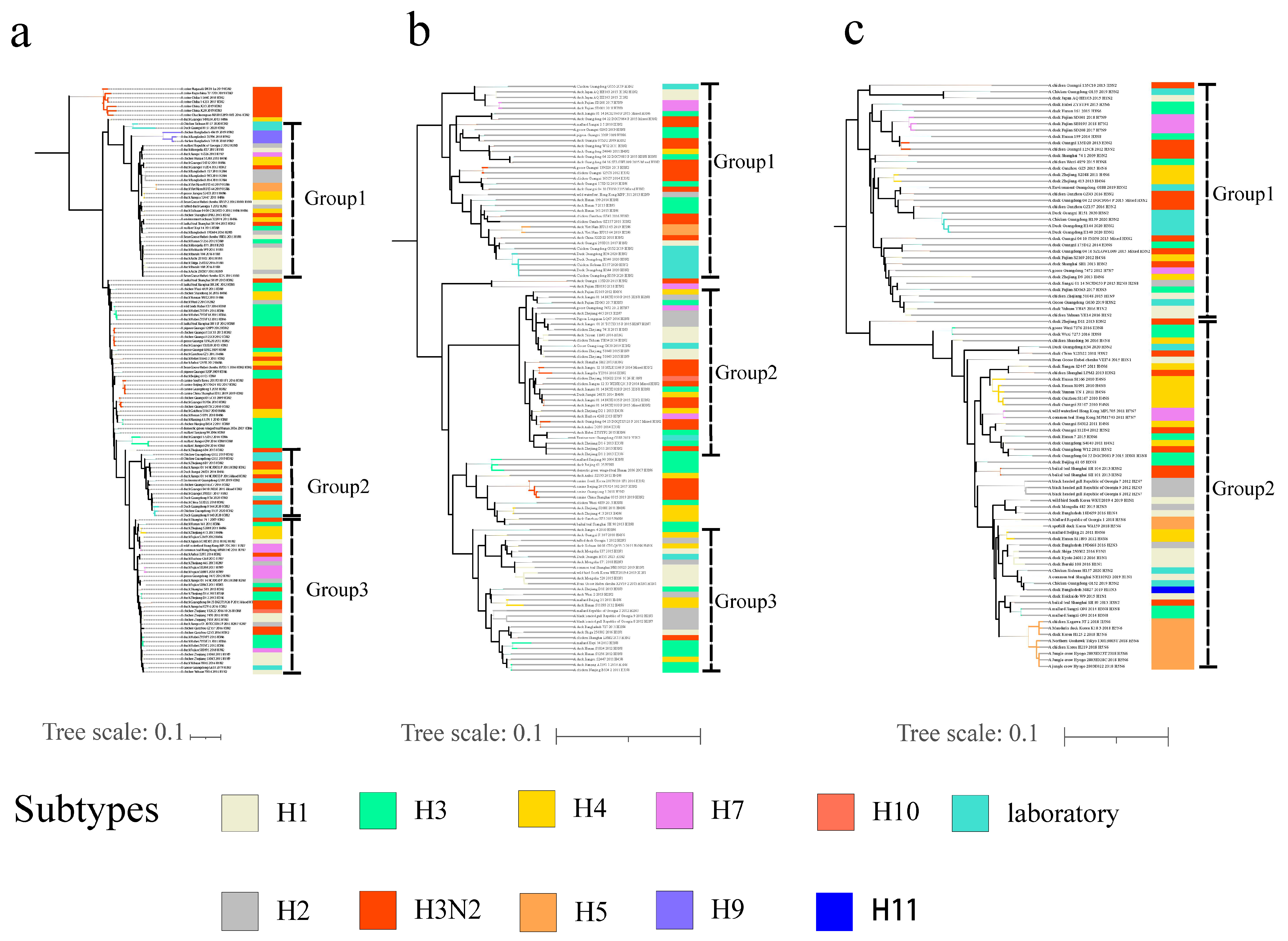
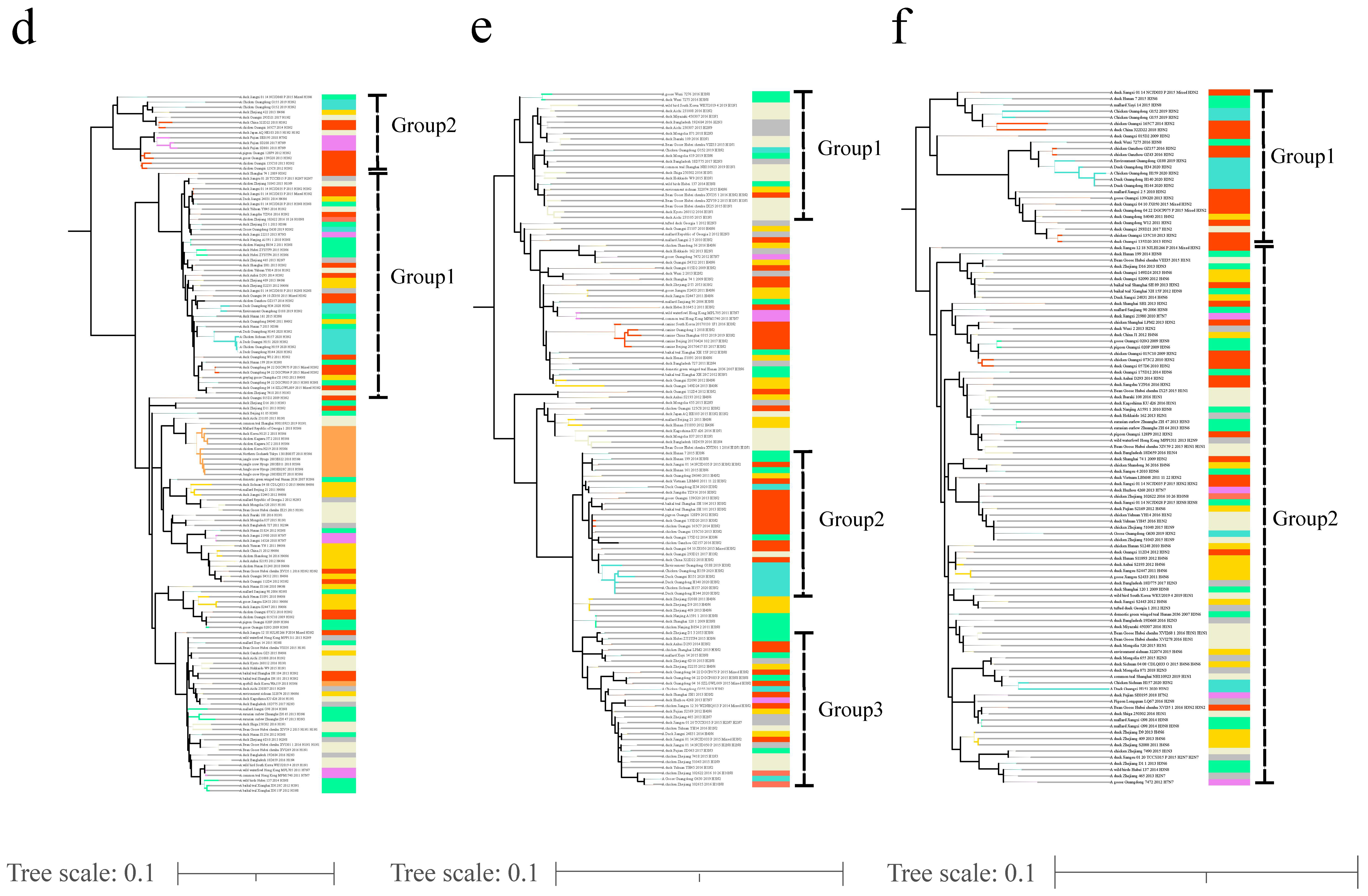
3.1. Phylogenetic Analyses of Viral Envelope-Encoding Genes
3.2. Phylogenetic Analyses of Internal Gene
3.3. Analysis of H3N2 AIVs Evolutionary Information
3.4. Key Amino Acids in Eight Proteins
3.5. Pathogenicity Test of H3N2 AIVs on BALB/c-Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bean, W.J.; Schell, M.; Katz, J.; Kawaoka, Y.; Naeve, C.; Gorman, O.; Webster, R.G. Evolution of the H3 influenza virus hemagglutinin from human and nonhuman hosts. J. Virol. 1992, 66, 1129–1167. [Google Scholar] [CrossRef] [PubMed]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef] [PubMed]
- Cockburn, W.C.; Delon, P.J.; Ferreira, W. Origin and progress of the 1968–69 Hong Kong influenza epidemic. Bull. World Health Organ. 1969, 41, 345–358. [Google Scholar] [PubMed]
- Scholtissek, C.; Rohde, W.; Von Hoyningen, V.; Rott, R. On the origin of the human influenza virus subtypes H2N2 and H3N2. Virology 1978, 87, 13–20. [Google Scholar] [CrossRef]
- Shen, H.X.; Li, X.; Yang, D.Q.; Ju, H.B.; Ge, F.F.; Wang, J.; Zhao, H.J. Phylogenetic analysis and evolutionary dynamics of H3N2 canine and feline influenza virus strains from 2006 to 2019. J. Med. Virol. 2021, 93, 3496–3507. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, G.; Shu, Y.; Chen, X.; Zhu, Y.; Yang, L.; Ma, G.; Kitamura, Y.; Liu, W. Genetic correlation between H3N2 human and swine influenza viruses. J. Clin. Virol. 2009, 44, 141–145. [Google Scholar] [CrossRef][Green Version]
- Lu, L.; Yin, Y.; Sun, Z.; Gao, L.; Gao, G.F.; Liu, S.; Sun, L.; Liu, W. Genetic correlation between current circulating H1N1 swine and human influenza viruses. J. Clin. Virol. 2010, 49, 186–191. [Google Scholar] [CrossRef]
- Zhou, N.N.; Senne, D.A.; Landgraf, J.S.; Swenson, S.L.; Erickson, G.; Rossow, K.; Liu, L.; Yoon, K.j.; Krauss, S.; Webster, R.G. Genetic Reassortment of Avian, Swine, and Human Influenza A Viruses in American Pigs. J. Virol. 1999, 73, 8851–8856. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, M.; Cui, X.; Xu, F.; Wen, F.; Pan, L.; Li, S.; Sun, H.; Zhu, X.; Lin, J.; et al. Pathogenicity and transmissibility of current H3N2 swine influenza virus in Southern China: A zoonotic potential. Transbound. Emerg. Dis. 2021, 69, 2052–2064. [Google Scholar] [CrossRef]
- de Jong, J.C.; Smith, D.J.; Lapedes, A.S.; Donatelli, I.; Campitelli, L.; Barigazzi, G.; Van Reeth, K.; Jones, T.C.; Rimmelzwaan, G.F.; Osterhaus, A.D.; et al. Antigenic and Genetic Evolution of Swine Influenza A (H3N2) Viruses in Europe. J. Virol. 2007, 81, 4315–4322. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, C.; Hou, Y.; Chen, Y.; Meng, F.; Zhuang, Y.; Liu, L.; Suzuki, Y.; Shi, J.; Deng, G.; et al. Pandemic threat posed by H3N2 avian influenza virus. Sci. China Life Sci. 2021, 64, 1984–1987. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Tan, D.; Shi, J.; Cui, P.; Jiang, Y.; Liu, L.; Tian, G.; Kawaoka, Y.; Li, C.; Chen, H. Complex Reassortment of Multiple Subtypes of Avian Influenza Viruses in Domestic Ducks at the Dongting Lake Region of China. J. Virol. 2013, 87, 9452–9462. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Deng, G.; Ma, S.; Zeng, X.; Yin, X.; Li, M.; Zhang, B.; Cui, P.; Chen, Y.; Yang, H.; et al. Rapid Evolution of H7N9 Highly Pathogenic Viruses that Emerged in China in 2017. Cell Host Microbe 2018, 24, 558–568.e7. [Google Scholar] [CrossRef] [PubMed]
- Subbarao, K.; Klimov, A.; Katz, J.; Regnery, H.; Lim, W.; Hall, H.; Perdue, M.; Swayne, D.; Bender, C.; Huang, J.; et al. Characterization of an Avian Influenza A (H5N1) Virus Isolated from a Child with a Fatal Respiratory Illness. Sci. Am. Assoc. Adv. Sci. 1998, 279, 393–396. [Google Scholar] [CrossRef]
- Song, D.; Kang, B.; Lee, C.; Jung, K.; Ha, G.; Kang, D.; Park, S.; Park, B.; Oh, J. Transmission of avian influenza virus (H3N2) to dogs. Emerg. Infect. Dis. 2008, 14, 741–746. [Google Scholar] [CrossRef]
- Zhao, J.; He, W.; Lu, M.; He, H.; Lai, A. Emergence and Characterization of a Novel Reassortant Canine Influenza Virus Isolated from Cats. Pathogens 2021, 10, 1320. [Google Scholar] [CrossRef]
- Bi, Y.; Chen, Q.; Wang, Q.; Chen, J.; Jin, T.; Wong, G.; Quan, C.; Liu, J.; Wu, J.; Yin, R.; et al. Genesis, Evolution and Prevalence of H5N6 Avian Influenza Viruses in China. Cell Host Microbe 2016, 20, 810–821. [Google Scholar] [CrossRef]
- Kwon, Y.K.; Lee, Y.J.; Choi, J.G.; Lee, E.K.; Jeon, W.J.; Jeong, O.M.; Kim, M.C.; Joh, S.J.; Kwon, J.H.; Kim, J.H. An outbreak of avian influenza subtype H9N8 among chickens in South Korea. Avian Pathol. 2006, 35, 443–447. [Google Scholar] [CrossRef]
- Li, C.; Yu, M.; Liu, L.; Sun, H. Characterization of a novel H3N2 influenza virus isolated from domestic ducks in China. Virus Genes 2016, 52, 568–630. [Google Scholar] [CrossRef]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A. viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.L.; Muench, H. A Simple Method of Estimating Fifty Per Cent Endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Li, X.; Shi, J.; Guo, J.; Deng, G.; Zhang, Q.; Wang, J.; He, X.; Wang, K.; Chen, J.; Li, Y.; et al. Genetics, receptor binding property, and transmissibility in mammals of naturally isolated H9N2 Avian Influenza viruses. PLoS Pathog. 2014, 10, e1004508. [Google Scholar] [CrossRef]
- Zhang, J.; Su, R.; Jian, X.; An, H.; Jiang, R.; Mok, C.K.P. The D253N Mutation in the Polymerase Basic 2 Gene in Avian Influenza(H9N2) Virus Contributes to the Pathogenesis of the Virus in Mammalian Hosts. Virol. Sin. 2018, 33, 531–537. [Google Scholar] [CrossRef]
- Kong, H.; Ma, S.; Wang, J.; Gu, C.; Wang, Z.; Shi, J.; Deng, G.; Guan, Y.; Chen, H. Identification of Key Amino Acids in the PB2 and M1 Proteins of H7N9 Influenza Virus That Affect Its Transmission in Guinea Pigs. J. Virol. 2019, 94, e01180–e01191. [Google Scholar] [CrossRef]
- Li, B.; Su, G.; Xiao, C.; Zhang, J.; Li, H.; Sun, N.; Lao, G.; Yu, Y.; Ren, X.; Qi, W.; et al. The PB2 co-adaptation of H10N8 avian influenza virus increases the pathogenicity to chickens and mice. Transbound Emerg. Dis. 2022, 69, 1794–1803. [Google Scholar] [CrossRef]
- Xiao, C.; Ma, W.; Sun, N.; Huang, L.; Li, Y.; Zeng, Z.; Wen, Y.; Zhang, Z.; Li, H.; Li, Q.; et al. PB2–588 V promotes the mammalian adaptation of H10N8, H7N9 and H9N2 avian influenza viruses. Sci. Rep. 2016, 6, 19474. [Google Scholar] [CrossRef]
- Czudai-Matwich, V.; Otte, A.; Matrosovich, M.; Gabriel, G.; Klenk, H.D. PB2 mutations D701N and S714R promote adaptation of an influenza H5N1 virus to a mammalian host. J. Virol. 2014, 88, 8735–8742. [Google Scholar] [CrossRef]
- Hulse-Post, D.J.; Franks, J.; Boyd, K.; Salomon, R.; Hoffmann, E.; Yen, H.L.; Webby, R.J.; Walker, D.; Nguyen, T.D.; Webster, R.G. Molecular changes in the polymerase genes (PA and PB1) associated with high pathogenicity of H5N1 influenza virus in mallard ducks. J. Virol. 2007, 81, 8515–8524. [Google Scholar] [CrossRef]
- Kamiki, H.; Franks, J.; Boyd, K.; Salomon, R.; Hoffmann, E.; Yen, H.L.; Webby, R.J.; Walker, D.; Nguyen, T.D.; Webster, R.G. A PB1-K577E Mutation in H9N2 Influenza Virus Increases Polymerase Activity and Pathogenicity in Mice. Viruses 2018, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Wang, Z.; Shi, J.; Deng, G.; Kong, H.; Tao, S.; Li, C.; Liu, L.; Guan, Y.; Chen, H. Glycine at Position 622 in PB1 Contributes to the Virulence of H5N1 Avian Influenza Virus in Mice. J. Virol. 2016, 90, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- Clements, A.L.; Sealy, J.E.; Peacock, T.P.; Sadeyen, J.R.; Hussain, S.; Lycett, S.J.; Shelton, H.; Digard, P.; Iqbal, M. Contribution of segment 3 to the acquisition of virulence in contemporary H9N2 avian influenza viruses. J. Virol. 2020, 94, e01173–e01193. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Feng, H.; Xu, J.; Zhao, D.; Shi, J.; Li, Y.; Deng, G.; Jiang, Y.; Li, X.; Zhu, P.; et al. The PA protein directly contributes to the virulence of H5N1 avian influenza viruses in domestic ducks. J. Virol. 2011, 85, 2180–2188. [Google Scholar] [CrossRef]
- Zhong, G.; Le, M.Q.; Lopes, T.J.S.; Halfmann, P.; Hatta, M.; Fan, S.; Neumann, G.; Kawaoka, Y. Mutations in the PA Protein of Avian H5N1 Influenza Viruses Affect Polymerase Activity and Mouse Virulence. J. Virol. 2018, 92, e01557-17. [Google Scholar] [CrossRef]
- Xu, G.; Zhang, X.; Gao, W.; Wang, C.; Wang, J.; Sun, H.; Sun, Y.; Guo, L.; Zhang, R.; Chang, K.C.; et al. Prevailing PA Mutation K356R in Avian Influenza H9N2 Virus Increases Mammalian Replication and Pathogenicity. J. Virol. 2016, 90, 8105–8114. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, B.; Shi, J.; Yin, X.; Wang, G.; Cui, P.; Liu, L.; Deng, G.; Jiang, Y.; Li, C.; et al. Amino Acid Mutations A286V and T437M in the Nucleoprotein Attenuate H7N9 Viruses in Mice. J. Virol. 2020, 94, e01530. [Google Scholar] [CrossRef]
- Zhu, W.; Feng, Z.; Chen, Y.; Yang, L.; Liu, J.; Li, X.; Liu, S.; Zhou, L.; Wei, H.; Gao, R.; et al. Mammalian-adaptive mutation NP-Q357K in Eurasian H1N1 Swine Influenza viruses determines the virulence phenotype in mice. Emerg. Microbes Infect. 2019, 8, 989–999. [Google Scholar] [CrossRef]
- Weinstock, D.M.; Gubareva, L.V.; Zuccotti, G. Prolonged shedding of multidrug-resistant influenza A virus in an immunocompromised patient. N. Engl. J. Med. 2003, 348, 867–868. [Google Scholar] [CrossRef]
- Kiso, M.; Mitamura, K.; Sakai-Tagawa, Y.; Shiraishi, K.; Kawakami, C.; Kimura, K.; Hayden, F.G.; Sugaya, N.; Kawaoka, Y. Resistant influenza A viruses in children treated with oseltamivir: Descriptive study. Lancet 2004, 364, 759–765. [Google Scholar] [CrossRef]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Shi, J.; Kong, X.; Ma, S.; Zhang, Y.; Yin, X.; He, X.; Liu, L.; Suzuki, Y.; Li, C.; et al. H3N2 avian influenza viruses detected in live poultry markets in China bind to human-type receptors and transmit in guinea pigs and ferrets. Emerg. Microbes Infect. 2019, 8, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology 2008, 384, 28–32. [Google Scholar] [CrossRef]
- Zhong, L.; Wang, X.; Li, Q.; Liu, D.; Chen, H.; Zhao, M.; Gu, X.; He, L.; Liu, X.; Gu, M.; et al. Molecular mechanism of the airborne transmissibility of H9N2 avian influenza A viruses in chickens. J. Virol. 2014, 88, 9568–9578. [Google Scholar] [CrossRef]
- He, W.; Zhang, W.; Yan, H.; Xu, H.; Xie, Y.; Wu, Q.; Wang, C.; Dong, G. Distribution and evolution of H1N1 influenza A viruses with adamantanes-resistant mutations worldwide from 1918 to 2019. J. Med. Virol. 2021, 93, 3473–3483. [Google Scholar] [CrossRef] [PubMed]
- WHO. Recommended Composition of Influenza Virus Vaccines for Use in the 2021–2022 Northern Hemisphere Influenza Season. 2021. Available online: https://www.who.int/publications/i/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2021-2022-northern-hemisphere-influenza-season (accessed on 6 May 2022).
- WHO. Recommended Composition of Influenza Virus Vaccines for Use in the 2022–2023 Northern Hemisphere Influenza Season. Available online: https://www.who.int/publications/m/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2022-2023-northern-hemisphere-influenza-season (accessed on 6 May 2022).
- Li, X.; Liu, B.; Ma, S.; Cui, P.; Liu, W.; Li, Y.; Guo, J.; Chen, H. High frequency of reassortment after co-infection of chickens with the H4N6 and H9N2 influenza A viruses and the biological characteristics of the reassortants. Vet. Microbiol. 2018, 222, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Tang, J.; Zhang, Y.; Liu, L.; Li, X.; Meng, Y.; Zhao, X.; Yang, L.; Shu, Y.; Wang, D. Molecular characterization of H3 subtype avian influenza viruses based on poultry-related environmental surveillance in China between 2014 and 2017. Virology 2020, 542, 8–19. [Google Scholar] [CrossRef]
- Hu, J.; Zha, Y.; Jin, X.; Wen, X.; Li, Z.; Wang, X.; Dai, Y.; Li, X.; Liao, M.; Jia, W. Continuous adaptation of the HA and NA gene of H3N2 subtypes of avian influenza virus in South China, 2017–2018. J. Infect. 2019, 79, 61–74. [Google Scholar] [CrossRef]
- Sun, H.; Cui, P.; Song, Y.; Qi, Y.; Li, X.; Qi, W.; Xu, C.; Jiao, P.; Liao, M. PB2 segment promotes high-pathogenicity of H5N1 avian influenza viruses in mice. Front Microbiol. 2015, 6, 73. [Google Scholar] [CrossRef]
- Sun, S.; Wang, Q.; Zhao, F.; Chen, W.; Li, Z. Glycosylation site alteration in the evolution of influenza A (H1N1) viruses. PLoS ONE 2011, 6, e22844. [Google Scholar] [CrossRef]
- Tsuchiya, E.; Sugawara, K.; Hongo, S.; Matsuzaki, Y.; Muraki, Y.; Li, Z.N.; Nakamura, K. Antigenic structure of the haemagglutinin of human influenza A/H2N2 virus. J. Gen. Virol. 2001, 82, 2475–2484. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, E.; Sugawara, K.; Hongo, S.; Matsuzaki, Y.; Muraki, Y.; Li, Z.N.; Nakamura, K. Effect of addition of new oligosaccharide chains to the globular head of influenza A/H2N2 virus haemagglutinin on the intracellular transport and biological activities of the molecule. J. Gen. Virol. 2002, 83, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Rao, P.; Krug, R.M. The active sites of the influenza cap-dependent endonuclease are on different polymerase subunits. EMBO J. 2001, 20, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Guilligay, D.; Tarendeau, F.; Resa-Infante, P.; Coloma, R.; Crepin, T.; Sehr, P.; Lewis, J.; Ruigrok, R.W.; Ortin, J.; Hart, D.J.; et al. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat. Struct. Mol. Biol. 2008, 15, 500–506. [Google Scholar] [CrossRef]
- Marklund, J.K.; Ye, Q.; Dong, J.; Tao, Y.J.; Krug, R.M. Sequence in the influenza A virus nucleoprotein required for viral polymerase binding and RNA synthesis. J. Virol. 2012, 86, 7292–7299. [Google Scholar] [CrossRef]
- Pu, J.; Wang, S.; Yin, Y.; Zhang, G.; Carter, R.A.; Wang, J.; Xu, G.; Sun, H.; Wang, M.; Wen, C.; et al. Evolution of the H9N2 influenza genotype that facilitated the genesis of the novel H7N9 virus. Proc. Natl. Acad. Sci. USA 2015, 112, 548–553. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Gene | Closest Virus | Homology (%) |
|---|---|---|---|
| H34 | HA | A/duck/Hunan/7/2015(H3N6) | 96.47 |
| NA | A/duck/China/322D22/2018(H3N2) | 98.02 | |
| M | A/duck/Vietnam/LBM48/2011(H3N2) | 98.07 | |
| NP | A/chicken/Ganzhou/GZ157/2016(H3N2) | 97.8 | |
| NS | A/chicken/Ganzhou/GZ43/2016(H3N2) | 98.2 | |
| PA | A/duck/China/322D22/2018(H3N2) | 98.03 | |
| PB1 | A/duck/China/322D22/2018(H3N2) | 97.95 | |
| PB2 | A/duck/Guangxi/293D21/2017(H1N2) | 98.85 | |
| H159 | HA | A/duck/Hunan/7/2015(H3N6) | 96.36 |
| NA | A/duck/Zhejiang/727042/2014(H6N2) | 96.45 | |
| M | A/duck/China/322D22/2018(H3N2) | 99.42 | |
| NP | A/duck/Guangdong/S4040/2011(H4N2) | 100 | |
| NS | A/chicken/Ganzhou/GZ43/2016(H3N2) | 97.53 | |
| PA | A/chicken/Ganzhou/GZ157/2016(H3N2) | 97.81 | |
| PB1 | A/duck/Guangxi/293D21/2017(H1N2) | 97.74 | |
| PB2 | A/duck/Guangxi/293D21/2017(H1N2) | 97.78 | |
| G188 | HA | A/duck/Hubei/ZYSYF18/2015(H3N6) | 97.51 |
| NA | A/chicken/Ganzhou/GZ43/2016(H3N2) | 98.08 | |
| M | A/duck/China/322D22/2018(H3N2) | 99.32 | |
| NP | A/chicken/Ganzhou/GZ157/2016(H3N2) | 97.93 | |
| NS | A/chicken/Ganzhou/GZ43/2016(H3N2) | 98.43 | |
| PA | A/chicken/Ganzhou/GZ43/2016(H3N2) | 97.54 | |
| PB1 | A/duck/Hubei/ZYSYF2/2015(H3N6) | 98.3 | |
| PB2 | A/chicken/Guangxi/165C7/2014(H3N2) | 97.31 | |
| G630 | HA | A/duck/Hubei/ZYSYF18/2015(H3N6) | 95.53 |
| NA | A/duck/Guangdong/8.30_DGCP036-C/2017(H6N2) | 98.09 | |
| M | A/chicken/Zhejiang/102622/2016(H10N8) | 99.29 | |
| NP | A/duck/Jiangxi/22215/2013(H7N3) | 99.4 | |
| NS | A/chicken/Zhejiang/51048/2015(H1N9) | 98.47 | |
| PA | A/chicken/Yuhuan/YH14/2016(H1N2) | 97.90 | |
| PB1 | A/chicken/Zhejiang/51048/2015(H1N9) | 97.5 | |
| PB2 | A/duck/Yuhuan/YH45/2016(H1N2) | 97.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, T.; Huang, Y.; Xie, S.; Xu, L.; Chen, J.; Qi, W.; Liao, M.; Jia, W. A Characterization and an Evolutionary and a Pathogenicity Analysis of Reassortment H3N2 Avian Influenza Virus in South China in 2019–2020. Viruses 2022, 14, 2574. https://doi.org/10.3390/v14112574
Liu T, Huang Y, Xie S, Xu L, Chen J, Qi W, Liao M, Jia W. A Characterization and an Evolutionary and a Pathogenicity Analysis of Reassortment H3N2 Avian Influenza Virus in South China in 2019–2020. Viruses. 2022; 14(11):2574. https://doi.org/10.3390/v14112574
Chicago/Turabian StyleLiu, Tengfei, Yuhao Huang, Shumin Xie, Lingyu Xu, Junhong Chen, Wenbao Qi, Ming Liao, and Weixin Jia. 2022. "A Characterization and an Evolutionary and a Pathogenicity Analysis of Reassortment H3N2 Avian Influenza Virus in South China in 2019–2020" Viruses 14, no. 11: 2574. https://doi.org/10.3390/v14112574
APA StyleLiu, T., Huang, Y., Xie, S., Xu, L., Chen, J., Qi, W., Liao, M., & Jia, W. (2022). A Characterization and an Evolutionary and a Pathogenicity Analysis of Reassortment H3N2 Avian Influenza Virus in South China in 2019–2020. Viruses, 14(11), 2574. https://doi.org/10.3390/v14112574