

Rift Valley Fever Virus Non-Structural Protein S Is Associated with Nuclear Translocation of Active Caspase-3 and Inclusion Body Formation

 , ,
, ,  , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus
2.2. Mouse Experiments and Sampling
2.3. Histology and Immunohistochemistry
2.4. Immunofluorescence
2.5. Statistical Analysis
3. Results
3.1. Clinical Signs
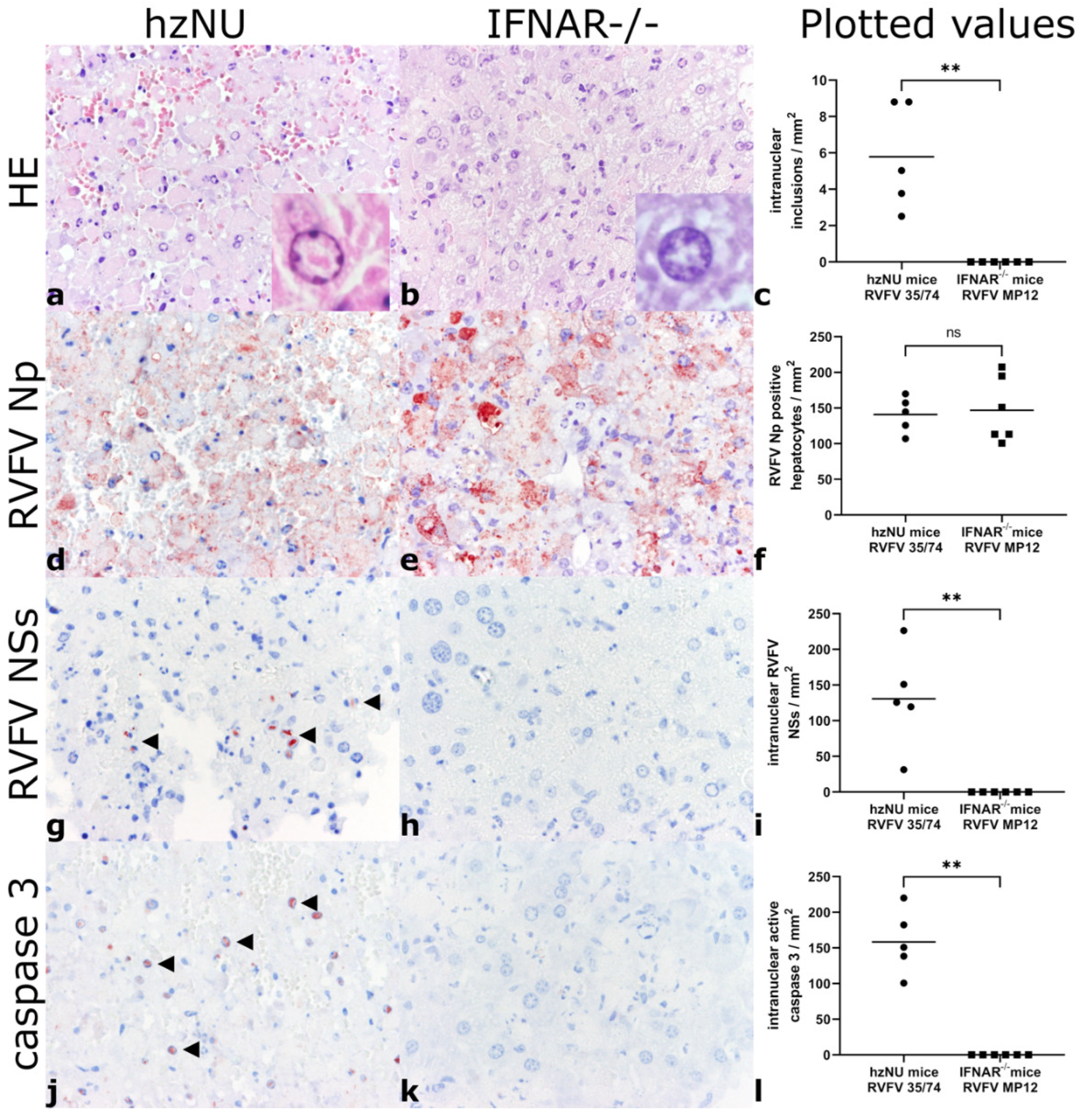
3.2. Histology and Immunohistochemistry
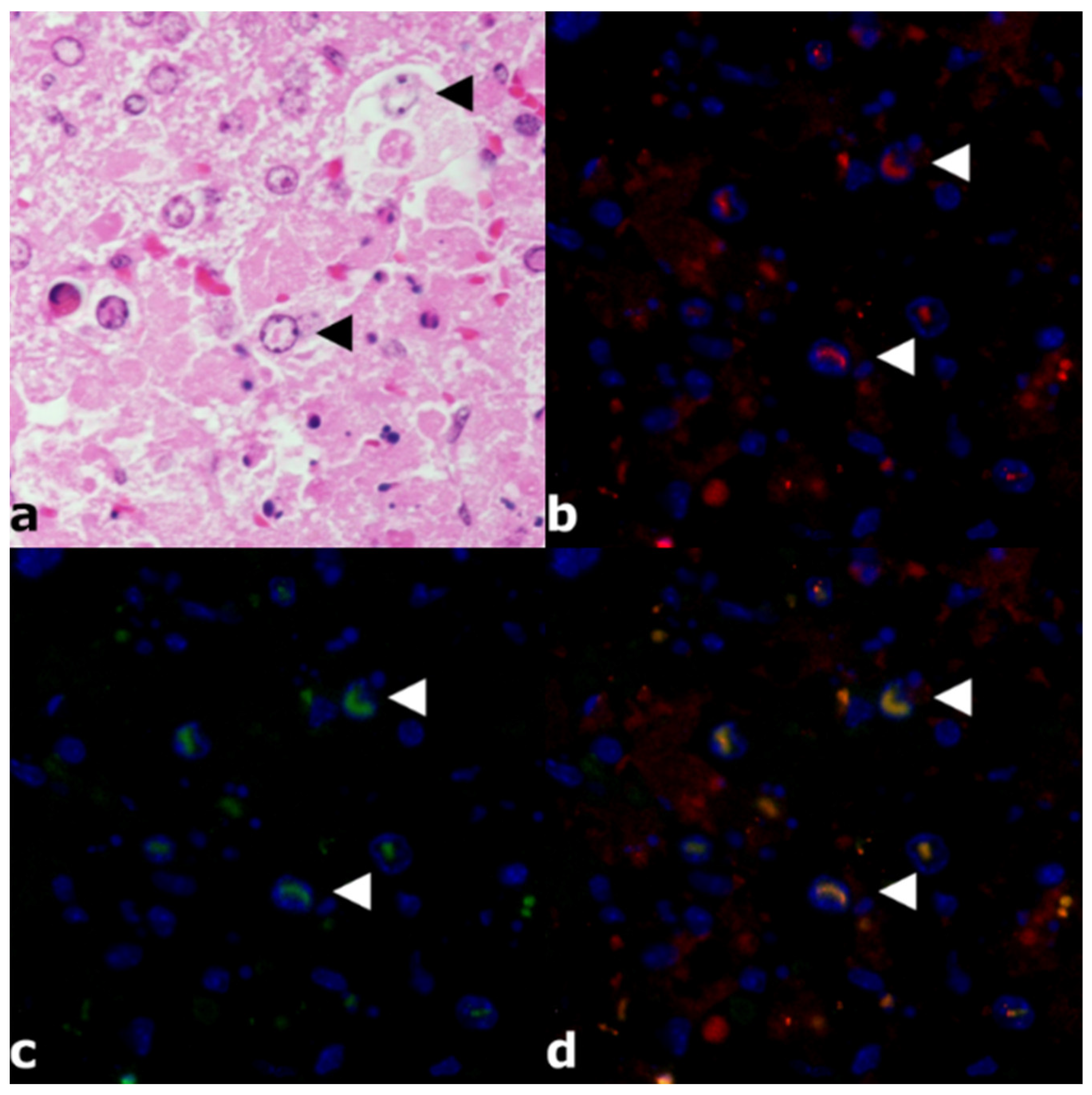
3.3. Immunofluorescence
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hartman, A. Rift Valley Fever. Clin. Lab. Med. 2017, 37, 285–301. [Google Scholar] [CrossRef]
- Ikegami, T.; Makino, S. The Pathogenesis of Rift Valley Fever. Viruses 2011, 3, 493–519. [Google Scholar] [CrossRef] [PubMed]
- Kwaśnik, M.; Rożek, W.; Rola, J. Rift Valley Fever—A Growing Threat to Humans and Animals. J. Vet. Res. 2021, 65, 7–14. [Google Scholar] [CrossRef]
- Al-Hazmi, M.; Ayoola, E.A.; Abdurahman, M.; Banzal, S.; Ashraf, J.; El-Bushra, A.; Hazmi, A.; Abdullah, M.; Abbo, H.; Elamin, A.; et al. Epidemic Rift Valley Fever in Saudi Arabia: A Clinical Study of Severe Illness in Humans. Clin. Infect. Dis. 2003, 36, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Linthicum, K.J.; Britch, S.C.; Anyamba, A. Rift Valley Fever: An Emerging Mosquito-Borne Disease. Annu. Rev. Entomol. 2016, 61, 395–415. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Indran, S.V.; Balaraman, V.; Wilson, W.C.; Richt, J.A. Molecular Aspects of Rift Valley Fever Virus and the Emergence of Reassortants. Virus Genes 2019, 55, 1–11. [Google Scholar] [CrossRef]
- Odendaal, L.; Clift, S.J.; Fosgate, G.T.; Davis, A.S. Ovine Fetal and Placental Lesions and Cellular Tropism in Natural Rift Valley Fever Virus Infections. Vet. Pathol. 2020, 57, 791–806. [Google Scholar] [CrossRef]
- Odendaal, L.; Clift, S.J.; Fosgate, G.T.; Davis, A.S. Lesions and Cellular Tropism of Natural Rift Valley Fever Virus Infection in Adult Sheep. Vet. Pathol. 2019, 56, 61–77. [Google Scholar] [CrossRef]
- Oymans, J.; Wichgers Schreur, P.J.; van Keulen, L.; Kant, J.; Kortekaas, J. Rift Valley Fever Virus Targets the Maternal-Foetal Interface in Ovine and Human Placentas. PLoS Negl. Trop. Dis. 2020, 14, e0007898. [Google Scholar] [CrossRef]
- Bird, B.H.; McElroy, A.K. Rift Valley Fever Virus: Unanswered Questions. Antiviral Res. 2016, 132, 274–280. [Google Scholar] [CrossRef]
- Nielsen, S.S.; Alvarez, J.; Bicout, D.J.; Calistri, P.; Depner, K.; Drewe, J.A.; Garin-Bastuji, B.; Rojas, J.L.G.; Schmidt, C.G.; Michel, V.; et al. Rift Valley Fever—Epidemiological Update and Risk of Introduction into Europe. EFSA J. 2020, 18, e06041. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Wilson, W.C.; Faburay, B.; Richt, J.; McVey, D.S.; Rowland, R.R. Multiplex Detection of Igg and Igm to Rift Valley Fever Virus Nucleoprotein, Nonstructural Proteins, and Glycoprotein in Ovine and Bovine. Vector Borne Zoonotic Dis. 2016, 16, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, G.; Lopez-Gil, E.; Warimwe, G.M.; Brun, A. Understanding Rift Valley Fever: Contributions of Animal Models to Disease Characterization and Control. Mol. Immunol. 2015, 66, 78–88. [Google Scholar] [CrossRef]
- Poueme, R.; Stoek, F.; Nloga, N.; Awah-Ndukum, J.; Rissmann, M.; Schulz, A.; Wade, A.; Kouamo, J.; Moctar, M.; Eisenbarth, A.; et al. Seroprevalence and Associated Risk Factors of Rift Valley Fever in Domestic Small Ruminants in the North Region of Cameroon. Vet. Med. Int. 2019, 2019, 8149897. [Google Scholar] [CrossRef]
- Rissmann, M.; Ulrich, R.; Schroder, C.; Hammerschmidt, B.; Hanke, D.; Mroz, C.; Groschup, M.H.; Eiden, M. Vaccination of Alpacas against Rift Valley Fever Virus: Safety, Immunogenicity and Pathogenicity of Mp-12 Vaccine. Vaccine 2017, 35, 655–662. [Google Scholar] [CrossRef]
- Ross, T.M.; Bhardwaj, N.; Bissel, S.J.; Hartman, A.L.; Smith, D.R. Animal Models of Rift Valley Fever Virus Infection. Virus Res. 2012, 163, 417–423. [Google Scholar] [CrossRef]
- Wichgers Schreur, P.J.; Oreshkova, N.; van Keulen, L.; Kant, J.; van de Water, S.; Soos, P.; Dehon, Y.; Kollar, A.; Penzes, Z.; Kortekaas, J. Safety and Efficacy of Four-Segmented Rift Valley Fever Virus in Young Sheep, Goats and Cattle. NPJ Vaccines 2020, 5, 65. [Google Scholar] [CrossRef]
- Wright, D.; Kortekaas, J.; Bowden, T.A.; Warimwe, G.M. Rift Valley Fever: Biology and Epidemiology. J. Gen. Virol. 2019, 100, 1187–1199. [Google Scholar] [CrossRef]
- Lozach, P.-Y.; Kühbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. Dc-Sign as a Receptor for Phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef]
- Lathan, R.; Simon-Chazottes, D.; Jouvion, G.; Godon, O.; Malissen, M.; Flamand, M.; Bruhns, P.; Panthier, J.J. Innate Immune Basis for Rift Valley Fever Susceptibility in Mouse Models. Sci. Rep. 2017, 7, 7096. [Google Scholar] [CrossRef]
- Thomson, B.J. Viruses and Apoptosis. Int. J. Exp. Pathol. 2001, 82, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Odendaal, L.; Davis, A.S.; Venter, E.H. Insights into the Pathogenesis of Viral Haemorrhagic Fever Based on Virus Tropism and Tissue Lesions of Natural Rift Valley Fever. Viruses 2021, 13, 709. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, New and Emerging Functions of Caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, I.; Alldinger, S.; Ulrich, R.; Baumgärtner, W. Spatio-Temporal Expression of Immediate Early Genes in the Central Nervous System of Sjl/J Mice. Int. J. Dev. Neurosci. 2005, 23, 637–649. [Google Scholar] [CrossRef]
- Gerhauser, I.; Baumgärtner, W.; Herden, C. Unusual Type of Reactive Astrocytes in the Feline Central Nervous System. Dtsch. Tierarztl. Wochenschr. 2007, 114, 124–128. [Google Scholar]
- Mirzayans, R.; Murray, D. Do Tunel and Other Apoptosis Assays Detect Cell Death in Preclinical Studies? Int. J. Mol. Sci. 2020, 21, 9090. [Google Scholar] [CrossRef]
- Porter, A.G.; Jänicke, R.U. Emerging Roles of Caspase-3 in Apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Snigdha, S.; Smith, E.D.; Prieto, G.A.; Cotman, C.W. Caspase-3 Activation as a Bifurcation Point between Plasticity and Cell Death. Neurosci. Bull. 2012, 28, 14–24. [Google Scholar] [CrossRef]
- Smith, D.R.; Steele, K.E.; Shamblin, J.; Honko, A.; Johnson, J.; Reed, C.; Kennedy, M.; Chapman, J.L.; Hensley, L.E. The Pathogenesis of Rift Valley Fever Virus in the Mouse Model. Virology 2010, 407, 256–267. [Google Scholar] [CrossRef]
- Ulrich, R. Rift Valley Fever: An Ancient Plague on Its Way out of Africa? Vet. Pathol. 2019, 56, 178–179. [Google Scholar] [CrossRef]
- Struthers, J.K.; Swanepoel, R. Identification of a Major Non-Structural Protein in the Nuclei of Rift Valley Fever Virus-Infected Cells. J. Gen. Virol. 1982, 60, 381–384. [Google Scholar] [CrossRef]
- Swanepoel, R.; Blackburn, N.K. Demonstration of Nuclear Immunofluorescence in Rift Valley Fever Infected Cells. J. Gen. Virol. 1977, 34, 557–561. [Google Scholar] [CrossRef]
- Yadani, F.Z.; Kohl, A.; Préhaud, C.; Billecocq, A.; Bouloy, M. The Carboxy-Terminal Acidic Domain of Rift Valley Fever Virus Nss Protein Is Essential for the Formation of Filamentous Structures but Not for the Nuclear Localization of the Protein. J. Virol. 1999, 73, 5018–5025. [Google Scholar] [CrossRef] [PubMed]
- Mroz, C.; Schmidt, K.M.; Reiche, S.; Groschup, M.H.; Eiden, M. Development of Monoclonal Antibodies to Rift Valley Fever Virus and Their Application in Antigen Detection and Indirect Immunofluorescence. J. Immunol. Methods 2018, 460, 36–44. [Google Scholar] [CrossRef]
- Michaely, L.M.; Rissmann, M.; Keller, M.; König, R.; von Arnim, F.; Eiden, M.; Rohn, K.; Baumgärtner, W.; Groschup, M.; Ulrich, R. Nsg-Mice Reveal the Importance of a Functional Innate and Adaptive Immune Response to Overcome Rvfv Infection. Viruses 2022, 14, 350. [Google Scholar] [CrossRef] [PubMed]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic Evidence for an Interferon-Antagonistic Function of Rift Valley Fever Virus Nonstructural Protein Nss. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Lang, Y.; Henningson, J.; Jasperson, D.; Li, Y.; Lee, J.; Ma, J.; Li, Y.; Cao, N.; Liu, H.; Wilson, W.; et al. Mouse Model for the Rift Valley Fever Virus Mp12 Strain Infection. Vet. Microbiol. 2016, 195, 70–77. [Google Scholar] [CrossRef]
- Hirasawa, T.; Yamashita, H.; Makino, S. Genetic Typing of the Mouse and Rat Nude Mutations by Pcr and Restriction Enzyme Analysis. Exp. Anim. 1998, 47, 63–67. [Google Scholar] [CrossRef][Green Version]
- Gregor, K.M.; Michaely, L.M.; Gutjahr, B.; Rissmann, M.; Keller, M.; Dornbusch, S.; Naccache, F.; Schön, K.; Jansen, S.; Heitmann, A.; et al. Rift Valley Fever Virus Detection in Susceptible Hosts with Special Emphasis in Insects. Sci. Rep. 2021, 11, 9822. [Google Scholar] [CrossRef]
- Gutjahr, B.; Keller, M.; Rissmann, M.; von Arnim, F.; Jäckel, S.; Reiche, S.; Ulrich, R.; Groschup, M.H.; Eiden, M. Two Monoclonal Antibodies against Glycoprotein Gn Protect Mice from Rift Valley Fever Challenge by Cooperative Effects. PLoS Negl. Trop. Dis. 2020, 14, e0008143. [Google Scholar] [CrossRef]
- Crabtree, M.B.; Kent Crockett, R.J.; Bird, B.H.; Nichol, S.T.; Erickson, B.R.; Biggerstaff, B.J.; Horiuchi, K.; Miller, B.R. Infection and Transmission of Rift Valley Fever Viruses Lacking the Nss and/or Nsm Genes in Mosquitoes: Potential Role for Nsm in Mosquito Infection. PLoS Negl. Trop. Dis. 2012, 6, e1639. [Google Scholar] [CrossRef] [PubMed]
- Jackel, S.; Eiden, M.; Dauber, M.; Balkema-Buschmann, A.; Brun, A.; Groschup, M.H. Generation and Application of Monoclonal Antibodies against Rift Valley Fever Virus Nucleocapsid Protein Np and Glycoproteins Gn and Gc. Arch. Virol. 2014, 159, 535–546. [Google Scholar] [CrossRef]
- Armando, F.; Gambini, M.; Corradi, A.; Becker, K.; Marek, K.; Pfankuche, V.M.; Mergani, A.E.; Brogden, G.; de Buhr, N.; von Köckritz-Blickwede, M.; et al. Mesenchymal to Epithelial Transition Driven by Canine Distemper Virus Infection of Canine Histiocytic Sarcoma Cells Contributes to a Reduced Cell Motility In Vitro. J. Cell. Mol. Med. 2020, 24, 9332–9348. [Google Scholar] [CrossRef] [PubMed]
- Krüger, N.; Rocha, C.; Runft, S.; Krüger, J.; Färber, I.; Armando, F.; Leitzen, E.; Brogden, G.; Gerold, G.; Pöhlmann, S.; et al. The Upper Respiratory Tract of Felids Is Highly Susceptible to SARS-CoV-2 Infection. Int. J. Mol. Sci. 2021, 22, 10636. [Google Scholar] [CrossRef] [PubMed]
- Odendaal, L.; Davis, A.S.; Fosgate, G.T.; Clift, S.J. Lesions and Cellular Tropism of Natural Rift Valley Fever Virus Infection in Young Lambs. Vet. Pathol. 2020, 57, 66–81. [Google Scholar] [CrossRef]
- Borrego, B.; Moreno, S.; de la Losa, N.; Weber, F.; Brun, A. The Change P82l in the Rift Valley Fever Virus Nss Protein Confers Attenuation in Mice. Viruses 2021, 13, 542. [Google Scholar] [CrossRef]
- Lihoradova, O.; Ikegami, T. Countermeasure Development for Rift Valley Fever: Deletion, Modification or Targeting of Major Virulence Factor Nss. Future Virol. 2014, 9, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Léger, P.; Nachman, E.; Richter, K.; Tamietti, C.; Koch, J.; Burk, R.; Kummer, S.; Xin, Q.; Stanifer, M.; Bouloy, M.; et al. Nss Amyloid Formation Is Associated with the Virulence of Rift Valley Fever Virus in Mice. Nat. Commun. 2020, 11, 3281. [Google Scholar] [CrossRef] [PubMed]
- Ly, H.J.; Ikegami, T. Rift Valley Fever Virus Nss Protein Functions and the Similarity to Other Bunyavirus Nss Proteins. Virol. J. 2016, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Billecocq, A.; Spiegel, M.; Vialat, P.; Kohl, A.; Weber, F.; Bouloy, M.; Haller, O. Nss Protein of Rift Valley Fever Virus Blocks Interferon Production by Inhibiting Host Gene Transcription. J. Virol. 2004, 78, 9798–9806. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Dubaele, S.; Proietti De Santis, L.; Billecocq, A.; Bouloy, M.; Egly, J.M. Tfiih Transcription Factor, a Target for the Rift Valley Hemorrhagic Fever Virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef]
- Habjan, M.; Pichlmair, A.; Elliott, R.M.; Overby, A.K.; Glatter, T.; Gstaiger, M.; Superti-Furga, G.; Unger, H.; Weber, F. Nss Protein of Rift Valley Fever Virus Induces the Specific Degradation of the Double-Stranded Rna-Dependent Protein Kinase. J. Virol. 2009, 83, 4365–4375. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Rift Valley Fever Virus Nss Protein Promotes Post-Transcriptional Downregulation of Protein Kinase Pkr and Inhibits Eif2alpha Phosphorylation. PLoS Pathog. 2009, 5, e1000287. [Google Scholar] [CrossRef] [PubMed]
- Kalveram, B.; Lihoradova, O.; Indran, S.V.; Lokugamage, N.; Head, J.A.; Ikegami, T. Rift Valley Fever Virus Nss Inhibits Host Transcription Independently of the Degradation of Dsrna-Dependent Protein Kinase Pkr. Virology 2013, 435, 415–424. [Google Scholar] [CrossRef]
- Austin, D.; Baer, A.; Lundberg, L.; Shafagati, N.; Schoonmaker, A.; Narayanan, A.; Popova, T.; Panthier, J.J.; Kashanchi, F.; Bailey, C.; et al. P53 Activation Following Rift Valley Fever Virus Infection Contributes to Cell Death and Viral Production. PLoS ONE 2012, 7, e36327. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Josse, T.; Gilleron, J.; Billecocq, A.; Leger, P.; Bouloy, M.; Bonnefoy, E. Nonstructural Nss Protein of Rift Valley Fever Virus Interacts with Pericentromeric DNA Sequences of the Host Cell, Inducing Chromosome Cohesion and Segregation Defects. J. Virol. 2010, 84, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Benferhat, R.; Josse, T.; Albaud, B.; Gentien, D.; Mansuroglu, Z.; Marcato, V.; Soues, S.; Le Bonniec, B.; Bouloy, M.; Bonnefoy, E. Large-Scale Chromatin Immunoprecipitation with Promoter Sequence Microarray Analysis of the Interaction of the Nss Protein of Rift Valley Fever Virus with Regulatory DNA Regions of the Host Genome. J. Virol. 2012, 86, 11333–11344. [Google Scholar] [CrossRef] [PubMed]
- Copeland, A.M.; Altamura, L.A.; Van Deusen, N.M.; Schmaljohn, C.S. Nuclear Relocalization of Polyadenylate Binding Protein During Rift Valley Fever Virus Infection Involves Expression of the Nss Gene. J. Virol. 2013, 87, 11659–11669. [Google Scholar] [CrossRef][Green Version]
- Baer, A.; Austin, D.; Narayanan, A.; Popova, T.; Kainulainen, M.; Bailey, C.; Kashanchi, F.; Weber, F.; Kehn-Hall, K. Induction of DNA Damage Signaling Upon Rift Valley Fever Virus Infection Results in Cell Cycle Arrest and Increased Viral Replication. J. Biol. Chem. 2012, 287, 7399–7410. [Google Scholar] [CrossRef] [PubMed]
- Won, S.; Ikegami, T.; Peters, C.J.; Makino, S. Nsm Protein of Rift Valley Fever Virus Suppresses Virus-Induced Apoptosis. J. Virol. 2007, 81, 13335–13345. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptotic DNA Fragmentation. Exp. Cell Res. 2000, 256, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Taimen, P.; Kallajoki, M. NuMA and Nuclear Lamins Behave Differently in Fas-Mediated Apoptosis. J. Cell Sci. 2003, 116, 571–583. [Google Scholar] [CrossRef][Green Version]
- Sinha, A.; Schalk, S.; Lager, K.M.; Wang, C.; Opriessnig, T. Singular PCV2a or PCV2b Infection Results in Apoptosis of Hepatocytes in Clinically Affected Gnotobiotic Pigs. Res. Vet. Sci. 2012, 92, 151–156. [Google Scholar] [CrossRef]
- Zuzarte-Luis, V.; Berciano, M.T.; Lafarga, M.; Hurle, J.M. Caspase Redundancy and Release of Mitochondrial Apoptotic Factors Characterize Interdigital Apoptosis. Apoptosis 2006, 11, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, J.; Oreshkova, N.; van Keulen, L.; Kant, J.; Bosch, B.J.; Bouloy, M.; Moulin, V.; Goovaerts, D.; Moormann, R.J. Comparative Efficacy of Two Next-Generation Rift Valley Fever Vaccines. Vaccine 2014, 32, 4901–4908. [Google Scholar] [CrossRef]
- Chawla-Sarkar, M.; Lindner, D.J.; Liu, Y.-F.; Williams, B.R.; Sen, G.C.; Silvermann, R.H.; Borden, E.C. Apoptosis and Interferons: Role of Interferon-Stimulated Genes as Mediators of Apoptosis. Apoptosis 2003, 8, 237–249. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| N | Mouse Strain | Infected with | Age | Clinical Signs | Sampling |
|---|---|---|---|---|---|
| 6 | IFNAR−/− | 1.43 × 103 TCID50 RVFV MP12 | 5–7 weeks | Ruffled fur, lack of movement, tachypnea, apathy | 3 dpi |
| 2 | IFNAR−/− | Placebo medium | 5–7 weeks | None | 14 dpi |
| 5 | hzNU | 1.4 × 103 TCID50 RVFV 35/74 | 5–7 weeks | Ruffled fur, lack of movement, tachypnea, apathy | 2–3 dpi |
| 2 | hzNU | Placebo medium | 5–7 weeks | None | 14 dpi |
| Target Antigen | Antibody | Species/Clonality | Pretreatment | Concentration | Source |
|---|---|---|---|---|---|
| NP | S24NP | Sheep, pc | None | 1:2000 | FLI [34,39,40] |
| NSs | NSs5F12 | Mouse, mc | Hcb | 1:100 | FLI [34,39,40] |
| Active (cleaved) Caspase-3 | Active®® Caspase-3 Antibody | Rabbit, pc | None | 1:200 | R&D systems |
| Mouse Strain | Infected with | Hepatocellular Necrosis | RVFV NP | Intranuclear Inclusions | Intranuclear NSs | Intranuclear Active Caspase-3 |
|---|---|---|---|---|---|---|
| IFNAR−/− | 1.43 × 103 TCID50 RVFV MP12 | Severe | 132.08 [100.63–207.55] | 0 | 0 | 0 |
| hzNU | 1.4 × 103 TCID50 RVFV 35/74 | Severe | 144.65 [106.92–169.81] | 5.03 [2.52–8.81] | 125.79 [31.45–226.42] | 150.94 [100.63–220.13] |
| IFNAR−/− | Placebo medium | None | 0 | 0 | 0 | 0 |
| hzNU | Placebo medium | None | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michaely, L.M.; Rissmann, M.; Armando, F.; von Arnim, F.; Keller, M.; Eiden, M.; König, R.; Gutjahr, B.; Baumgärtner, W.; Groschup, M.H.; et al. Rift Valley Fever Virus Non-Structural Protein S Is Associated with Nuclear Translocation of Active Caspase-3 and Inclusion Body Formation. Viruses 2022, 14, 2487. https://doi.org/10.3390/v14112487
Michaely LM, Rissmann M, Armando F, von Arnim F, Keller M, Eiden M, König R, Gutjahr B, Baumgärtner W, Groschup MH, et al. Rift Valley Fever Virus Non-Structural Protein S Is Associated with Nuclear Translocation of Active Caspase-3 and Inclusion Body Formation. Viruses. 2022; 14(11):2487. https://doi.org/10.3390/v14112487
Chicago/Turabian StyleMichaely, Lukas Mathias, Melanie Rissmann, Federico Armando, Felicitas von Arnim, Markus Keller, Martin Eiden, Rebecca König, Benjamin Gutjahr, Wolfgang Baumgärtner, Martin H. Groschup, and et al. 2022. "Rift Valley Fever Virus Non-Structural Protein S Is Associated with Nuclear Translocation of Active Caspase-3 and Inclusion Body Formation" Viruses 14, no. 11: 2487. https://doi.org/10.3390/v14112487
APA StyleMichaely, L. M., Rissmann, M., Armando, F., von Arnim, F., Keller, M., Eiden, M., König, R., Gutjahr, B., Baumgärtner, W., Groschup, M. H., & Ulrich, R. (2022). Rift Valley Fever Virus Non-Structural Protein S Is Associated with Nuclear Translocation of Active Caspase-3 and Inclusion Body Formation. Viruses, 14(11), 2487. https://doi.org/10.3390/v14112487