
Innate Immunity, Inflammation, and Intervention in HBV Infection

 ,
,  and
and 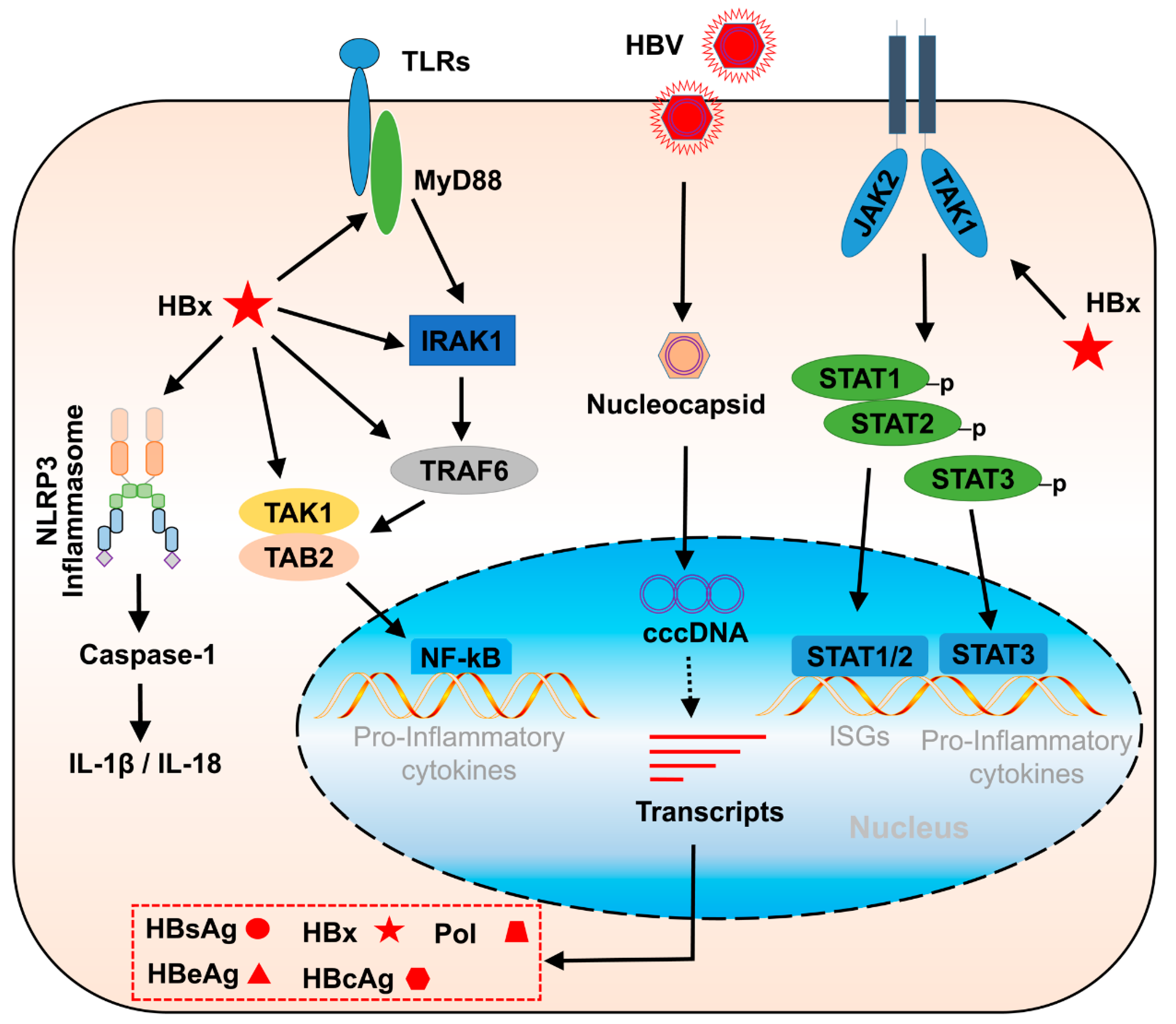{kind=link}
{kind=link}
Abstract
1. Introduction
2. Activation of Innate Immune and Inflammatory Response by Hepatitis B Virus
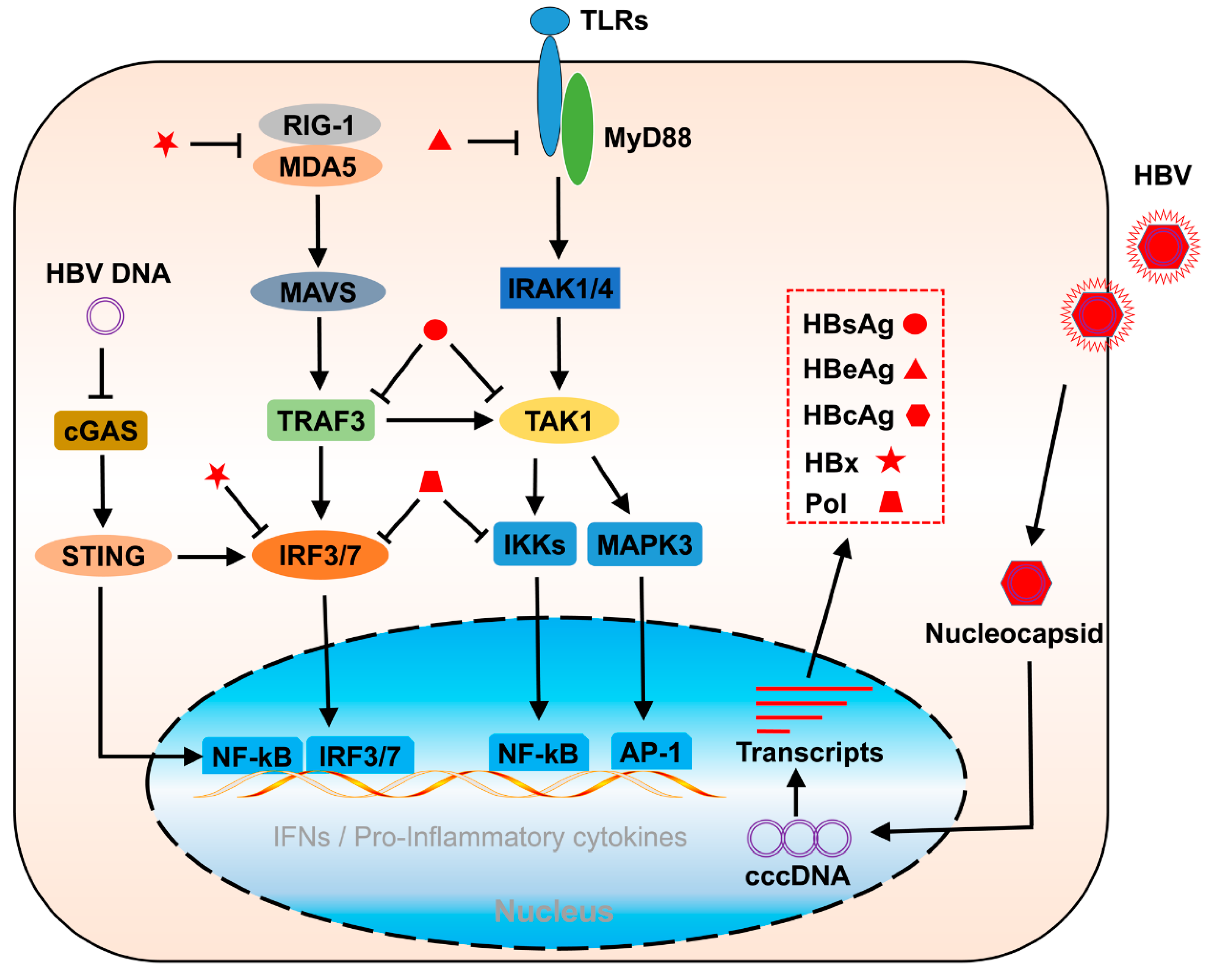
2.1. Toll-like Receptor Pathway Stimulation in HBV Infection
2.2. NLR-mediated Pathway Stimulation in HBV Infection
2.3. RLR-Mediated Pathway Stimulation in HBV Infection
2.4. JAK/STAT Pathway Stimulation in HBV Infection
2.5. NF-κB Signal Stimulation in HBV Infection
3. Mechanism of Immunosuppression in Hepatitis B Virus Infection
3.1. The Immunoregulatory Roles of Hepatitis B Virus e Antigen (HBeAg) and Surface Antigen (HBsAg)
3.2. The regulatory Role of HBV Core Antigen (HBcAg) in Innate Immunity and Inflammation
3.3. The Negative Regulatory Activities of HBx in Innate Immune Response
3.4. Inhibition of Innate Immunity by HBV Polymerase (Pol)
4. Drugs and Therapeutic Strategies Targeting Innate Immunity or Inflammation for HBV Infection
4.1. Antiviral Strategies via Targeting PRRs
4.2. IFN-Based Inhibition of HBV Infection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Sallberg, M.; Pasetto, A. Liver, Tumor and viral hepatitis: Key players in the complex balance between tolerance and Immune Activation. Front. Immunol. 2020, 11, 552. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Xu, Z.; Zhao, K.; Zhong, Y.; Wang, J.; Zhao, L.; Zheng, Z.; Hou, W.; Zhu, C.; Chen, X.; et al. Novel function of SART1 in HNF4alpha transcriptional regulation contributes to its antiviral role during HBV infection. J. Hepatol. 2021, 75, 1072–1082. [Google Scholar] [CrossRef]
- Pattyn, J.; Hendrickx, G.; Vorsters, A.; Van Damme, P. Hepatitis B vaccines. J. Infect. Dis. 2021, 224, S343–S351. [Google Scholar] [CrossRef]
- Fang, Z.L.; Harrison, T.J.; Yang, J.Y.; Chen, Q.Y.; Wang, X.Y.; Mo, J.J. Prevalence of hepatitis B virus infection in a highly endemic area of southern China after catch-up immunization. J. Med. Virol. 2012, 84, 878–884. [Google Scholar] [CrossRef]
- Huang, H.L.; Jeng, K.S.; Hu, C.P.; Tsai, C.H.; Lo, S.J.; Chang, C. Identification and characterization of a structural protein of hepatitis B virus: A polymerase and surface fusion protein encoded by a spliced RNA. Virology 2000, 275, 398–410. [Google Scholar] [CrossRef]
- Kayesh, M.E.H.; Kohara, M.; Tsukiyama-Kohara, K. Toll-like receptor response to hepatitis B virus infection and potential of TLR agonists as immunomodulators for treating chronic hepatitis B: An overview. Int. J. Mol. Sci. 2021, 22, 10462. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Spangenberg, H.C.; Thimme, R.; Blum, H.E. Tracking cccDNA in chronic HBV infection. Hepatology 2004, 39, 1736–1738. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Heptatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef]
- Rouse, B.T.; Sehrawat, S. Immunity and immunopathology to viruses: What decides the outcome? Nat. Rev. Immunol. 2010, 10, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B.; Thimme, R. Insights from antiviral therapy into immune responses to hepatitis B and C virus infection. Gastroenterology 2019, 156, 369–383. [Google Scholar] [CrossRef]
- Kwon, H.; Lok, A.S. Hepatitis B therapy. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 275–284. [Google Scholar] [CrossRef]
- Yousfi, N.; Hattaf, K.; Tridane, A. Modeling the adaptive immune response in HBV infection. J. Math. Biol. 2011, 63, 933–957. [Google Scholar] [CrossRef]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Gut 2012, 61, 1754–1764. [Google Scholar] [CrossRef]
- Chang, J.; Block, T.M.; Guo, J.T. The innate immune response to hepatitis B virus infection: Implications for pathogenesis and therapy. Antiviral Res. 2012, 96, 405–413. [Google Scholar] [CrossRef]
- Katze, M.G.; Fornek, J.L.; Palermo, R.E.; Walters, K.A.; Korth, M.J. Innate immune modulation by RNA viruses: Emerging insights from functional genomics. Nat. Rev. Immunol. 2008, 8, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2002, 99, 15669–15674. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like receptors and the control of immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Li, Q.; Wang, J.; Islam, H.; Kirschning, C.; Lu, H.; Hoffmann, D.; Dittmer, U.; Lu, M. Hepatitis B virus particles activate B cells through the TLR2-MyD88-mTOR axis. Cell Death Dis. 2021, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Zhang, Y.; Yang, X.; Li, M.; Hu, H.; Xiong, J.; Wang, N.; Jin, J.; Zhang, Y.; Song, Y.; et al. Hepatitis B core antigen impairs the polarization while promoting the production of inflammatory cytokines of M2 macrophages via the TLR2 pathway. Front. Immunol. 2020, 11, 535. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Zhu, N.; Yuan, W.J. Effects of hepatitis B virus X gene on apoptosis and expression of immune molecules of human proximal tubular epithelial cells. Arch Virol. 2013, 158, 2479–2485. [Google Scholar] [CrossRef]
- Xiang, W.Q.; Feng, W.F.; Ke, W.; Sun, Z.; Chen, Z.; Liu, W. Hepatitis B virus X protein stimulates IL-6 expression in hepatocytes via a MyD88-dependent pathway. J. Hepatol. 2011, 54, 26–33. [Google Scholar] [CrossRef]
- Liu, S.; Peng, N.; Xie, J.; Hao, Q.; Zhang, M.; Zhang, Y.; Xia, Z.; Xu, G.; Zhao, F.; Wang, Q.; et al. Human hepatitis B virus surface and e antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol. 2015, 62, 1015–1023. [Google Scholar] [CrossRef]
- Tan, G.; Song, H.; Xu, F.; Cheng, G. When hepatitis B virus meets interferons. Front. Microbiol. 2018, 9, 1611. [Google Scholar] [CrossRef]
- Cavlar, T.; Ablasser, A.; Hornung, V. Induction of type I IFNs by intracellular DNA-sensing pathways. Immunol. Cell Biol. 2012, 90, 474–482. [Google Scholar] [CrossRef]
- Zhou, R.; Liu, L.; Wang, Y. Viral proteins recognized by different TLRs. J. Med. Virol. 2021, 93, 6116–6123. [Google Scholar] [CrossRef]
- Nie, L.; Cai, S.Y.; Shao, J.Z.; Chen, J. Toll-like receptors, associated biological roles, and signaling networks in non-mammals. Front. Immunol. 2018, 9, 1523. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, S.; Ma, J.W.; Lei, Z.; Zhu, H.F.; Lei, P.; Yang, Z.S.; Zhang, B.; Yao, X.X.; Shi, C.; et al. Hepatitis B virus protein X-induced expression of the CXC chemokine IP-10 is mediated through activation of NF-kappaB and increases migration of leukocytes. J. Biol. Chem. 2010, 285, 12159–12168. [Google Scholar] [CrossRef]
- Wu, H.; Yang, T.Y.; Li, Y.; Ye, W.L.; Liu, F.; He, X.S.; Wang, J.R.; Gan, W.J.; Li, X.M.; Zhang, S.; et al. Tumor necrosis factor receptor-associated factor 6 promotes hepatocarcinogenesis by interacting with histone deacetylase 3 to enhance c-Myc gene expression and protein stability. Hepatology 2020, 71, 148–163. [Google Scholar] [CrossRef]
- Chen, W.N.; Liu, L.L.; Jiao, B.Y.; Lin, W.S.; Lin, X.J.; Lin, X. Hepatitis B virus X protein increases the IL-1beta-induced NF-kappaB activation via interaction with evolutionarily conserved signaling intermediate in Toll pathways (ECSIT). Virus Res. 2015, 195, 236–245. [Google Scholar] [CrossRef]
- Kaparakis, M.; Philpott, D.J.; Ferrero, R.L. Mammalian NLR proteins; discriminating foe from friend. Immunol. Cell Biol. 2007, 85, 495–502. [Google Scholar] [CrossRef]
- Meunier, E.; Broz, P. Evolutionary Convergence and Divergence in NLR Function and Structure. Trends Immunol. 2017, 38, 744–757. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Ding, X.; Lei, Q.; Li, T.; Li, L.; Qin, B. Hepatitis B core antigen can regulate NLRP3 inflammasome pathway in HepG2 cells. J. Med. Virol. 2019, 91, 1528–1536. [Google Scholar] [CrossRef]
- Xie, W.H.; Ding, J.; Xie, X.X.; Yang, X.H.; Wu, X.F.; Chen, Z.X.; Guo, Q.L.; Gao, W.Y.; Wang, X.Z.; Li, D. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm. Res. 2020, 69, 683–696. [Google Scholar] [CrossRef]
- Okamoto, Y.; Shinjo, K.; Shimizu, Y.; Sano, T.; Yamao, K.; Gao, W.; Fujii, M.; Osada, H.; Sekido, Y.; Murakami, S.; et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 2014, 146, 562–572. [Google Scholar] [CrossRef]
- Yoshino, H.; Saitoh, T.; Kozakai, M.; Kashiwakura, I. Effects of ionizing radiation on retinoic acid-inducible gene-I-like receptors. Biomed. Rep. 2015, 3, 59–62. [Google Scholar] [CrossRef]
- Eisenacher, K.; Krug, A. Regulation of RLR-mediated innate immune signaling—It is all about keeping the balance. Eur. J. Cell Biol. 2012, 91, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.L.; Liao, F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J. Immunol. 2013, 191, 3264–3276. [Google Scholar] [CrossRef]
- Kong, F.; You, H.; Zheng, K.; Tang, R.; Zheng, C. The crosstalk between pattern-recognition receptor signaling and calcium signaling. Int. J. Biol. Macromol. 2021, 192, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.Z.; Bonner, J.A. Bridging radiotherapy to immunotherapy: The IFN-JAK-STAT axis. Int. J. Mol. Sci. 2021, 22, 12295. [Google Scholar] [CrossRef] [PubMed]
- You, H.; Qin, S.; Zhang, F.; Hu, W.; Li, X.; Liu, D.; Kong, F.; Pan, X.; Zheng, K.; Tang, R. Regulation of Pattern-Recognition Receptor Signaling by HBX during Hepatitis B Virus Infection. Front. Immunol. 2022, 13, 829923. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Yun, Y. HBx protein of hepatitis B virus activates Jak1-STAT signaling. J. Biol. Chem. 1998, 273, 25510–25515. [Google Scholar] [CrossRef]
- Quetier, I.; Brezillon, N.; Duriez, M.; Massinet, H.; Giang, E.; Ahodantin, J.; Lamant, C.; Brunelle, M.N.; Soussan, P.; Kremsdorf, D. Hepatitis B virus HBx protein impairs liver regeneration through enhanced expression of IL-6 in transgenic mice. J. Hepatol. 2013, 59, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Zhou, K.; Zhu, T.; Lian, Q.; Tao, Y.; Li, N.; Tu, T.; Bi, Y.; Yang, X.; Pan, X.; et al. Interleukin-34 mediated by hepatitis B virus X protein via CCAAT/enhancer-binding protein alpha contributes to the proliferation and migration of hepatoma cells. Cell Prolif. 2019, 52, e12703. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Su, F.; Schneider, R.J. Hepatitis B virus HBx protein activates transcription factor NF-kappaB by acting on multiple cytoplasmic inhibitors of rel-related proteins. J. Virol. 1996, 70, 4558–4566. [Google Scholar] [CrossRef]
- Bui-Nguyen, T.M.; Pakala, S.B.; Sirigiri, R.D.; Xia, W.; Hung, M.C.; Sarin, S.K.; Kumar, V.; Slagle, B.L.; Kumar, R. NF-kappaB signaling mediates the induction of MTA1 by hepatitis B virus transactivator protein HBx. Oncogene 2010, 29, 1179–1189. [Google Scholar] [CrossRef]
- Jiang, J.; Tang, H. Mechanism of inhibiting type I interferon induction by hepatitis B virus X protein. Protein Cell 2010, 1, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Choi, H.S.; Park, Y.K.; Park, E.S.; Shin, G.C.; Kim, D.H.; Ahn, S.H.; Kim, K.H. HBx-induced NF-kappaB signaling in liver cells is potentially mediated by the ternary complex of HBx with p22-FLIP and NEMO. PLoS ONE 2013, 8, e57331. [Google Scholar]
- Luo, M.X.; Wong, S.H.; Chan, M.T.; Yu, L.; Yu, S.S.; Wu, F.; Xiao, Z.; Wang, X.; Zhang, L.; Cheng, A.S.; et al. Autophagy mediates HhBx-induced nuclear factor-kappaB activation and release of IL-6, IL-8, and CXCL2 in hepatocytes. J. Cell. Physiol. 2015, 230, 2382–2389. [Google Scholar] [CrossRef]
- Yen, C.J.; Lin, Y.J.; Yen, C.S.; Tsai, H.W.; Tsai, T.F.; Chang, K.Y.; Huang, W.C.; Lin, P.W.; Chiang, C.W.; Chang, T.T. Hepatitis B virus X protein upregulates mTOR signaling through IKKbeta to increase cell proliferation and VEGF production in hepatocellular carcinoma. PLoS ONE 2012, 7, e41931. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, J.C.; Kim, J.K.; Kim, H.J.; Lee, H.M.; Choi, M.S.; Maeng, P.J.; Ahn, J.K. Hepatitis B virus X protein enhances NFkappaB activity through cooperating with VBP1. BMB Rep. 2008, 41, 158–163. [Google Scholar] [CrossRef]
- Jiao, B.Y.; Lin, W.S.; She, F.F.; Chen, W.N.; Lin, X. Hepatitis B virus X protein enhances activation of nuclear factor kappaB through interaction with valosin-containing protein. Arch. Virol. 2011, 156, 2015–2021. [Google Scholar] [CrossRef]
- Hong, A.; Han, D.D.; Wright, C.J.; Burch, T.; Piper, J.; Osiowy, C.; Gao, C.; Chiang, S.; Magill, T.; Dick, K.; et al. The interaction between hepatitis B virus X protein and AIB1 oncogene is required for the activation of NFkappaB signal transduction. Biochem. Biophys. Res. Commun. 2012, 423, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.; Yuan, Z. New insights into how HBV manipulates the innate immune response to establish acute and persistent infection. Antivir. Ther. 2013, 18, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, M.; Woziwodzka, A.; Romanowski, T.; Stalke, P.; Dreczewski, M.; Bielawski, K.P. Differences in sequences between HBV-relaxed circular DNA and covalently closed circular DNA. Emerg. Microbes Infect. 2017, 6, e55. [Google Scholar] [CrossRef]
- Mohd-Ismail, N.K.; Lim, Z.; Gunaratne, J.; Tan, Y.J. Mapping the interactions of HBV cccDNA with host factors. Int. J. Mol. Sci. 2019, 20, 4276. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Gummuluru, S.; Hu, J. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 2007, 81, 4465–4472. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.W.; Imam, H.; Khan, M.; Siddiqui, A. N(6)-Methyladenosine modification of hepatitis B and C viral RNAs attenuates host innate immunity via RIG-I signaling. J. Biol. Chem. 2020, 295, 13123–13133. [Google Scholar]
- Kim, G.W.; Imam, H.; Khan, M.; Mir, S.A.; Kim, S.J.; Yoon, S.K.; Hur, W.; Siddiqui, A. HBV-induced increased N6 methyladenosine modification of PTEN RNA affects innate immunity and contributes to HCC. Hepatology 2021, 73, 533–547. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; He, R.; Fang, P.; Li, M.; Yu, H.; Wang, Q.; Yu, Y.; Wang, F.; Zhang, Y.; Chen, A.; et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat. Commun. 2021, 12, 98. [Google Scholar] [CrossRef]
- Verrier, E.R.; Yim, S.A.; Heydmann, L.; El Saghire, H.; Bach, C.; Turon-Lagot, V.; Mailly, L.; Durand, S.C.; Lucifora, J.; Durantel, D.; et al. Hepatitis B virus evasion from cyclic guanosine monophosphate-adenosine monophosphate synthase sensing in human hepatocytes. Hepatology 2018, 68, 1695–1709. [Google Scholar] [PubMed]
- Lauterbach-Riviere, L.; Bergez, M.; Monch, S.; Qu, B.; Riess, M.; Vondran, F.W.R.; Liese, J.; Hornung, V.; Urban, S.; Konig, R. Hepatitis B Virus DNA is a substrate for the cGAS/STING pathway but is not sensed in infected hepatocytes. Viruses 2020, 12, 592. [Google Scholar] [CrossRef]
- Chen, J.; Xu, W.; Chen, Y.; Xie, X.; Zhang, Y.; Ma, C.; Yang, Q.; Han, Y.; Zhu, C.; Xiong, Y.; et al. Matrix metalloproteinase 9 facilitates hepatitis B virus replication through binding with type I interferon (IFN) receptor 1 to repress IFN/JAK/STAT signaling. J. Virol. 2017, 91, e01824-16. [Google Scholar] [CrossRef]
- Bai, L.; Zhang, W.; Tan, L.; Yang, H.; Ge, M.; Zhu, C.; Zhang, R.; Cao, Y.; Chen, J.; Luo, Z.; et al. Hepatitis B virus hijacks CTHRC1 to evade host immunity and maintain replication. J. Mol. Cell Biol. 2015, 7, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhu, J.; Yang, J.; Li, X.; Yuan, J.; Wu, J.; Liu, Z. GP73 facilitates hepatitis B virus replication by repressing the NF-kappaB signaling pathway. J. Med. Virol. 2020, 92, 3327–3335. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Gu, C.; Shen, Z.; Liu, Y.; Wang, W.; Tao, S.; Cui, X.; Liu, J.; Xie, Y. Hepatocyte nuclear factor 1alpha downregulates HBV gene expression and replication by activating the NF-kappaB signaling pathway. PLoS ONE 2017, 12, e0174017. [Google Scholar]
- Lin, S.; Wu, M.; Xu, Y.; Xiong, W.; Yi, Z.; Zhang, X.; Zhenghong, Y. Inhibition of hepatitis B virus replication by MyD88 is mediated by nuclear factor-kappaB activation. Biochim. Biophys. Acta. 2007, 1772, 1150–1157. [Google Scholar] [CrossRef]
- Ren, S.; Wang, J.; Chen, T.L.; Li, H.Y.; Wan, Y.S.; Peng, N.F.; Gui, X.E.; Zhu, Y. Hepatitis B virus stimulated fibronectin facilitates viral maintenance and replication through two distinct mechanisms. PLoS ONE 2016, 11, e0152721. [Google Scholar] [CrossRef]
- Ma, Z.; Cao, Q.; Xiong, Y.; Zhang, E.; Lu, M. Interaction between Hepatitis B virus and Toll-like receptors: Current status and potential therapeutic use for chronic hepatitis B. Vaccines 2018, 6, 6. [Google Scholar] [CrossRef]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef]
- Wu, S.; Kanda, T.; Imazeki, F.; Arai, M.; Yonemitsu, Y.; Nakamoto, S.; Fujiwara, K.; Fukai, K.; Nomura, F.; Yokosuka, O. Hepatitis B virus e antigen downregulates cytokine production in human hepatoma cell lines. Viral Immunol. 2010, 23, 467–476. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, L.; Yang, G.; Zhan, J.; Guo, L.; Chen, Y.; Fan, C.; Liu, D.; Guo, D. Hepatitis B e antigen inhibits NF-kappaB activity by interrupting K63-linked ubiquitination of NEMO. J. Virol. 2019, 93, e00667-18. [Google Scholar]
- Chen, Z.; Cheng, Y.; Xu, Y.; Liao, J.; Zhang, X.; Hu, Y.; Zhang, Q.; Wang, J.; Zhang, Z.; Shen, F.; et al. Expression profiles and function of Toll-like receptors 2 and 4 in peripheral blood mononuclear cells of chronic hepatitis B patients. Clin. Immunol. 2008, 128, 400–408. [Google Scholar] [CrossRef]
- Cheng, J.; Imanishi, H.; Morisaki, H.; Liu, W.; Nakamura, H.; Morisaki, T.; Hada, T. Recombinant HBsAg inhibits LPS-induced COX-2 expression and IL-18 production by interfering with the NFkappaB pathway in a human monocytic cell line, THP-1. J. Hepatol. 2005, 43, 465–471. [Google Scholar] [CrossRef]
- Muller, C.; Zielinski, C.C. Impaired lipopolysaccharide-inducible tumor necrosis factor production in vitro by peripheral blood monocytes of patients with viral hepatitis. Hepatology 1990, 12, 1118–1124. [Google Scholar]
- Wang, S.; Chen, Z.; Hu, C.; Qian, F.; Cheng, Y.; Wu, M.; Shi, B.; Chen, J.; Hu, Y.; Yuan, Z. Hepatitis B virus surface antigen selectively inhibits TLR2 ligand-induced IL-12 production in monocytes/macrophages by interfering with JNK activation. J. Immunol. 2013, 190, 5142–5151. [Google Scholar]
- Jiang, M.; Broering, R.; Trippler, M.; Poggenpohl, L.; Fiedler, M.; Gerken, G.; Lu, M.; Schlaak, J.F. Toll-like receptor-mediated immune responses are attenuated in the presence of high levels of hepatitis B virus surface antigen. J. Viral Hepat. 2014, 21, 860–872. [Google Scholar] [CrossRef]
- Op den Brouw, M.L.; Binda, R.S.; van Roosmalen, M.H.; Protzer, U.; Janssen, H.L.; van der Molen, R.G.; Woltman, A.M. Hepatitis B virus surface antigen impairs myeloid dendritic cell function: A possible immune escape mechanism of hepatitis B virus. Immunology 2009, 126, 280–289. [Google Scholar] [CrossRef]
- Vincent, I.E.; Zannetti, C.; Lucifora, J.; Norder, H.; Protzer, U.; Hainaut, P.; Zoulim, F.; Tommasino, M.; Trepo, C.; Hasan, U.; et al. Hepatitis B virus impairs TLR9 expression and function in plasmacytoid dendritic cells. PLoS ONE 2011, 6, e26315. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, Y.; Shi, B.; Zhang, X.; Wang, J.; Zhang, Z.; Shen, F.; Zhang, Q.; Sun, S.; Yuan, Z. HBsAg inhibits TLR9-mediated activation and IFN-alpha production in plasmacytoid dendritic cells. Mol. Immunol. 2009, 46, 2640–2646. [Google Scholar] [CrossRef]
- Ouaguia, L.; Leroy, V.; Dufeu-Duchesne, T.; Durantel, D.; Decaens, T.; Hubert, M.; Valladeau-Guilemond, J.; Bendriss-Vermare, N.; Chaperot, L.; Aspord, C. Circulating and hepatic BDCA1+, BDCA2+, and BDCA3+ dendritic cells are differentially subverted in patients with chronic HBV infection. Front. Immunol. 2019, 10, 112. [Google Scholar] [CrossRef]
- Tout, I.; Gomes, M.; Ainouze, M.; Marotel, M.; Pecoul, T.; Durantel, D.; Vaccarella, S.; Dubois, B.; Loustaud-Ratti, V.; Walzer, T.; et al. Hepatitis B virus blocks the CRE/CREB complex and prevents TLR9 transcription and function in human B cells. J. Immunol. 2018, 201, 2331–2344. [Google Scholar] [CrossRef]
- Deng, F.; Xu, G.; Cheng, Z.; Huang, Y.; Ma, C.; Luo, C.; Yu, C.; Wang, J.; Xu, X.; Liu, S.; et al. Hepatitis B surface antigen suppresses the activation of nuclear factor kappa B pathway via interaction with the TAK1-TAB2 complex. Front. Immunol. 2021, 12, 618196. [Google Scholar] [CrossRef]
- Han, Q.; Zhang, C.; Zhang, J.; Tian, Z. The role of innate immunity in HBV infection. Semin. Immunopathol. 2013, 35, 23–38. [Google Scholar] [CrossRef]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.P.; Dittmer, U.; Yang, D.; et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2009, 49, 1132–1140. [Google Scholar] [CrossRef]
- Twu, J.S.; Lee, C.H.; Lin, P.M.; Schloemer, R.H. Hepatitis B virus suppresses expression of human beta-interferon. Proc. Natl. Acad. Sci. USA 1988, 85, 252–256. [Google Scholar] [CrossRef]
- Rosmorduc, O.; Sirma, H.; Soussan, P.; Gordien, E.; Lebon, P.; Horisberger, M.; Br Chot, C.; Kremsdorf, D. Inhibition of interferon-inducible MxA protein expression by hepatitis B virus capsid protein. J. Gen. Virol. 1999, 80 Pt 5, 1253–1262. [Google Scholar] [CrossRef]
- Li, T.; Ke, Z.; Liu, W.; Xiong, Y.; Zhu, Y.; Liu, Y. Human Hepatitis B Virus Core Protein Inhibits IFNalpha-Induced IFITM1 Expression by Interacting with BAF200. Viruses 2019, 11, 427. [Google Scholar] [CrossRef]
- Soussan, P.; Garreau, F.; Zylberberg, H.; Ferray, C.; Brechot, C.; Kremsdorf, D. In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J. Clin. Investig. 2000, 105, 55–60. [Google Scholar] [CrossRef]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef]
- Lee, S.; Goyal, A.; Perelson, A.S.; Ishida, Y.; Saito, T.; Gale, M., Jr. Suppression of hepatitis B virus through therapeutic activation of RIG-I and IRF3 signaling in hepatocytes. iScience 2021, 24, 101969. [Google Scholar] [CrossRef]
- Chen, X.; Qian, Y.; Yan, F.; Tu, J.; Yang, X.; Xing, Y.; Chen, Z. 5′-triphosphate-siRNA activates RIG-I-dependent type I interferon production and enhances inhibition of hepatitis B virus replication in HepG2.2.15 cells. Eur. J. Pharmacol. 2013, 721, 86–95. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Mao, A.; Li, C.; Li, Y.; Tien, P. Hepatitis B virus X protein suppresses virus-triggered IRF3 activation and IFN-beta induction by disrupting the VISA-associated complex. Cell Mol. Immunol. 2010, 7, 341–348. [Google Scholar] [CrossRef]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 2016, 12, e1005693. [Google Scholar] [CrossRef] [PubMed]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellaker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef]
- Wang, L.; Sun, Y.; Song, X.; Wang, Z.; Zhang, Y.; Zhao, Y.; Peng, X.; Zhang, X.; Li, C.; Gao, C.; et al. Hepatitis B virus evades immune recognition via RNA adenosine deaminase ADAR1-mediated viral RNA editing in hepatocytes. Cell Mol. Immunol. 2021, 18, 1871–1882. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, I.; Das, D.; Singh, S.P.; Chakravarty, R.; Das, C. Host transcription factor Speckled 110 kDa (Sp110), a nuclear body protein, is hijacked by hepatitis B virus protein X for viral persistence. J. Biol. Chem. 2017, 292, 20379–20393. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.R.; Oh, M.; Koh, S.S.; Malilas, W.; Srisuttee, R.; Jhun, B.H.; Pellegrini, S.; Fuchs, S.Y.; Chung, Y.H. Hepatitis B virus X protein inhibits extracellular IFN-alpha-mediated signal transduction by downregulation of type I IFN receptor. Int. J. Mol. Med. 2012, 29, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Park, E.S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.H.; Kim, C.W.; et al. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5′-UTR of TRIM22. Gut 2018, 67, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.L.; Liu, L.L.; Lu, S.X.; Luo, R.Z.; Wang, C.H.; Wang, H.; Cai, S.H.; Yang, X.; Xie, D.; Zhang, C.Z.; et al. HBx-mediated decrease of AIM2 contributes to hepatocellular carcinoma metastasis. Mol. Oncol. 2017, 11, 1225–1240. [Google Scholar] [CrossRef]
- Liu, D.; Wu, A.; Cui, L.; Hao, R.; Wang, Y.; He, J.; Guo, D. Hepatitis B virus polymerase suppresses NF-kappaB signaling by inhibiting the activity of IKKs via interaction with Hsp90beta. PLoS ONE 2014, 9, e91658. [Google Scholar]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ryu, W.S. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: Implications for immune evasion. PLoS Pathog. 2010, 6, e1000986. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar]
- McMahon, B.J. The natural history of chronic hepatitis B virus infection. Hepatology 2009, 49, S45–S55. [Google Scholar] [CrossRef] [PubMed]
- Isogawa, M.; Robek, M.D.; Furuichi, Y.; Chisari, F.V. Toll-like receptor signaling inhibits hepatitis B virus replication in vivo. J. Virol. 2005, 79, 7269–7272. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lu, M.; Meng, Z.; Trippler, M.; Broering, R.; Szczeponek, A.; Krux, F.; Dittmer, U.; Roggendorf, M.; Gerken, G.; et al. Toll-like receptor-mediated control of HBV replication by nonparenchymal liver cells in mice. Hepatology 2007, 46, 1769–1778. [Google Scholar] [CrossRef]
- Du, K.; Liu, J.; Broering, R.; Zhang, X.; Yang, D.; Dittmer, U.; Lu, M. Recent advances in the discovery and development of TLR ligands as novel therapeutics for chronic HBV and HIV infections. Expert. Opin. Drug Discov. 2018, 13, 661–670. [Google Scholar] [CrossRef]
- Zhang, X.; Kraft, A.; Broering, R.; Schlaak, J.F.; Dittmer, U.; Lu, M. Preclinical development of TLR ligands as drugs for the treatment of chronic viral infections. Expert. Opin. Drug Discov. 2012, 7, 597–611. [Google Scholar] [CrossRef]
- Ma, Z.; Zhang, E.; Yang, D.; Lu, M. Contribution of Toll-like receptors to the control of hepatitis B virus infection by initiating antiviral innate responses and promoting specific adaptive immune responses. Cell Mol. Immunol. 2015, 12, 273–282. [Google Scholar] [CrossRef][Green Version]
- Lanford, R.E.; Guerra, B.; Chavez, D.; Giavedoni, L.; Hodara, V.L.; Brasky, K.M.; Fosdick, A.; Frey, C.R.; Zheng, J.; Wolfgang, G.; et al. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology 2013, 144, 1508–1517. [Google Scholar] [CrossRef] [PubMed]
- Niu, C.; Li, L.; Daffis, S.; Lucifora, J.; Bonnin, M.; Maadadi, S.; Salas, E.; Chu, R.; Ramos, H.; Livingston, C.M.; et al. Toll-like receptor 7 agonist GS-9620 induces prolonged inhibition of HBV via a type I interferon-dependent mechanism. J. Hepatol. 2018, 68, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Ebert, G.; Poeck, H.; Lucifora, J.; Baschuk, N.; Esser, K.; Esposito, I.; Hartmann, G.; Protzer, U. 5′ Triphosphorylated small interfering RNAs control replication of hepatitis B virus and induce an interferon response in human liver cells and mice. Gastroenterology 2011, 141, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Zhang, C.; Han, Q.; Zhang, J.; Tian, Z. Therapeutic recovery of hepatitis B virus (HBV)-induced hepatocyte-intrinsic immune defect reverses systemic adaptive immune tolerance. Hepatology 2013, 58, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Korolowicz, K.E.; Iyer, R.P.; Czerwinski, S.; Suresh, M.; Yang, J.; Padmanabhan, S.; Sheri, A.; Pandey, R.K.; Skell, J.; Marquis, J.K.; et al. Antiviral Efficacy and Host Innate Immunity Associated with SB 9200 Treatment in the Woodchuck Model of Chronic Hepatitis B. PLoS ONE 2016, 11, e0161313. [Google Scholar]
- Li, J.; Lin, S.; Chen, Q.; Peng, L.; Zhai, J.; Liu, Y.; Yuan, Z. Inhibition of hepatitis B virus replication by MyD88 involves accelerated degradation of pregenomic RNA and nuclear retention of pre-S/S RNAs. J. Virol. 2010, 84, 6387–6399. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jiang, D.; Ma, D.; Chang, J.; Dougherty, A.M.; Cuconati, A.; Block, T.M.; Guo, J.T. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J. Virol. 2009, 83, 847–858. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Q.; Zhang, X.; You, L.; Wang, W.; Liu, W.; Han, Y.; Ma, C.; Xu, W.; Chen, J.; et al. HoxA10 facilitates SHP-1-catalyzed dephosphorylation of p38 MAPK/STAT3 to repress hepatitis B virus replication by a feedback regulatory mechanism. J. Virol. 2019, 93, e01607-18. [Google Scholar] [CrossRef]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef]
- Chen, J.; Yuan, Z. Interplay between hepatitis B virus and the innate immune responses: Implications for new therapeutic strategies. Virol. Sin. 2014, 29, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Allweiss, L.; Dandri, M. Experimental in vitro and in vivo models for the study of human hepatitis B virus infection. J. Hepatol. 2016, 64, S17–S31. [Google Scholar] [CrossRef]
- Allweiss, L.; Volz, T.; Lutgehetmann, M.; Giersch, K.; Bornscheuer, T.; Lohse, A.W.; Petersen, J.; Ma, H.; Klumpp, K.; Fletcher, S.P.; et al. Immune cell responses are not required to induce substantial hepatitis B virus antigen decline during pegylated interferon-alpha administration. J. Hepatol. 2014, 60, 500–507. [Google Scholar] [CrossRef]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Shang, J.; Zhang, W.; Gong, G.; Li, Y.; Chen, X.; Jiang, J.; Xie, Q.; Dou, X.; Sun, Y.; et al. HBsAg loss with peg-interferon alfa-2a in hepatitis B patients with partial response to nucleos(t)ide analog: New switch study. J. Clin. Transl. Hepatol. 2018, 6, 25–34. [Google Scholar] [CrossRef]
- Ye, J.; Chen, J. Interferon and hepatitis B: Current and future perspectives. Front. Immunol. 2021, 12, 733364. [Google Scholar] [CrossRef] [PubMed]
- Gordien, E.; Rosmorduc, O.; Peltekian, C.; Garreau, F.; Brechot, C.; Kremsdorf, D. Inhibition of hepatitis B virus replication by the interferon-inducible MxA protein. J. Virol. 2001, 75, 2684–2691. [Google Scholar] [CrossRef]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.-T.; Guo, H. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013, 9, e1003494. [Google Scholar] [CrossRef]
- Mao, R.; Dong, M.; Shen, Z.; Zhang, H.; Liu, Y.; Cai, D.; Mitra, B.; Zhang, J.; Guo, H. RNA helicase DDX17 inhibits hepatitis B virus replication by blocking viral pregenomic RNA encapsidation. J. Virol. 2021, 95, e0044421. [Google Scholar] [CrossRef]
- Pagliaccetti, N.E.; Chu, E.N.; Bolen, C.R.; Kleinstein, S.H.; Robek, M.D. Lambda and alpha interferons inhibit hepatitis B virus replication through a common molecular mechanism but with different in vivo activities. Virology 2010, 401, 197–206. [Google Scholar] [CrossRef]
- Koh, C.; Da, B.L.; Glenn, J.S. HBV/HDV Coinfection: A Challenge for therapeutics. Clin. Liver Dis. 2019, 23, 557–572. [Google Scholar] [CrossRef]
- Cao, Y.; Zhang, R.; Zhang, W.; Zhu, C.; Yu, Y.; Song, Y.; Wang, Q.; Bai, L.; Liu, Y.; Wu, K.; et al. IL-27, a cytokine, and IFN-lambda1, a type III IFN, are coordinated to regulate virus replication through type I IFN. J. Immunol. 2014, 192, 691–703. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Wan, P.; Zhang, Y.; Tan, Q.; Qudus, M.S.; Yue, Z.; Luo, W.; Zhang, W.; Ouyang, J.; Li, Y.; et al. Innate Immunity, Inflammation, and Intervention in HBV Infection. Viruses 2022, 14, 2275. https://doi.org/10.3390/v14102275
Yang G, Wan P, Zhang Y, Tan Q, Qudus MS, Yue Z, Luo W, Zhang W, Ouyang J, Li Y, et al. Innate Immunity, Inflammation, and Intervention in HBV Infection. Viruses. 2022; 14(10):2275. https://doi.org/10.3390/v14102275
Chicago/Turabian StyleYang, Ge, Pin Wan, Yaru Zhang, Qiaoru Tan, Muhammad Suhaib Qudus, Zhaoyang Yue, Wei Luo, Wen Zhang, Jianhua Ouyang, Yongkui Li, and et al. 2022. "Innate Immunity, Inflammation, and Intervention in HBV Infection" Viruses 14, no. 10: 2275. https://doi.org/10.3390/v14102275
APA StyleYang, G., Wan, P., Zhang, Y., Tan, Q., Qudus, M. S., Yue, Z., Luo, W., Zhang, W., Ouyang, J., Li, Y., & Wu, J. (2022). Innate Immunity, Inflammation, and Intervention in HBV Infection. Viruses, 14(10), 2275. https://doi.org/10.3390/v14102275