
HIV-1 Nef Protein Affects Cytokine and Extracellular Vesicles Production in the GEN2.2 Plasmacytoid Dendritic Cell Line

 , , ,
, , , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Isolation and Culture
2.2. Recombinant Nef Protein Preparations and Reagents
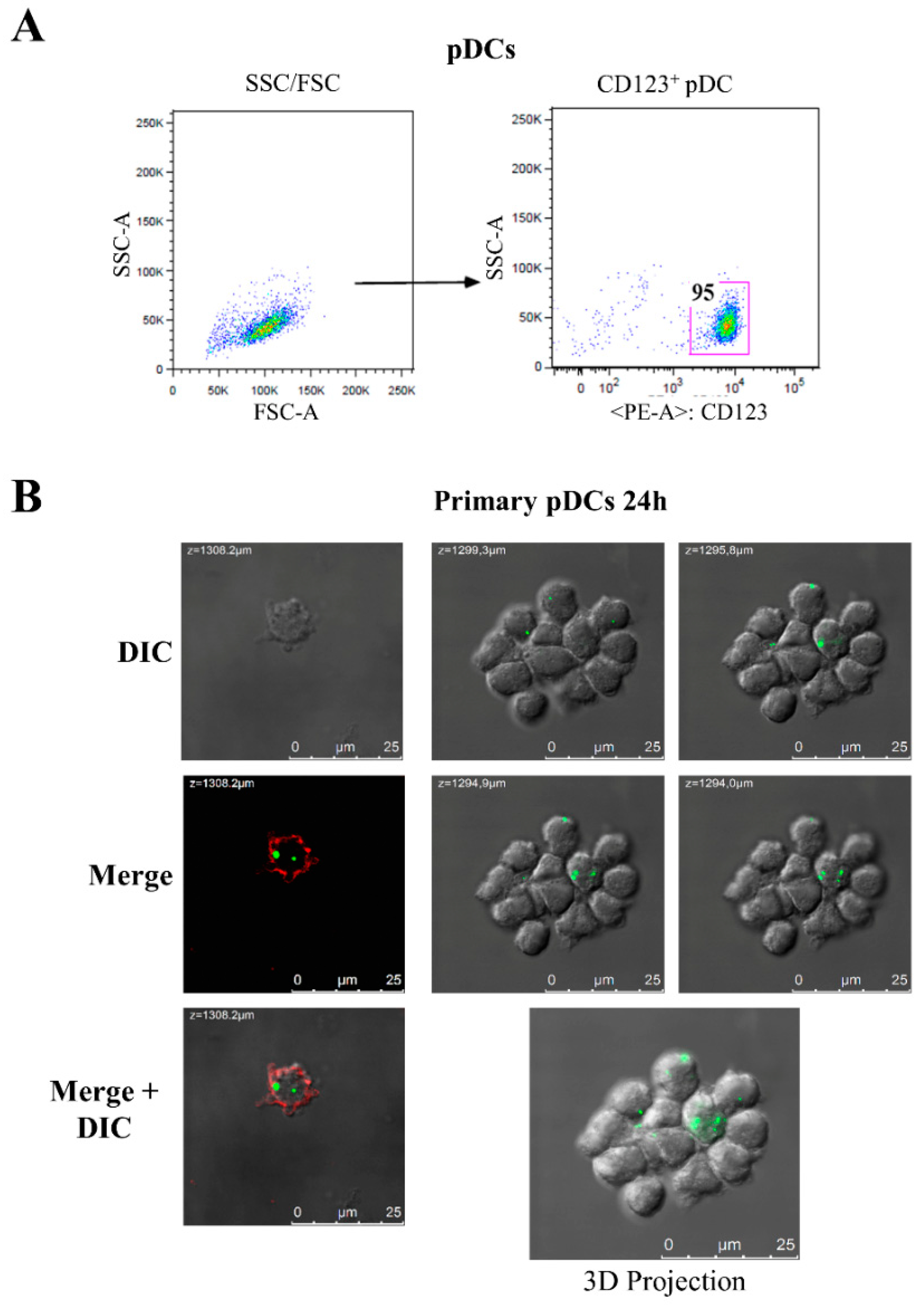
2.3. Flow Cytometry Analysis
2.4. Bodipy FL C16 Reconstitution and Cell Labelling
2.5. Extracellular Vesicle Purification
2.6. Quantification of Vesicles by Flow Cytometry
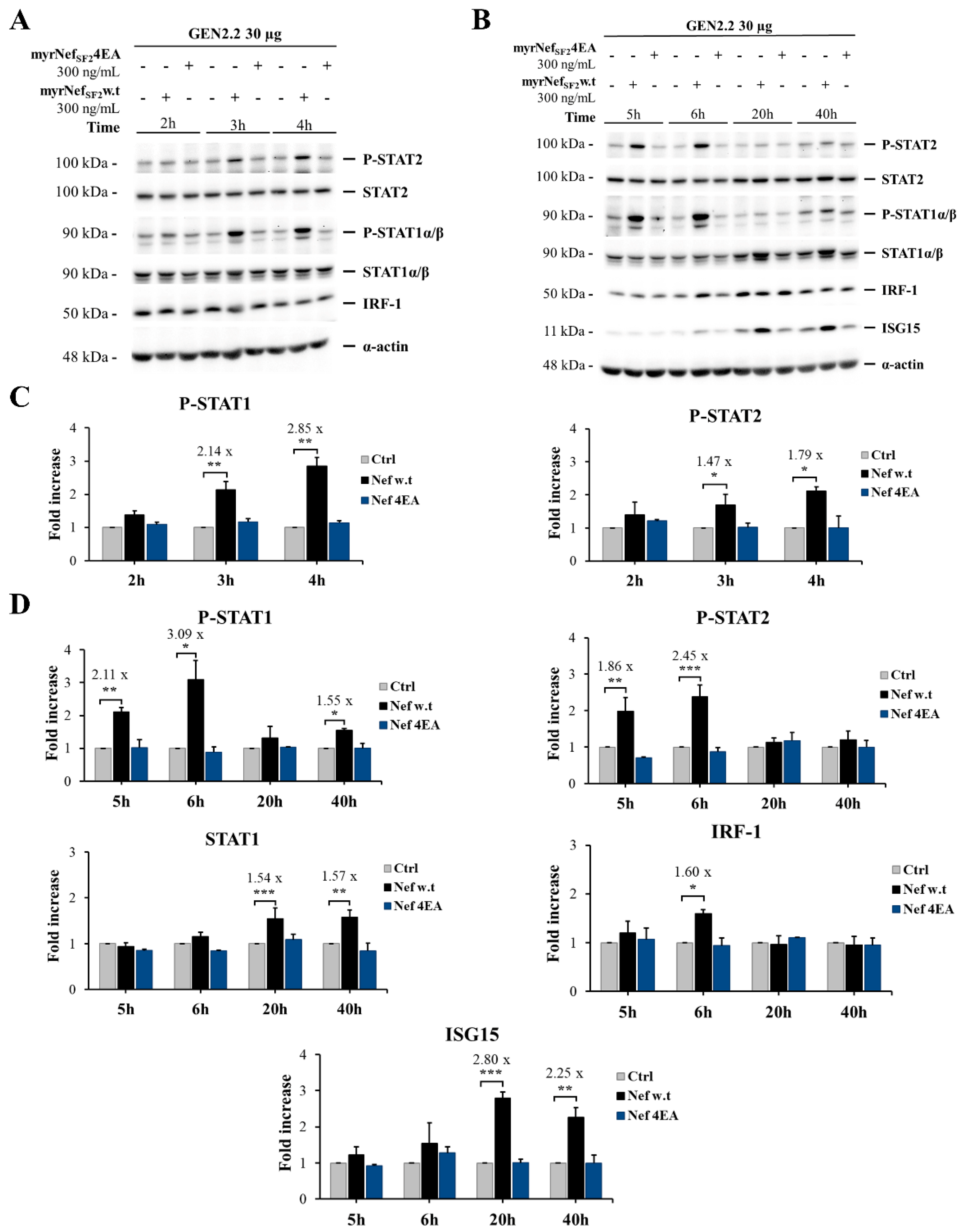
2.7. Western Blot Assay
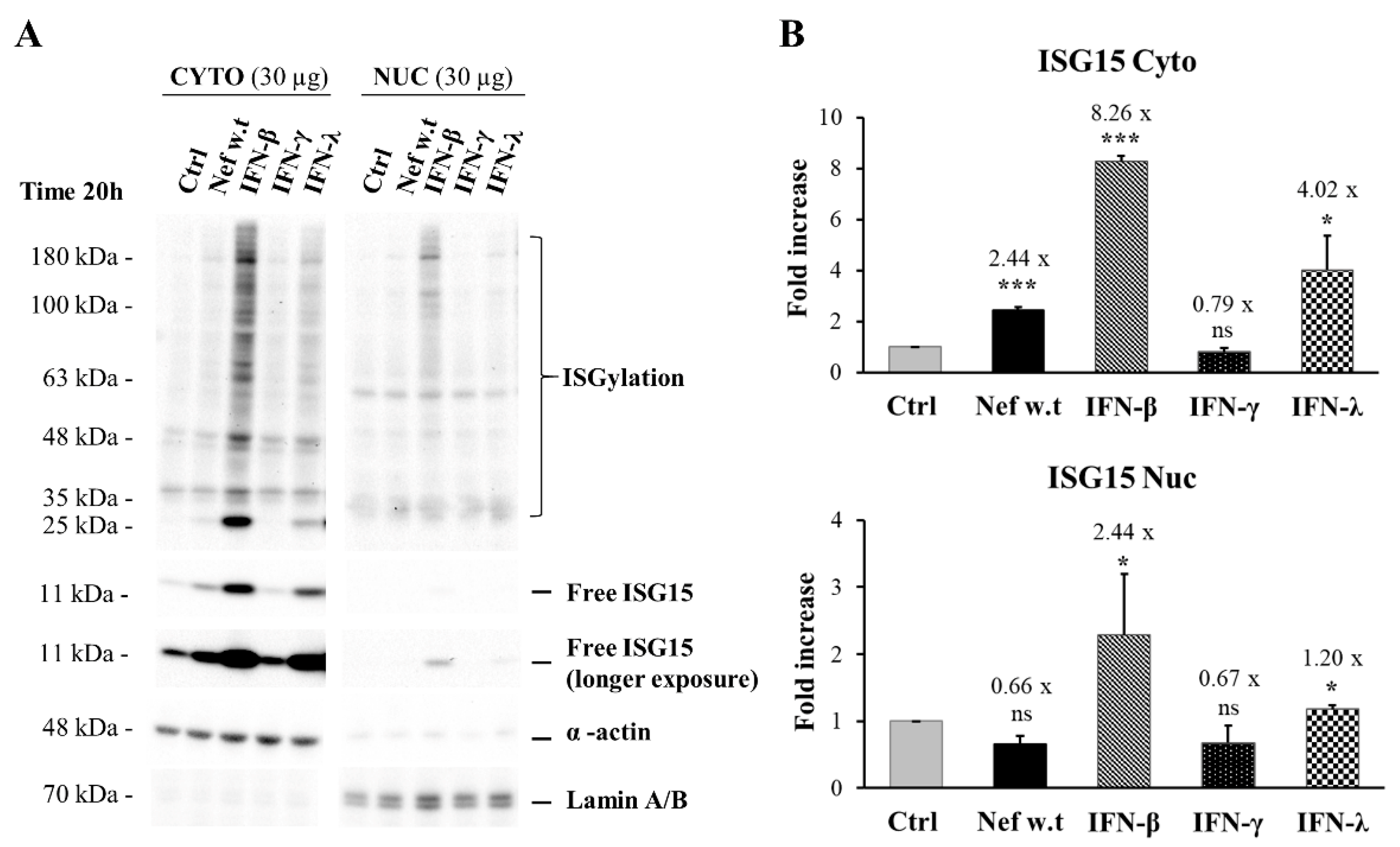
2.8. Nuclear and Cytoplasmic Extract Preparation
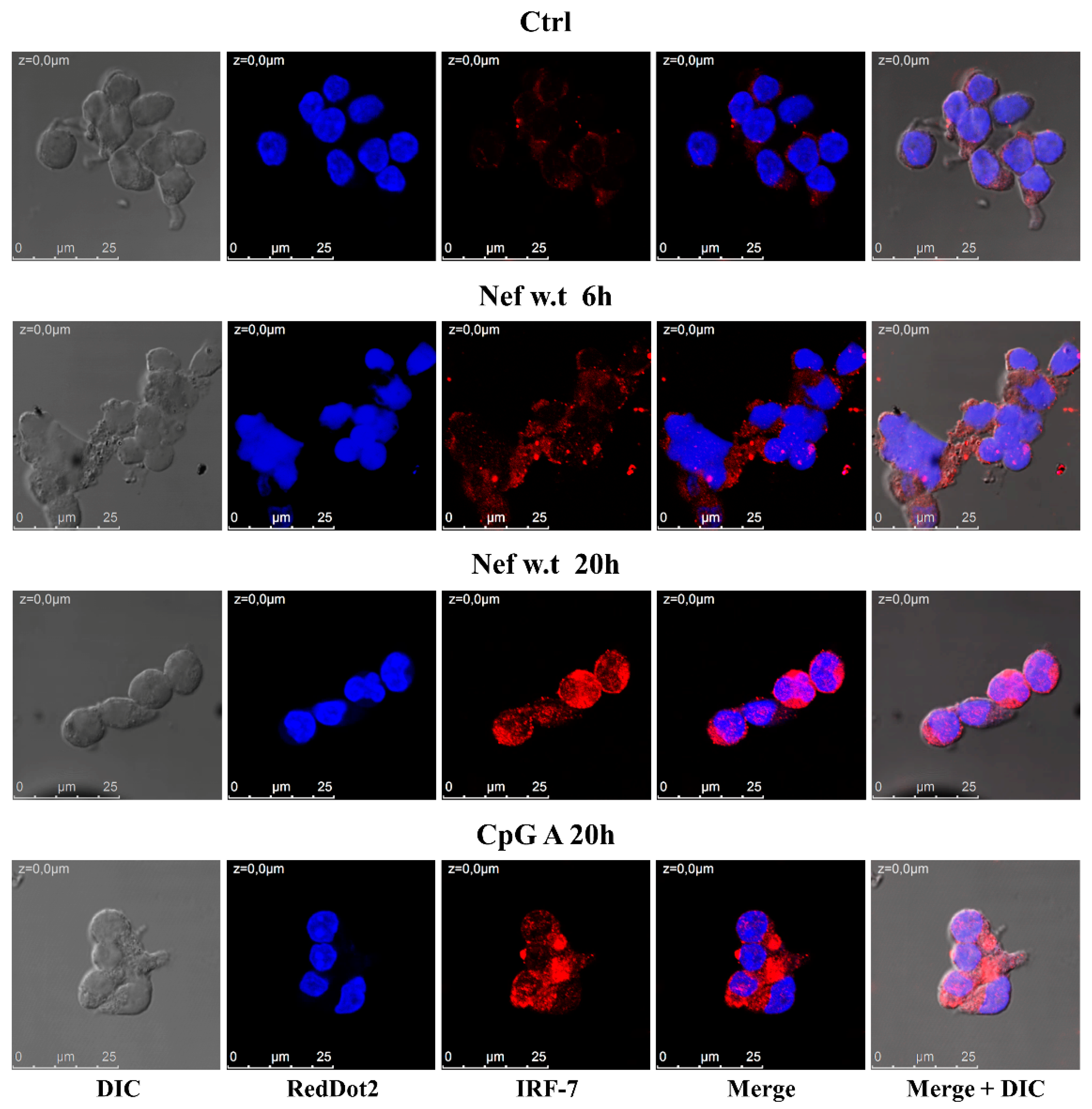
2.9. Confocal Microscopy
2.10. RNA Extraction and Quantitative RT-PCR Analysis
2.11. Bio-Plex Analysis
2.12. Statistical Analysis
3. Results
3.1. myrNefSF2 Induces the Tyrosine Phosphorylation of STAT1 in Human PBLs but Not in PBLs Depleted of pDCs, and Increases mxA Expression
3.2. Nef Induces the Increase and Nuclear Translocation of IRF-7 in Primary pDCs
3.3. GEN2.2 Cell Line as a Model System for Studying the Effects Induced by HIV-1 Nef on pDCs
3.4. HIV-1 myrNefSF2w.t Protein Induces STAT-1 and -2 Tyrosine Phosphorylation and Increases the Expression of Proteins Transcriptionally Regulated by Their Activation
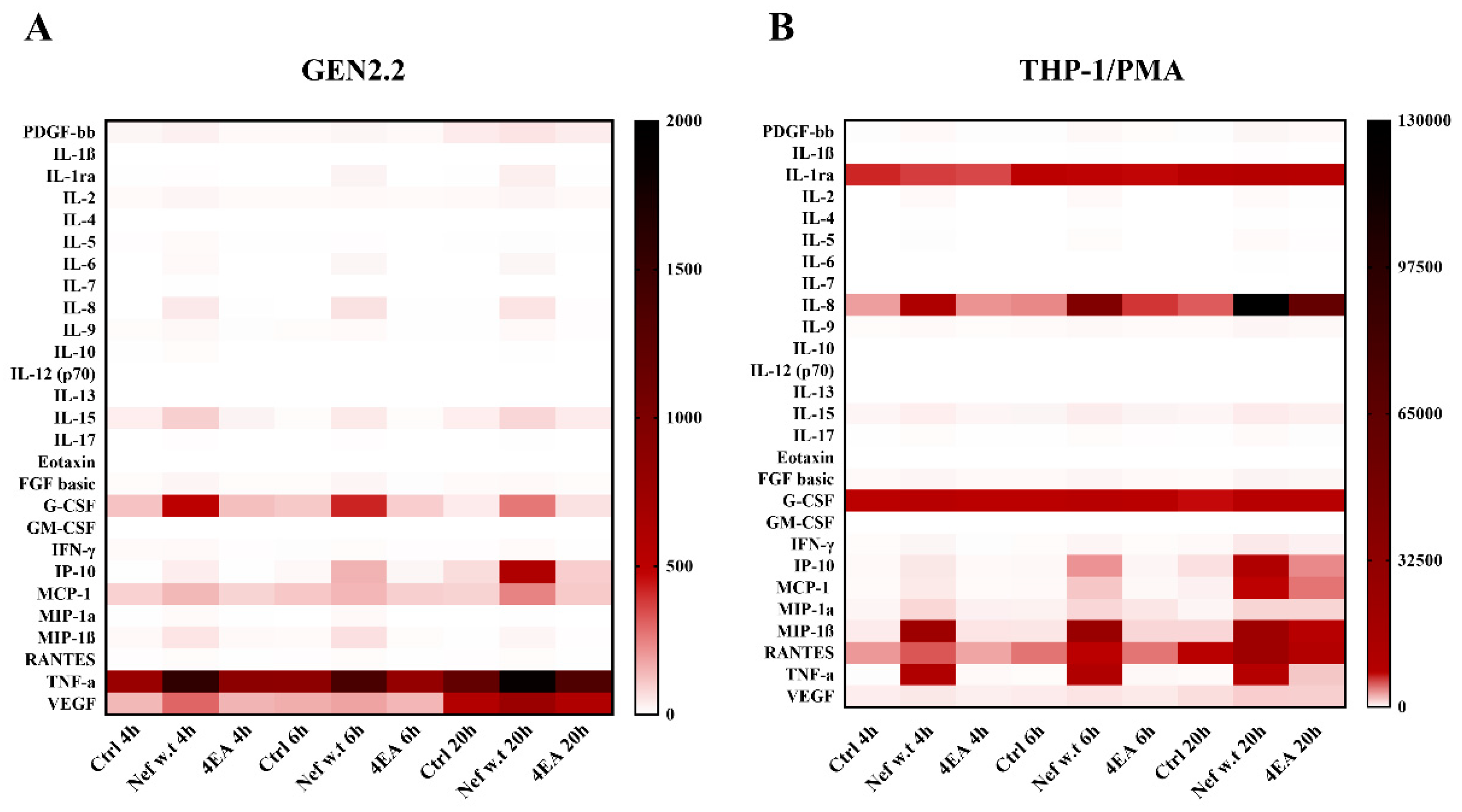
3.5. Wild-Type Nef Induces the Production of a Different Pattern of Cytokines/Chemokines in GEN2.2 Cells Compared to Differentiated THP-1 Cells
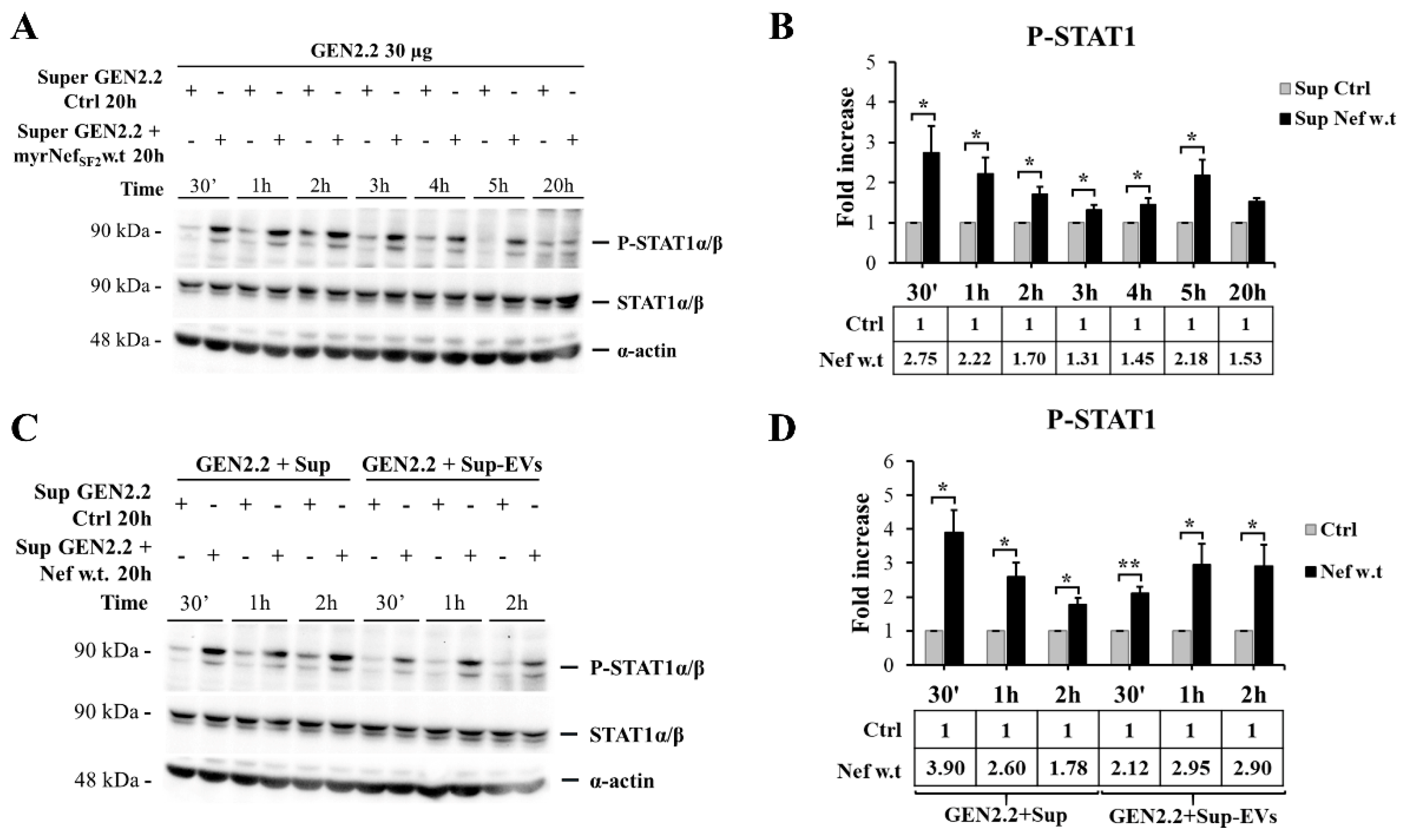
3.6. Supernatant of GEN2.2 Cells Treated with myrNefSF2w.t Stimulates an Early Response of Untreated GEN2.2 Cell Population Independently from the Extracellular Vesicle (EV) Content
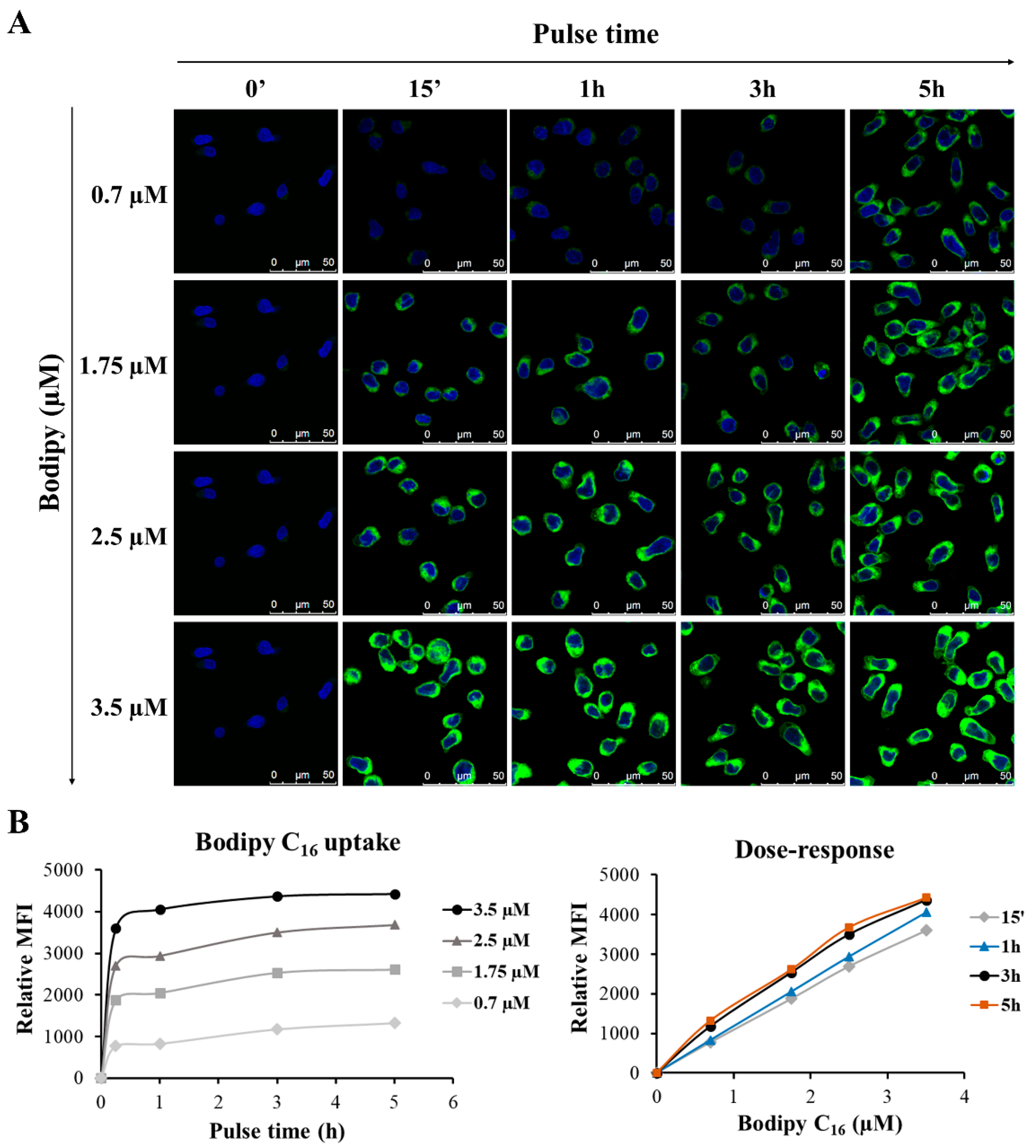
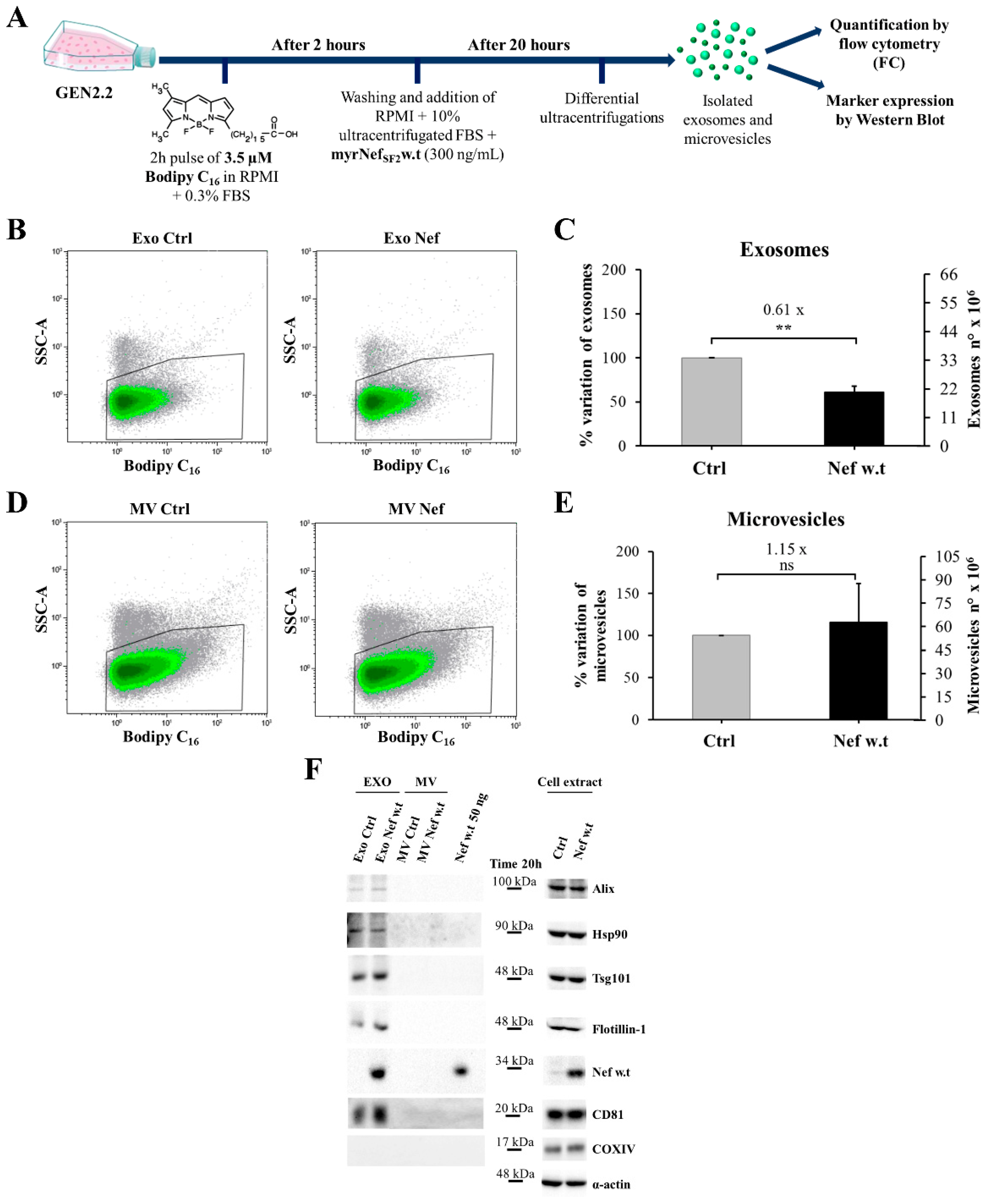
3.7. Set Up of the Protocol for GEN2.2 Cell Labelling with Bodipy C16
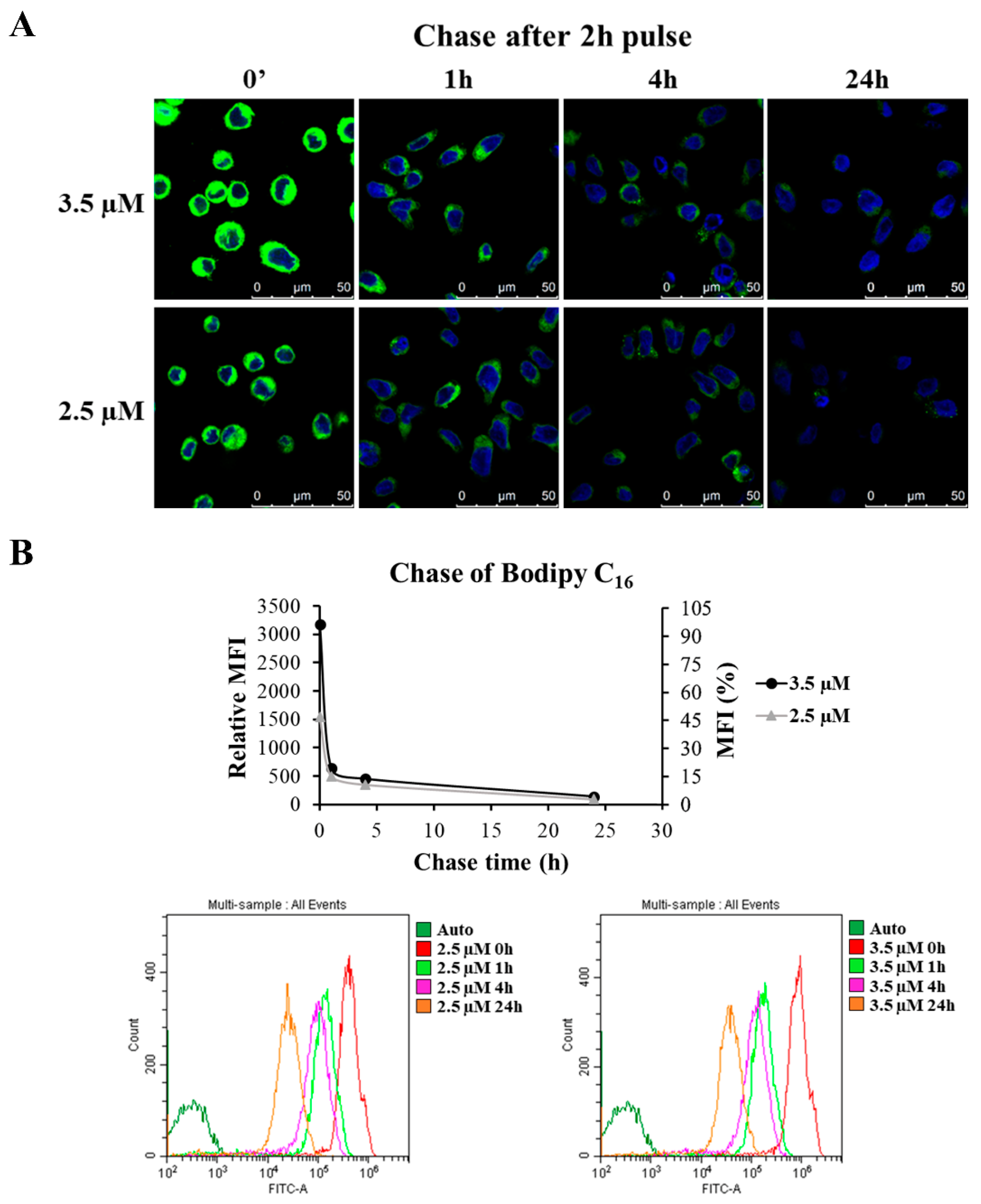
3.8. Nef Reduces the Exosome Production and Is Found Associated with the Exosomal Fraction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lande, R.; Gilliet, M. Plasmacytoid Dendritic Cells: Key Players in the Initiation and Regulation of Immune Responses. Ann. N. Y. Acad. Sci. 2010, 1183, 89–103. [Google Scholar] [CrossRef]
- Swiecki, M.; Colonna, M. The Multifaceted Biology of Plasmacytoid Dendritic Cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Fitzgerald-Bocarsly, P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönnblom, L.; Eloranta, M.-L. The Interferon Signature in Autoimmune Diseases. Curr. Opin. Rheumatol. 2013, 25, 248–253. [Google Scholar] [CrossRef]
- Cao, W. Pivotal Functions of Plasmacytoid Dendritic Cells in Systemic Autoimmune Pathogenesis. J. Clin. Cell. Immunol. 2014, 5, 212. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, M.; Manches, O.; Bhardwaj, N. Plasmacytoid Dendritic Cells in HIV Infection. Adv. Exp. Med. Biol. 2013, 762, 71–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Estes, J.D.; Schlievert, P.M.; Duan, L.; Brosnahan, A.J.; Southern, P.J.; Reilly, C.S.; Peterson, M.L.; Schultz-Darken, N.; Brunner, K.G.; et al. Glycerol Monolaurate Prevents Mucosal SIV Transmission. Nature 2009, 458, 1034–1038. [Google Scholar] [CrossRef]
- Reszka-Blanco, N.J.; Sivaraman, V.; Zhang, L.; Su, L. HIV-1 Env and Nef Cooperatively Contribute to Plasmacytoid Dendritic Cell Activation via CD4-Dependent Mechanisms. J. Virol. 2015, 89, 7604–7611. [Google Scholar] [CrossRef] [Green Version]
- Haupt, S.; Donhauser, N.; Chaipan, C.; Schuster, P.; Puffer, B.; Daniels, R.S.; Greenough, T.C.; Kirchhoff, F.; Schmidt, B. CD4 Binding Affinity Determines Human Immunodeficiency Virus Type 1-Induced Alpha Interferon Production in Plasmacytoid Dendritic Cells. J. Virol. 2008, 82, 8900–8905. [Google Scholar] [CrossRef] [Green Version]
- Beignon, A.-S. Endocytosis of HIV-1 Activates Plasmacytoid Dendritic Cells via Toll-like Receptor- Viral RNA Interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef] [Green Version]
- Pritschet, K.; Donhauser, N.; Schuster, P.; Ries, M.; Haupt, S.; Kittan, N.A.; Korn, K.; Pöhlmann, S.; Holland, G.; Bannert, N.; et al. CD4- and Dynamin-Dependent Endocytosis of HIV-1 into Plasmacytoid Dendritic Cells. Virology 2012, 423, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Aiello, A.; Giannessi, F.; Percario, Z.A.; Affabris, E. The Involvement of Plasmacytoid Cells in HIV Infection and Pathogenesis. Cytokine Growth Factor Rev. 2018, 40, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Munn, D.; Fallahi, A.; Lifson, J.; Chaperot, L.; Plumas, J.; Bhardwaj, N. HIV-Activated Human Plasmacytoid DCs Induce Tregs through an Indoleamine 2,3-Dioxygenase-Dependent Mechanism. J. Clin. Investig. 2008, 118, 3431–3439. [Google Scholar] [CrossRef]
- Hanna, Z.; Kay, D.G.; Rebai, N.; Guimond, A.; Jothy, S.; Jolicoeur, P. Nef Harbors a Major Determinant of Pathogenicity for an AIDS-like Disease Induced by HIV-1 in Transgenic Mice. Cell 1998, 95, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Arold, S.T.; Baur, A.S. Dynamic Nef and Nef Dynamics: How Structure Could Explain the Complex Activities of This Small HIV Protein. Trends Biochem. Sci. 2001, 26, 356–363. [Google Scholar] [CrossRef]
- Geyer, M.; Fackler, O.T.; Peterlin, B.M. Structure—Function Relationships in HIV-1 Nef. EMBO Rep. 2001, 2, 580–585. [Google Scholar] [CrossRef]
- Swingler, S.; Mann, A.; Jacqué, J.; Brichacek, B.; Sasseville, V.G.; Williams, K.; Lackner, A.A.; Janoff, E.N.; Wang, R.; Fisher, D.; et al. HIV-1 Nef Mediates Lymphocyte Chemotaxis and Activation by Infected Macrophages. Nat. Med. 1999, 5, 997–1003. [Google Scholar] [CrossRef]
- Mangino, G.; Percario, Z.A.; Fiorucci, G.; Vaccari, G.; Manrique, S.; Romeo, G.; Federico, M.; Geyer, M.; Affabris, E. In Vitro Treatment of Human Monocytes/Macrophages with Myristoylated Recombinant Nef of Human Immunodeficiency Virus Type 1 Leads to the Activation of Mitogen-Activated Protein Kinases, IκB Kinases, and Interferon Regulatory Factor 3 and to the Release of Beta Interferon. J. Virol. 2007, 81, 2777–2791. [Google Scholar] [CrossRef] [Green Version]
- Mangino, G.; Percario, Z.A.; Fiorucci, G.; Vaccari, G.; Acconcia, F.; Chiarabelli, C.; Leone, S.; Noto, A.; Horenkamp, F.A.; Manrique, S.; et al. HIV-1 Nef Induces Proinflammatory State in Macrophages through Its Acidic Cluster Domain: Involvement of TNF Alpha Receptor Associated Factor 2. PLoS ONE 2011, 6, e22982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Federico, M.; Percario, Z.; Olivetta, E.; Fiorucci, G.; Muratori, C.; Micheli, A.; Romeo, G.; Affabris, E. HIV-1 Nef Activates STAT1 in Human Monocytes/Macrophages through the Release of Soluble Factors. Blood 2001, 98, 2752–2761. [Google Scholar] [CrossRef] [Green Version]
- Olivetta, E.; Percario, Z.; Fiorucci, G.; Mattia, G.; Schiavoni, I.; Dennis, C.; Jäger, J.; Harris, M.; Romeo, G.; Affabris, E.; et al. HIV-1 Nef Induces the Release of Inflammatory Factors from Human Monocyte/Macrophages: Involvement of Nef Endocytotic Signals and NF-Kappa B Activation. J. Immunol. 2003, 170, 1716–1727. [Google Scholar] [CrossRef]
- Percario, Z.; Olivetta, E.; Fiorucci, G.; Mangino, G.; Peretti, S.; Romeo, G.; Affabris, E.; Federico, M. Human Immunodeficiency Virus Type 1 (HIV-1) Nef Activates STAT3 in Primary Human Monocyte/Macrophages through the Release of Soluble Factors: Involvement of Nef Domains Interacting with the Cell Endocytotic Machinery. J. Leukoc. Biol. 2003, 74, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Muratori, C.; Cavallin, L.E.; Krätzel, K.; Tinari, A.; De Milito, A.; Fais, S.; D’Aloja, P.; Federico, M.; Vullo, V.; Fomina, A.; et al. Massive Secretion by T Cells Is Caused by HIV Nef in Infected Cells and by Nef Transfer to Bystander Cells. Cell Host Microbe 2009, 6, 218–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenassi, M.; Cagney, G.; Liao, M.; Vaupotic, T.; Bartholomeeusen, K.; Cheng, Y.; Krogan, N.J.; Plemenitas, A.; Peterlin, B.M. HIV Nef Is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4+ T Cells. Traffic Cph. Den. 2010, 11, 110–122. [Google Scholar] [CrossRef] [Green Version]
- Pužar Dominkuš, P.; Ferdin, J.; Plemenitaš, A.; Peterlin, B.M.; Lenassi, M. Nef Is Secreted in Exosomes from Nef.GFP-Expressing and HIV-1-Infected Human Astrocytes. J. Neurovirol. 2017, 23, 713–724. [Google Scholar] [CrossRef]
- Giannessi, F.; Aiello, A.; Franchi, F.; Percario, Z.A.; Affabris, E. The Role of Extracellular Vesicles as Allies of HIV, HCV and SARS Viruses. Viruses 2020, 12, 571. [Google Scholar] [CrossRef] [PubMed]
- Arenaccio, C.; Chiozzini, C.; Columba-Cabezas, S.; Manfredi, F.; Affabris, E.; Baur, A.; Federico, M. Exosomes from Human Immunodeficiency Virus Type 1 (HIV-1)-Infected Cells License Quiescent CD4+ T Lymphocytes to Replicate HIV-1 through a Nef- and ADAM17-Dependent Mechanism. J. Virol. 2014, 88, 11529–11539. [Google Scholar] [CrossRef] [Green Version]
- Arenaccio, C.; Chiozzini, C.; Columba-Cabezas, S.; Manfredi, F.; Federico, M. Cell Activation and HIV-1 Replication in Unstimulated CD4+ T Lymphocytes Ingesting Exosomes from Cells Expressing Defective HIV-1. Retrovirology 2014, 11, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arenaccio, C.; Anticoli, S.; Manfredi, F.; Chiozzini, C.; Olivetta, E.; Federico, M. Latent HIV-1 Is Activated by Exosomes from Cells Infected with Either Replication-Competent or Defective HIV-1. Retrovirology 2015, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Ostalecki, C.; Wittki, S.; Lee, J.-H.; Geist, M.M.; Tibroni, N.; Harrer, T.; Schuler, G.; Fackler, O.T.; Baur, A.S. HIV Nef- and Notch1-Dependent Endocytosis of ADAM17 Induces Vesicular TNF Secretion in Chronic HIV Infection. EBioMedicine 2016, 13, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Raymond, A.D.; Diaz, P.; Chevelon, S.; Agudelo, M.; Yndart-Arias, A.; Ding, H.; Kaushik, A.; Jayant, R.D.; Nikkhah-Moshaie, R.; Roy, U.; et al. Microglia-Derived HIV Nef+ Exosome Impairment of the Blood-Brain Barrier Is Treatable by Nanomedicine-Based Delivery of Nef Peptides. J. Neurovirol. 2016, 22, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Baur, A.S. HIV-Nef and AIDS Pathogenesis: Are We Barking up the Wrong Tree? Trends Microbiol. 2011, 19, 435–440. [Google Scholar] [CrossRef]
- Campbell, T.D.; Khan, M.; Huang, M.-B.; Bond, V.C.; Powell, M.D. HIV-1 Nef Protein Is Secreted into Vesicles That Can Fuse with Target Cells and Virions. Ethn. Dis. 2008, 18, S2. [Google Scholar] [PubMed]
- McNamara, R.P.; Costantini, L.M.; Myers, T.A.; Schouest, B.; Maness, N.J.; Griffith, J.D.; Damania, B.A.; MacLean, A.G.; Dittmer, D.P. Nef Secretion into Extracellular Vesicles or Exosomes Is Conserved across Human and Simian Immunodeficiency Viruses. mBio 2018, 9, e02344-17. [Google Scholar] [CrossRef] [Green Version]
- Raymond, A.D.; Campbell-Sims, T.C.; Khan, M.; Lang, M.; Huang, M.B.; Bond, V.C.; Powell, M.D. HIV Type 1 Nef Is Released from Infected Cells in CD45(+) Microvesicles and Is Present in the Plasma of HIV-Infected Individuals. AIDS Res. Hum. Retrovir. 2011, 27, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, Y.; Otake, K.; Tashiro, M.; Adachi, A. Soluble Nef Antigen of HIV-1 Is Cytotoxic for Human CD4+ T Cells. FEBS Lett. 1996, 393, 93–96. [Google Scholar] [CrossRef] [Green Version]
- Ameisen, J.C.; Guy, B.; Chamaret, S.; Loche, M.; Mouton, Y.; Neyrinck, J.L.; Khalife, J.; Leprevost, C.; Beaucaire, G.; Boutillon, C. Antibodies to the Nef Protein and to Nef Peptides in HIV-1-Infected Seronegative Individuals. AIDS Res. Hum. Retrovir. 1989, 5, 279–291. [Google Scholar] [CrossRef]
- Chaperot, L.; Blum, A.; Manches, O.; Lui, G.; Angel, J.; Molens, J.-P.; Plumas, J. Virus or TLR Agonists Induce TRAIL-Mediated Cytotoxic Activity of Plasmacytoid Dendritic Cells. J. Immunol. 2006, 176, 248–255. [Google Scholar] [CrossRef]
- Lund, M.E.; To, J.; O’Brien, B.A.; Donnelly, S. The Choice of Phorbol 12-Myristate 13-Acetate Differentiation Protocol Influences the Response of THP-1 Macrophages to a pro-Inflammatory Stimulus. J. Immunol. Methods 2016, 430, 64–70. [Google Scholar] [CrossRef]
- Breuer, S.; Gerlach, H.; Kolaric, B.; Urbanke, C.; Opitz, N.; Geyer, M. Biochemical Indication for Myristoylation-Dependent Conformational Changes in HIV-1 Nef. Biochemistry 2006, 45, 2339–2349. [Google Scholar] [CrossRef]
- Coscia, C.; Parolini, I.; Sanchez, M.; Biffoni, M.; Boussadia, Z.; Zanetti, C.; Fiani, M.L.; Sargiacomo, M. Generation, Quantification, and Tracing of Metabolically Labeled Fluorescent Exosomes. In Lentiviral Vectors and Exosomes as Gene and Protein Delivery Tools; Federico, M., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; Volume 1448, pp. 217–235. ISBN 978-1-4939-3751-6. [Google Scholar]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal Information for Studies of Extracellular Vesicles 2018 (MISEV2018): A Position Statement of the International Society for Extracellular Vesicles and Update of the MISEV2014 Guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Haller, O.; Kochs, G. Human MxA Protein: An Interferon-Induced Dynamin-like GTPase with Broad Antiviral Activity. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2011, 31, 79–87. [Google Scholar] [CrossRef]
- Zhou, J.-H.; Wang, Y.-N.; Chang, Q.-Y.; Ma, P.; Hu, Y.; Cao, X. Type III Interferons in Viral Infection and Antiviral Immunity. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 51, 173–185. [Google Scholar] [CrossRef] [PubMed]
- McKenna, K.; Beignon, A.-S.; Bhardwaj, N. Plasmacytoid Dendritic Cells: Linking Innate and Adaptive Immunity. J. Virol. 2005, 79, 17–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Sáez, P.; Varela, N.; Luque, M.J.; Toro-Domínguez, D.; Martorell-Marugan, J.; Alarcón-Riquelme, M.E.; Marañón, C. Metagene Projection Characterizes GEN2.2 and CAL-1 as Relevant Human Plasmacytoid Dendritic Cell Models. Bioinform. Oxf. Engl. 2017, 33, 3691–3695. [Google Scholar] [CrossRef] [Green Version]
- Perng, Y.C.; Lenschow, D.J. ISG15 in Antiviral Immunity and Beyond. Nat. Rev. Microbiol. 2018, 16, 423–439. [Google Scholar] [CrossRef] [PubMed]
- Villarroya-Beltri, C.; Baixauli, F.; Mittelbrunn, M.; Fernández-Delgado, I.; Torralba, D.; Moreno-Gonzalo, O.; Baldanta, S.; Enrich, C.; Guerra, S.; Sánchez-Madrid, F. ISGylation Controls Exosome Secretion by Promoting Lysosomal Degradation of MVB Proteins. Nat. Commun. 2016, 7, 13588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Wang, X.; Zhou, Y.; Zhou, R.H.; Ho, W.-Z.; Li, J.L. Exosomes Contribute to the Transmission of Anti-HIV Activity from TLR3-Activated Brain Microvascular Endothelial Cells to Macrophages. Antivir. Res. 2016, 134, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Fitzgerald, W.; Freeman, M.L.; Lederman, M.M.; Vasilieva, E.; Romero, R.; Margolis, L. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Sci. Rep. 2018, 8, 8973. [Google Scholar] [CrossRef] [Green Version]
- Aiello, A.; Giannessi, F.; Percario, Z.A.; Affabris, E. An Emerging Interplay between Extracellular Vesicles and Cytokines. Cytokine Growth Factor Rev. 2020, 51, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Konadu, K.A.; Chu, J.; Huang, M.B.; Amancha, P.K.; Armstrong, W.; Powell, M.D.; Villinger, F.; Bond, V.C. Association of Cytokines with Exosomes in the Plasma of HIV-1-Seropositive Individuals. J. Infect. Dis. 2015, 211, 1712–1716. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological Properties of Extracellular Vesicles and Their Physiological Functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, M.V.S.; Costa, C.S.; da Silva, L.L.P. The Ambiguous Roles of Extracellular Vesicles in HIV Replication and Pathogenesis. Front. Microbiol. 2018, 9, 2411. [Google Scholar] [CrossRef] [Green Version]
- Pérez, P.S.; Romaniuk, M.A.; Duette, G.A.; Zhao, Z.; Huang, Y.; Martin-Jaular, L.; Witwer, K.W.; Théry, C.; Ostrowski, M. Extracellular Vesicles and Chronic Inflammation during HIV Infection. J. Extracell. Vesicles 2019, 8, 1687275. [Google Scholar] [CrossRef] [PubMed]
- Zuidscherwoude, M.; Worah, K.; van der Schaaf, A.; Buschow, S.I.; van Spriel, A.B. Differential Expression of Tetraspanin Superfamily Members in Dendritic Cell Subsets. PLoS ONE 2017, 12, e0184317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Santini, P.A.; Sullivan, J.S.; He, B.; Shan, M.; Ball, S.C.; Dyer, W.B.; Ketas, T.J.; Chadburn, A.; Cohen-Gould, L.; et al. HIV-1 Evades Virus-Specific IgG2 and IgA Responses by Targeting Systemic and Intestinal B Cells via Long-Range Intercellular Conduits. Nat. Immunol. 2009, 10, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Percario, Z.A.; Ali, M.; Mangino, G.; Affabris, E. Nef, the Shuttling Molecular Adaptor of HIV, Influences the Cytokine Network. Cytokine Growth Factor Rev. 2015, 26, 159–173. [Google Scholar] [CrossRef]
- Wang, Z.; Shang, H.; Jiang, Y. Chemokines and Chemokine Receptors: Accomplices for Human Immunodeficiency Virus Infection and Latency. Front. Immunol. 2017, 8, 1274. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in Infectious Diseases Pathogenesis and Potential Therapeutic Implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Takano, S.; Ishikawa, E.; Matsuda, M.; Yamamoto, T.; Matsumura, A. Interferon-β Inhibits Glioma Angiogenesis through Downregulation of Vascular Endothelial Growth Factor and Upregulation of Interferon Inducible Protein 10. Int. J. Oncol. 2014, 45, 1837–1846. [Google Scholar] [CrossRef] [Green Version]
- Padovan, E.; Spagnoli, G.C.; Ferrantini, M.; Heberer, M. IFN-Alpha2a Induces IP-10/CXCL10 and MIG/CXCL9 Production in Monocyte-Derived Dendritic Cells and Enhances Their Capacity to Attract and Stimulate CD8+ Effector T Cells. J. Leukoc. Biol. 2002, 71, 669–676. [Google Scholar] [PubMed]
- Lei, J.; Yin, X.; Shang, H.; Jiang, Y. IP-10 Is Highly Involved in HIV Infection. Cytokine 2019, 115, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sankapal, S.; Gupta, P.; Ratner, D.; Ding, M.; Shen, C.; Sanyal, A.; Stolz, D.; Cu-Uvin, S.; Ramratnam, B.; Chen, Y. HIV Exposure to the Epithelia in Ectocervical and Colon Tissues Induces Inflammatory Cytokines without Tight Junction Disruption. AIDS Res. Hum. Retrovir. 2016, 32, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, R.P.; Scully, E.P.; Groden, E.E.; Arnold, K.B.; Chang, J.J.; Lane, K.; Lifson, J.; Rosenberg, E.; Lauffenburger, D.A.; Altfeld, M. HIV-1 Infection Induces Strong Production of IP-10 through TLR7/9-Dependent Pathways. AIDS Lond. Engl. 2013, 27, 2505–2517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, R.; Yao, H.; Dhillon, N.K.; Buch, S.J. HIV-1 Tat Co-Operates with IFN-Gamma and TNF-Alpha to Increase CXCL10 in Human Astrocytes. PLoS ONE 2009, 4, e5709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Zhang, L.-L.; Yin, L.-B.; Fu, Y.-J.; Jiang, Y.-J.; Ding, H.-B.; Chu, Z.-X.; Shang, H.; Zhang, Z.-N. Deregulated MicroRNA-21 Expression in Monocytes from HIV-Infected Patients Contributes to Elevated IP-10 Secretion in HIV Infection. Front. Immunol. 2017, 8, 1122. [Google Scholar] [CrossRef] [Green Version]
- Costa, L.J.; Chen, N.; Lopes, A.; Aguiar, R.S.; Tanuri, A.; Plemenitas, A.; Peterlin, B.M. Interactions between Nef and AIP1 Proliferate Multivesicular Bodies and Facilitate Egress of HIV-1. Retrovirology 2006, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Stumptner-Cuvelette, P.; Jouve, M.; Helft, J.; Dugast, M.; Glouzman, A.-S.; Jooss, K.; Raposo, G.; Benaroch, P. Human Immunodeficiency Virus-1 Nef Expression Induces Intracellular Accumulation of Multivesicular Bodies and Major Histocompatibility Complex Class II Complexes: Potential Role of Phosphatidylinositol 3-Kinase. Mol. Biol. Cell 2003, 14, 4857–4870. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Huang, M.-B.; Campbell, P.E.; Roth, W.W.; Campbell, T.; Khan, M.; Newman, G.; Villinger, F.; Powell, M.D.; Bond, V.C. Genetic Characterization of HIV Type 1 Nef-Induced Vesicle Secretion. AIDS Res. Hum. Retrovir. 2010, 26, 173–192. [Google Scholar] [CrossRef]
- Shelton, M.N.; Huang, M.-B.; Ali, S.A.; Powell, M.D.; Bond, V.C. Secretion Modification Region-Derived Peptide Disrupts HIV-1 Nef’s Interaction with Mortalin and Blocks Virus and Nef Exosome Release. J. Virol. 2012, 86, 406–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivetta, E.; Arenaccio, C.; Manfredi, F.; Anticoli, S.; Federico, M. The Contribution of Extracellular Nef to HIV-Induced Pathogenesis. Curr. Drug Targets 2016, 17, 46–53. [Google Scholar] [CrossRef]
- Dreux, M.; Garaigorta, U.; Boyd, B.; Décembre, E.; Chung, J.; Whitten-Bauer, C.; Wieland, S.; Chisari, F.V. Short-Range Exosomal Transfer of Viral RNA from Infected Cells to Plasmacytoid Dendritic Cells Triggers Innate Immunity. Cell Host Microbe 2012, 12, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Gray, L.R.; Gabuzda, D.; Cowley, D.; Ellett, A.; Chiavaroli, L.; Wesselingh, S.L.; Churchill, M.J.; Gorry, P.R. CD4 and MHC Class 1 Down-Modulation Activities of Nef Alleles from Brain- and Lymphoid Tissue-Derived Primary HIV-1 Isolates. J. Neurovirol. 2011, 17, 82–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.-H.; Schierer, S.; Blume, K.; Dindorf, J.; Wittki, S.; Xiang, W.; Ostalecki, C.; Koliha, N.; Wild, S.; Schuler, G.; et al. HIV-Nef and ADAM17-Containing Plasma Extracellular Vesicles Induce and Correlate with Immune Pathogenesis in Chronic HIV Infection. EBioMedicine 2016, 6, 103–113. [Google Scholar] [CrossRef] [Green Version]
- Mukhamedova, N.; Hoang, A.; Dragoljevic, D.; Dubrovsky, L.; Pushkarsky, T.; Low, H.; Ditiatkovski, M.; Fu, Y.; Ohkawa, R.; Meikle, P.J.; et al. Exosomes Containing HIV Protein Nef Reorganize Lipid Rafts Potentiating Inflammatory Response in Bystander Cells. PLoS Pathog. 2019, 15, e1007907. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Constitutive Expression | Cytokines/ Chemokines | Fold Increase vs. Ctrl | p Value | |||
|---|---|---|---|---|---|---|
| Nef w.t | Nef 4EA | Nef w.t vs. Ctrl | Nef w.t vs. 4EA | 4EA vs. Ctrl | ||
| Time 4 h | ||||||
| High | MCP-1 (91.47 ± 12.07) | 1.54 ± 0.11 | 0.95 ± 0.12 | 0.021 | 0.0169 | ns |
| TNF-α (773.32 ± 365) | 2.07 ± 0.13 | 1.19 ± 0.01 | 0.0015 | 0.0026 | ns | |
| G-CSF (118.98 ± 37.13) | 4.23 ± 0.39 | 1.19 ± 0.09 | 0.0016 | 0.0020 | ns | |
| VEGF (139.53 ± 11.56) | 2.20 ± 1.15 | 1.28 ± 0.10 | ns | ns | ns | |
| Low | IL-8 (1.44 ± 0.78) | 30.40 ± 0.06 | 1.22 ± 0.32 | <0.0001 | <0.0001 | ns |
| IP-10 (3.84 ± 1.32) | 5.39 ± 1.98 | 0.98 ± 0.03 | ns | ns | ns | |
| MIP-1β (10.13 ± 1.14) | 4.94 ± 1.96 | 1.08 ± 0.05 | ns | ns | ns | |
| IL-15 (34.87 ± 35.53) | 2.46 ± 0.85 | 1.02 ± 0.03 | ns | ns | ns | |
| Time 6 h | ||||||
| High | MCP-1 (109.83 ± 36.81) | 1.35 ± 0.17 | 0.85 ± 0.01 | ns | 0.0294 | ns |
| TNF-α (868.11 ± 447.62) | 1.58 ± 0.02 | 0.95 ± 0.09 | 0.0035 | 0.0028 | ns | |
| G-CSF (107.80 ± 28.67) | 4.18 ± 2.21 | 0.86 ± 0.00 | ns | ns | ns | |
| VEGF (163.75 ± 36.01) | 1.09 ± 0.14 | 0.86 ± 0.04 | ns | ns | ns | |
| Low | IL-8 (1.77 ± 0.70) | 30.23 ± 0.64 | 1.33 ±0.00 | <0.0001 | <0.0001 | ns |
| IP-10 (14.88 ± 3.02) | 10.99 ± 6.63 | 0.99 ± 0.19 | ns | ns | ns | |
| MIP-1β (9.56 ± 3.46) | 5.74 ± 3.60 | 0.81 ±0.14 | ns | ns | ns | |
| IL-15 (10.48 ± 3.22) | 3.40 ±3.48 | 0.95 ± 0.07 | ns | ns | ns | |
| Time 20 h | ||||||
| High | MCP-1 (87.92 ± 27.87) | 2.83 ± 0.23 | 1.10 ± 0.04 | 0.0017 | 0.0020 | ns |
| TNF-α (1200 ± 383.55) | 1.61 ± 0.25 | 1.12 ± 0.06 | ns | ns | ns | |
| G-CSF (39.67 ± 23.04) | 6.02 ± 2.45 | 1.16 ± 0.32 | ns | ns | ns | |
| IP-10 (69.37 ± 25.49) | 9.05 ± 2.22 | 1.17 ± 0.00 | 0.0166 | 0.0176 | ns | |
| VEGF (573.09 ± 141.74) | 1.43 ± 0.80 | 0.94 ± 0.18 | ns | ns | ns | |
| Low | IL-8 (2.14 ± 1.31) | 30.80 ± 14.09 | 2.09 ± 0.86 | ns | ns | ns |
| MIP-1β (3.61 ± 0.10) | 5.13 ± 0.54 | 1.15 ± 0.07 | 0.0020 | 0.0022 | ns | |
| IL-15 (39.02 ± 38.18) | 1.44 ± 0.76 | 0.97 ± 0.07 | ns | ns | ns | |
| Constitutive Expression | Cytokines/ Chemokines | Fold Increase vs. Ctrl | p Value | |||
|---|---|---|---|---|---|---|
| Nef w.t | Nef 4EA | Nef w.t vs. Ctrl | Nef w.t vs. 4EA | 4EA vs. Ctrl | ||
| Time 4 h | ||||||
| High | IL-8 (1144.8 ± 302.8) | 11.00 ± 1.97 | 1.07 ± 0.24 | 0.0065 | 0.0067 | ns |
| IL-15 (115.2 ± 7.1) | 1.83 ± 0.32 | 1.04 ± 0.20 | ns | ns | ns | |
| G-CSF (3047.1 ± 1297.9) | 1.63 ± 0.19 | 1.21 ± 0.16 | 0.0441 | ns | ns | |
| MIP-1α (120.81 ± 33.9) | 4.00 ± 0.85 | 1.51 ± 0.57 | 0.0299 | 0.0487 | ns | |
| MIP-1β (235.5 ± 60.9) | 99.25 ± 0.01 | 1.37 ± 0.53 | <0.0001 | <0.0001 | ns | |
| RANTES(1218.7 ± 103.2) | 1.60 ± 0.28 | 0.90 ± 0.00 | ns | 0.0468 | ns | |
| Low | FGF basic (59.7 ± 6.4) | 1.89 ± 0.26 | 1.08 ± 0.11 | 0.0248 | 0.0316 | ns |
| IFN-γ (35.3 ± 21.3) | 2.72 ± 0.02 | 0.89 ± 0.13 | 0.0005 | 0.0004 | ns | |
| IP-10 (66.7 ± 27.1) | 4.14 ± 0.37 | 0.96 ± 0.08 | 0.0015 | 0.0015 | ns | |
| MCP-1 (49.37 ± 1.7) | 5.11 ± 0.16 | 1.05 ± 0.21 | 0.0002 | 0.0002 | ns | |
| TNF-α (34.84 ± 9.8) | 257.41 ± 95.33 | 1.72 ± 0.88 | 0.0376 | 0.0379 | ns | |
| Time 6 h | ||||||
| High | IL-8 (1409.8 ± 436.8) | 30.55 ± 2.90 | 1.63 ± 0.19 | 0.0009 | 0.0009 | ns |
| IL-15 (126.4 ± 1.65) | 1.84 ± 0.08 | 1.08 ± 0.03 | 0.0012 | 0.0015 | ns | |
| G-CSF (3368.5 ± 1157.7) | 1.64 ± 0.23 | 1.27 ± 0.10 | 0.0428 | ns | ns | |
| MIP-1α (156.8 ± 31.2) | 3.13 ± 0.04 | 1.97 ± 0.67 | 0.0242 | ns | ns | |
| MIP-1β (296.7 ± 39.7) | 84.60 ± 0.85 | 1.67 ± 0.39 | <0.0001 | <0.0001 | ns | |
| RANTES (1644.3 ± 221.3) | 2.26 ± 0.09 | 1.01 ± 0.16 | 0.0028 | 0.0028 | ns | |
| Low | FGF basic (64.8 ± 9.4) | 1.92 ± 0.02 | 1.11 ± 0.02 | <0.0001 | <0.0001 | 0.0142 |
| IFN-γ (42.8 ± 25.8) | 3.05 ± 0.36 | 1.06 ± 0.09 | 0.0048 | 0.0052 | ns | |
| IP-10 (84.2 ± 15.1) | 15.19 ± 0.44 | 1.45 ± 0.49 | <0.0001 | <0.0001 | ns | |
| MCP-1 (70.3 ±6.3) | 9.55 ± 0.36 | 1.33 ± 0.18 | <0.0001 | <0.0001 | ns | |
| TNF-α (40.4 ± 19.1) | 247.33 ± 18.29 | 2.11 ± 0.58 | 0.0004 | 0.0004 | ns | |
| Time 20 h | ||||||
| High | IL-8 (1926.2 ± 37.0) | 65.46 ± 72.21 | 31.21 ± 36.79 | ns | ns | ns |
| IL-15 (112.7 ± 10.6) | 2.18 ± 0.12 | 1.63 ± 0.46 | 0.0476 | ns | ns | |
| G-CSF (2778.9 ± 1397.5) | 2.05 ± 0.07 | 1.95 ± 0.22 | 0.0086 | ns | 0.011 | |
| MIP-1α (125.8 ± 56.2) | 4.18 ± 0.96 | 4.25 ± 1.34 | ns | ns | ns | |
| MIP-1β (483.1 ± 160.3) | 47.01 ± 0.86 | 13.15 ± 15.77 | 0.0297 | ns | ns | |
| RANTES (4473.3 ± 1893.8) | 4.82 ± 1.80 | 2.26 ± 1.08 | ns | ns | ns | |
| Low | FGF basic (55.6 ± 0.86) | 2.33 ± 0.47 | 1.78 ± 0.68 | ns | ns | ns |
| IFN-γ (60.8 ± 31.3) | 4.21 ± 0.15 | 2.37 ± 1.37 | ns | ns | ns | |
| IP-10 (368.4 ± 168.3) | 25.52 ± 1.16 | 3.43 ± 1.60 | 0.0005 | 0.0007 | ns | |
| MCP-1 (176.4 ± 27.7) | 16.74 ± 0.48 | 10.16 ± 11.40 | ns | ns | ns | |
| TNF-α (51.4 ± 15.9) | 123.00 ± 21.50 | 11.06 ± 12.54 | 0.0070 | 0.0090 | ns | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiello, A.; Giannessi, F.; Percario, Z.A.; Fecchi, K.; Arenaccio, C.; Leone, S.; Carollo, M.; D’Aversa, E.; Chaperot, L.; Gambari, R.; et al. HIV-1 Nef Protein Affects Cytokine and Extracellular Vesicles Production in the GEN2.2 Plasmacytoid Dendritic Cell Line. Viruses 2022, 14, 74. https://doi.org/10.3390/v14010074
Aiello A, Giannessi F, Percario ZA, Fecchi K, Arenaccio C, Leone S, Carollo M, D’Aversa E, Chaperot L, Gambari R, et al. HIV-1 Nef Protein Affects Cytokine and Extracellular Vesicles Production in the GEN2.2 Plasmacytoid Dendritic Cell Line. Viruses. 2022; 14(1):74. https://doi.org/10.3390/v14010074
Chicago/Turabian StyleAiello, Alessandra, Flavia Giannessi, Zulema Antonia Percario, Katia Fecchi, Claudia Arenaccio, Stefano Leone, Maria Carollo, Elisabetta D’Aversa, Laurence Chaperot, Roberto Gambari, and et al. 2022. "HIV-1 Nef Protein Affects Cytokine and Extracellular Vesicles Production in the GEN2.2 Plasmacytoid Dendritic Cell Line" Viruses 14, no. 1: 74. https://doi.org/10.3390/v14010074
APA StyleAiello, A., Giannessi, F., Percario, Z. A., Fecchi, K., Arenaccio, C., Leone, S., Carollo, M., D’Aversa, E., Chaperot, L., Gambari, R., Sargiacomo, M., & Affabris, E. (2022). HIV-1 Nef Protein Affects Cytokine and Extracellular Vesicles Production in the GEN2.2 Plasmacytoid Dendritic Cell Line. Viruses, 14(1), 74. https://doi.org/10.3390/v14010074