
Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays


 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Plasmids
2.2. Small Molecules
2.3. Pseudoviruses Preparation
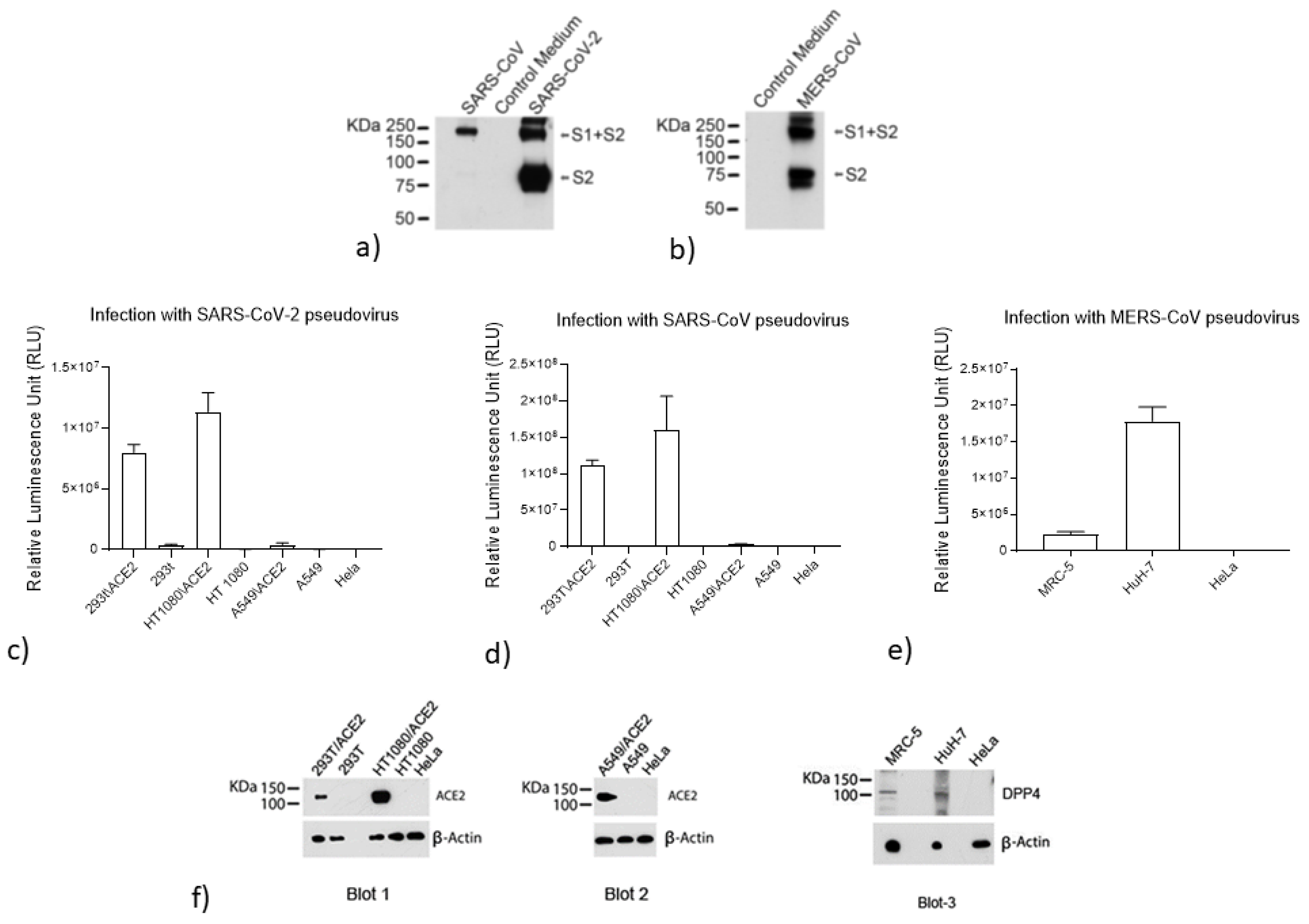
2.4. Analysis of S Protein Incorporation into the SARS-CoV-2, SARS-CoV, and MERS-CoV Pseudoviruses
2.5. Evaluation of ACE2 and DPP4 (CD26) Expression
2.6. Measurement of Antiviral Activity
2.7. SARS-CoV-2 Microneutralization Assay
2.8. Evaluation of Cytotoxicity
2.9. Drug Sensitivity of Spike-Mutated Pseudovirus
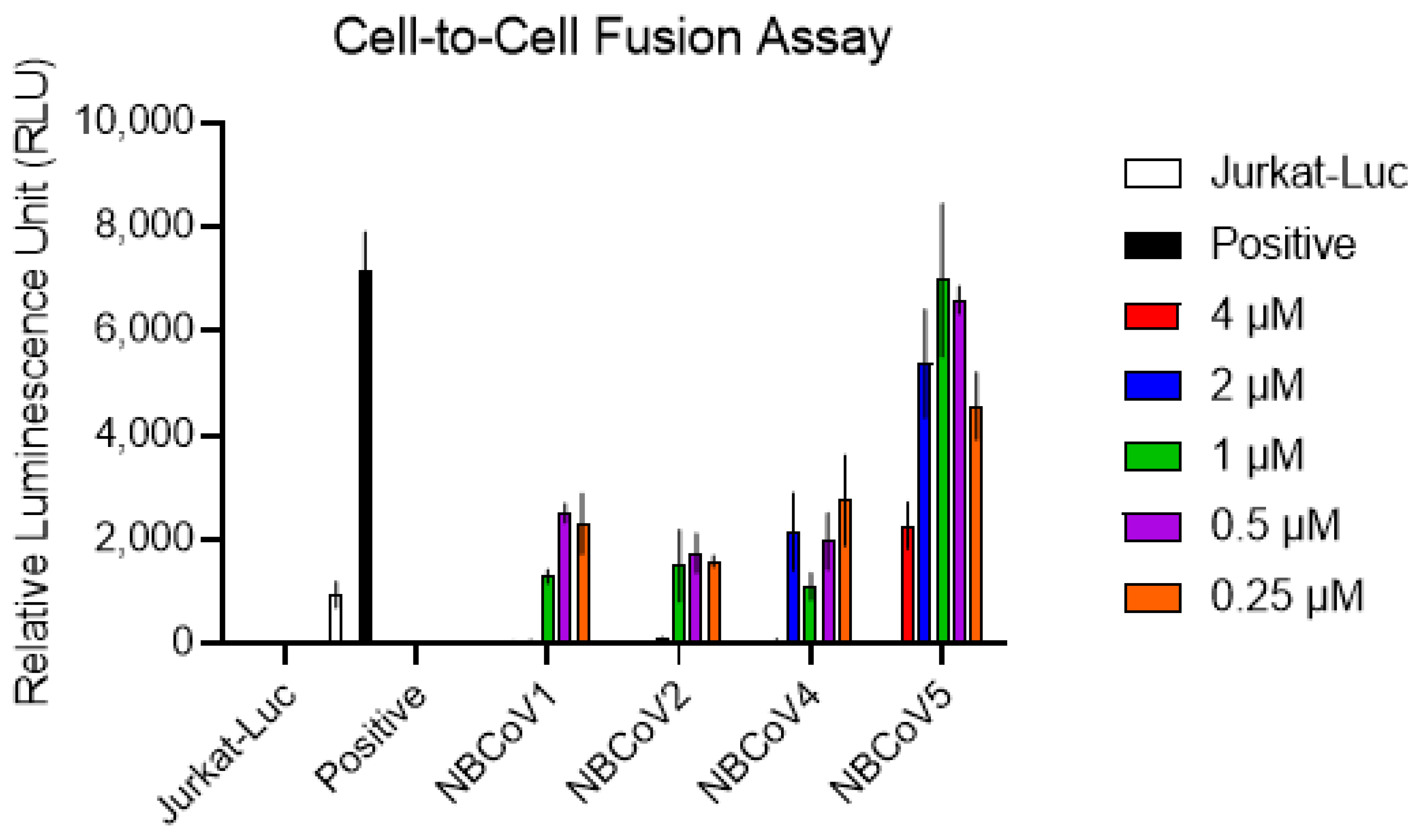
2.10. Cell-to-Cell Fusion Inhibition Assay
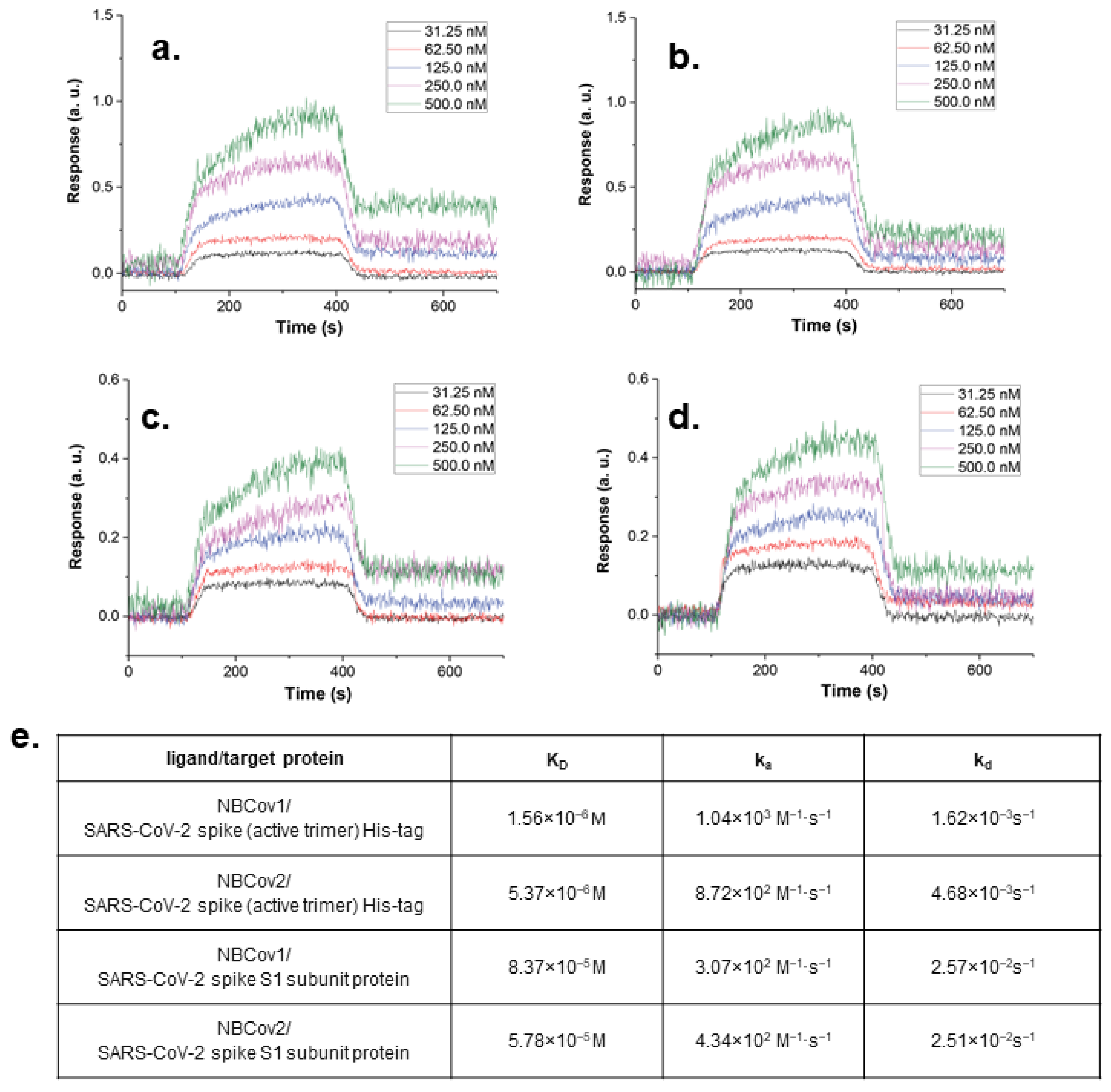
2.11. Binding Analysis by SPR
2.12. In Vitro ADME Study
2.13. In Vivo Pharmacokinetics in Rats
3. Results
3.1. Rationale for Screening HIV-1 Fusion Inhibitors as Possible Pan-CoV Inhibitors
3.2. Validation of the Pseudoviruses
3.3. Antiviral Activity and Cytotoxicity of the NBCoV Small Molecules in a Pseudovirus Assay
3.4. NBCoV Small Molecules Inhibit a Replication-Competent Authentic SARS-CoV-2 (US_WA-1/2020)
3.5. NBCoV Small Molecules Neutralize the SARS-CoV-2 Variants B.1.1.7 UK (Alpha), B.1.351 RSA (Beta), and B.1.617.2 India (Delta)
3.6. Binding Affinity of the Two Most Potent Inhibitors by SPR Analysis
3.7. NBCoV Small Molecules Inhibit SARS-CoV-2–Mediated Cell-to-Cell Fusion
3.8. In Vitro ADME Assessment
3.9. In Vivo Pharmacokinetics of NBCoV1 and NBCoV2
3.10. Confirmation That NBCoVs Are Not Promiscuous Binders or Aggregators
3.10.1. The Antiviral Activities of Ene-Rhodanine Derivatives Are Not Due to Luciferase Activity Inhibition or Direct Interference with Luminescence Measurements
3.10.2. Target Specificity Measured by a Direct Binding Study by SPR
3.10.3. The Inhibitors Specifically Bind the Viral S Protein
- (1).
- The timing of compound addition is critical for antiviral activity.
- (2).
- Cellular toxicity vs. antiviral activity.
3.10.4. The NBCoV Small Molecules Are Not Promiscuous Aggregation-Based Inhibitors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phelan, A.L.; Katz, R.; Gostin, L.O. The Novel Coronavirus Originating in Wuhan, China: Challenges for Global Health Governance. JAMA 2020, 323, 709–710. [Google Scholar] [CrossRef] [Green Version]
- Koslap-Petraco, M. Vaccine hesitancy: Not a new phenomenon, but a new threat. J. Am. Assoc. Nurse Pract. 2019, 31, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.F.; Buffarini, R.; Bertoldi, A.D.; Santos, I.S.; Barros, A.J.D.; Matijasevich, A.; Menezes, A.M.B.; Goncalves, H.; Horta, B.L.; Barros, F.C.; et al. The emergence of vaccine hesitancy among upper-class Brazilians: Results from four birth cohorts, 1982–2015. Vaccine 2020, 38, 482–488. [Google Scholar] [CrossRef] [PubMed]
- The Lancet Child Adolescent, H. Vaccine hesitancy: A generation at risk. Lancet Child Adolesc. Health 2019, 3, 281. [Google Scholar] [CrossRef]
- Butt, A.A.; Khan, T.; Yan, P.; Shaikh, O.S.; Omer, S.B.; Mayr, F. Rate and risk factors for breakthrough SARS-CoV-2 infection after vaccination. J. Infect. 2021, 83, 237–279. [Google Scholar] [CrossRef]
- Shastri, J.; Parikh, S.; Aggarwal, V.; Agrawal, S.; Chatterjee, N.; Shah, R.; Devi, P.; Mehta, P.; Pandey, R. Severe SARS-CoV-2 Breakthrough Reinfection With Delta Variant After Recovery From Breakthrough Infection by Alpha Variant in a Fully Vaccinated Health Worker. Front. Med. 2021, 8, 737007. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Lauring, A.S.; Malani, P.N. Variants of SARS-CoV-2. JAMA 2021, 326, 880. [Google Scholar] [CrossRef]
- Focosi, D.; Maggi, F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based COVID-19 therapeutics and vaccines. Rev. Med. Virol. 2021, 31, 1–21. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef]
- Mahase, E. Delta variant: What is happening with transmission, hospital admissions, and restrictions? BMJ 2021, 373, n1513. [Google Scholar] [CrossRef] [PubMed]
- Reardon, S. How the Delta variant achieves its ultrafast spread. Nature 2021, 21, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamorano Cuervo, N.; Grandvaux, N. ACE2: Evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. eLife 2020, 9, e61390. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Gao, J.; Lu, G.; Qi, J.; Li, Y.; Wu, Y.; Deng, Y.; Geng, H.; Li, H.; Wang, Q.; Xiao, H.; et al. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 13134–13140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. Domains and functions of spike protein in SARS-CoV-2 in the context of vaccine design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Gangadevi, S.; Badavath, V.N.; Thakur, A.; Yin, N.; De Jonghe, S.; Acevedo, O.; Jochmans, D.; Leyssen, P.; Wang, K.; Neyts, J.; et al. Kobophenol A inhibits binding of host ACE2 receptor with spike RBD domain of SARS-CoV-2, a lead compound for blocking COVID-19. J. Phys. Chem. Lett. 2021, 12, 1793–1802. [Google Scholar] [CrossRef]
- Larue, R.C.; Xing, E.; Kenney, A.D.; Zhang, Y.; Tuazon, J.A.; Li, J.; Yount, J.S.; Li, P.K.; Sharma, A. Rationally designed ACE2-derived peptides inhibit SARS-CoV-2. Bioconjug. Chem. 2021, 32, 215–223. [Google Scholar] [CrossRef]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Barnes, C.O.; Hsiang, T.Y.; Esser-Nobis, K.; Yu, K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, 208–1214. [Google Scholar] [CrossRef]
- Sitthiyotha, T.; Chunsrivirot, S. Computational design of 25-mer peptide binders of SARS-CoV-2. J. Phys. Chem. B 2020, 124, 10930–10942. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.K.; Hillyer, C.D.; Debnath, A.K. Stapled peptides based on human angiotensin-converting enzyme 2 (ACE2) potently inhibit SARS-CoV-2 infection in vitro. MBio 2020, 11, e02451-20. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klasse, P.J.; Moore, J.P. Antibodies to SARS-CoV-2 and their potential for therapeutic passive immunization. eLife 2020, 9, e57877. [Google Scholar] [CrossRef]
- Shah, M.; Ahmad, B.; Choi, S.; Woo, H.G. Mutations in the SARS-CoV-2 spike RBD are responsible for stronger ACE2 binding and poor anti-SARS-CoV mAbs cross-neutralization. Comput. Struct. Biotechnol. J. 2020, 18, 3402–3414. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.; Burgers, W.A. SARS-CoV-2 evolution and vaccines: Cause for concern? Lancet Respir. Med. 2021, 9, 333–335. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.H.; Michailidis, E.; Lorenzi, J.C.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020, 217, e20201181. [Google Scholar] [CrossRef]
- Chang, L.J.; Urlacher, V.; Iwakuma, T.; Cui, Y.; Zucali, J. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 1999, 6, 715–728. [Google Scholar] [CrossRef] [Green Version]
- Landau, N.R.; Page, K.A.; Littman, D.R. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency virus host range. J. Virol. 1991, 65, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Choe, S.; Walker, R.; Di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J.Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef] [Green Version]
- Katritzky, A.R.; Tala, S.R.; Lu, H.; Vakulenko, A.V.; Chen, Q.Y.; Sivapackiam, J.; Pandya, K.; Jiang, S.; Debnath, A.K. Design, synthesis, and structure-activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009, 52, 7631–7639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Nie, J.; Li, Q.; Wu, J.; Zhao, C.; Hao, H.; Liu, H.; Zhang, L.; Nie, L.; Qin, H.; Wang, M.; et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 680–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Schafer, A.; Kulkarni, S.S.; Liu, X.; Martinez, D.R.; Chen, C.; Sun, Z.; Leist, S.R.; Drelich, A.; Zhang, L.; et al. High potency of a bivalent human VH domain in SARS-CoV-2 animal models. Cell 2020, 183, 429–441.e16. [Google Scholar] [CrossRef]
- Du, L.; Zhao, G.; Yang, Y.; Qiu, H.; Wang, L.; Kou, Z.; Tao, X.; Yu, H.; Sun, S.; Tseng, C.T.; et al. A conformation-dependent neutralizing monoclonal antibody specifically targeting receptor-binding domain in Middle East respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 7045–7053. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.S.; Ying, T.; Tao, X.; Garron, T.; Algaissi, A.; Wang, Y.; Wang, L.; Peng, B.H.; Jiang, S.; Dimitrov, D.S.; et al. Passive transfer of a germline-like neutralizing human monoclonal antibody protects transgenic mice against lethal middle east respiratory syndrome coronavirus infection. Sci. Rep. 2016, 6, 31629. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Xie, S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput. Methods Programs Biomed. 2010, 99, 306–314. [Google Scholar] [CrossRef]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Melikyan, G.B.; Markosyan, R.M.; Hemmati, H.; Delmedico, M.K.; Lambert, D.M.; Cohen, F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell. Biol. 2000, 151, 413–424. [Google Scholar] [CrossRef]
- Liu, S.; Xiao, G.; Chen, Y.; He, Y.; Niu, J.; Escalante, C.R.; Xiong, H.; Farmar, J.; Debnath, A.K.; Tien, P.; et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: Implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet 2004, 363, 938–947. [Google Scholar] [CrossRef] [Green Version]
- Duquerroy, S.; Vigouroux, A.; Rottier, P.J.; Rey, F.A.; Bosch, B.J. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology 2005, 335, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Liu, S.; Li, J.; Lu, H.; Qi, Z.; Liu, Z.; Debnath, A.K.; Jiang, S. Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J. Virol. 2008, 82, 11129–11139. [Google Scholar] [CrossRef] [Green Version]
- Yu, D.; Zhu, Y.; Yan, H.; Wu, T.; Chong, H.; He, Y. Pan-coronavirus fusion inhibitors possess potent inhibitory activity against HIV-1, HIV-2, and simian immunodeficiency virus. Emerg. Microbes Infect. 2021, 10, 810–821. [Google Scholar] [CrossRef]
- Tomasic, T.; Peterlin Masic, L. Rhodanine as a scaffold in drug discovery: A critical review of its biological activities and mechanisms of target modulation. Expert Opin. Drug. Discov. 2012, 7, 49–560. [Google Scholar] [CrossRef]
- Jasial, S.; Gilberg, E.; Blaschke, T.; Bajorath, J. Machine learning distinguishes with high accuracy between pan-assay interference compounds that are promiscuous or represent dark chemical matter. J. Med. Chem. 2018, 61, 10255–10264. [Google Scholar] [CrossRef] [PubMed]
- Bolz, S.N.; Adasme, M.F.; Schroeder, M. Toward an understanding of pan-assay interference compounds and promiscuity: A structural perspective on binding modes. J. Chem. Inf. Model. 2021, 61, 2248–2262. [Google Scholar] [CrossRef] [PubMed]
- Mendgen, T.; Steuer, C.; Klein, C.D. Privileged scaffolds or promiscuous binders: A comparative study on rhodanines and related heterocycles in medicinal chemistry. J. Med. Chem. 2012, 55, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.R.F.; Patel, A.; Ramos, S.; Elwood, D.; Zhu, X.; Yan, J.; Gary, E.N.; Walker, S.N.; Schultheis, K.; Purwar, M.; et al. Immunogenicity of a DNA vaccine candidate for COVID-19. Nat. Commun. 2020, 11, 2601. [Google Scholar] [CrossRef]
- Whittaker, G.R.; Millet, J.K. Biochemical characterization of middle east respiratory syndrome coronavirus spike protein proteolytic processing. Methods Mol. Biol. 2020, 2099, 21–37. [Google Scholar]
- Koch, J.; Uckeley, Z.M.; Doldan, P.; Stanifer, M.; Boulant, S.; Lozach, P.Y. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. EMBO J. 2021, 40, e107821. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.J.; Urwin, L.; Nicklin, M.J.H.; James, D.C.; Green, L.R.; Monk, P.N. ACE2-Independent Interaction of SARS-CoV-2 Spike Protein with Human Epithelial Cells Is Inhibited by Unfractionated Heparin. Cells 2021, 10, 1419. [Google Scholar] [CrossRef]
- Ma, D.; Chen, C.B.; Jhanji, V.; Xu, C.; Yuan, X.L.; Liang, J.J.; Huang, Y.; Cen, L.P.; Ng, T.K. Expression of SARS-CoV-2 receptor ACE2 and TMPRSS2 in human primary conjunctival and pterygium cell lines and in mouse cornea. Eye 2020, 34, 1212–1219. [Google Scholar] [CrossRef]
- Hayashi, H.; Kubo, Y.; Izumida, M.; Takahashi, E.; Kido, H.; Sato, K.; Yamaya, M.; Nishimura, H.; Nakayama, K.; Matsuyama, T. Enterokinase Enhances Influenza A Virus Infection by Activating Trypsinogen in Human Cell Lines. Front. Cell. Infect. Microbiol. 2018, 8, 91. [Google Scholar] [CrossRef]
- Wu, C.Y.; Lin, Y.S.; Yang, Y.H.; Shu, L.H.; Cheng, Y.C.; Liu, H.T. Potential Simultaneous Inhibitors of Angiotensin-Converting Enzyme 2 and Transmembrane Protease, Serine 2. Front. Pharmacol. 2020, 11, 584158. [Google Scholar] [CrossRef]
- Rasmussen, I.; Vilhardt, F. Macropinocytosis is the entry mechanism of amphotropic murine leukemia virus. J. Virol. 2015, 89, 1851–1866. [Google Scholar] [CrossRef] [Green Version]
- Franco, E.J.; Rodriquez, J.L.; Pomeroy, J.J.; Hanrahan, K.C.; Brown, A.N. The effectiveness of antiviral agents with broad-spectrum activity against chikungunya virus varies between host cell lines. Antivir. Chem. Chemother. 2018, 26, 2040206618807580. [Google Scholar] [CrossRef]
- Smith, E.C.; Denison, M.R. Coronaviruses as DNA wannabes: A new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013, 9, e1003760. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef] [PubMed]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID-19’s next move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, Y.; Liu, J.; Zhang, X.; Zou, J.; Fontes-Garfias, C.R.; Xia, H.; Swanson, K.A.; Cutler, M.; Cooper, D.; et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021, 27, 620–621. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhao, J.; Nguyen, L.N.T.; Adkins, J.L.; Schank, M.; Khanal, S.; Nguyen, L.N.; Dang, X.; Cao, D.; Thakuri, B.K.C.; et al. Blockade of SARS-CoV-2 spike protein-mediated cell-cell fusion using COVID-19 convalescent plasma. Sci. Rep. 2021, 11, 5558. [Google Scholar] [CrossRef]
- Hornich, B.F.; Grosskopf, A.K.; Schlagowski, S.; Tenbusch, M.; Kleine-Weber, H.; Neipel, F.; Stahl-Hennig, C.; Hahn, A.S. SARS-CoV-2 and SARS-CoV spike-mediated cell-cell fusion differ in their requirements for receptor expression and proteolytic activation. J. Virol. 2021, 95, e00002-21. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Kennedy, T. Managing the drug discovery/development interface. Drug Discov. Today 1997, 2, 436–444. [Google Scholar] [CrossRef]
- Bharate, S.S. Recent developments in pharmaceutical salts: FDA approvals from 2015 to 2019. Drug Discov. Today 2021, 26, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Varghese Gupta, S. Salts of therapeutic agents: Chemical, physicochemical, and biological considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Lin, J.H.; Lu, A.Y. Metabolism-based drug-drug interactions: What determines individual variability in cytochrome P450 induction? Drug. Metab. Dispos. 2005, 33, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef] [PubMed]
- Turpeinen, M.; Zanger, U.M. Cytochrome P450 2B6: Function, genetics, and clinical relevance. Drug Metabol. Drug Interact. 2012, 27, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Kerns, H.E. Cytochrome P450 inhibition. In Drug-Like Properties: Concepts, Structure Design, and Methods; Associated Press: San Diego, CA, USA, 2016; pp. 229–242. [Google Scholar]
- Auld, D.S.; Southall, N.T.; Jadhav, A.; Johnson, R.L.; Diller, D.J.; Simeonov, A.; Austin, C.P.; Inglese, J. Characterization of chemical libraries for luciferase inhibitory activity. J. Med. Chem. 2008, 51, 2372–2386. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiarova, A.; Taslimi, P.; Elliman, S.J.; Kosinski, P.A.; Hubbard, B.; Kavana, M.; Kemp, D.M. Resveratrol inhibits firefly luciferase. Biochem. Biophys. Res. Commun. 2006, 351, 481–484. [Google Scholar] [CrossRef]
- Aldrich, C.; Bertozzi, C.; Georg, G.I.; Kiessling, L.; Lindsley, C.; Liotta, D.; Merz, K.M., Jr.; Schepartz, A.; Wang, S. The ecstasy and agony of assay interference compounds. ACS Med. Chem. Lett. 2017, 8, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Baell, J.; Walters, M.A. Chemistry: Chemical con artists foil drug discovery. Nature 2014, 513, 481–483. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Ryan, A.J.; Gray, N.M.; Lowe, P.N.; Chung, C.W. Effect of detergent on "promiscuous" inhibitors. J. Med. Chem. 2003, 46, 3448–3451. [Google Scholar] [CrossRef] [PubMed]
- Coan, K.E.; Shoichet, B.K. Stoichiometry and physical chemistry of promiscuous aggregate-based inhibitors. J. Am. Chem. Soc. 2008, 130, 9606–9612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGovern, S.L.; Helfand, B.T.; Feng, B.; Shoichet, B.K. A specific mechanism of nonspecific inhibition. J. Med. Chem. 2003, 46, 4265–4272. [Google Scholar] [CrossRef] [PubMed]
- Sassano, M.F.; Doak, A.K.; Roth, B.L.; Shoichet, B.K. Colloidal aggregation causes inhibition of G protein-coupled receptors. J. Med. Chem. 2013, 56, 2406–2414. [Google Scholar] [CrossRef]
- Shoichet, B.K. Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem. 2006, 49, 7274–7277. [Google Scholar] [CrossRef]
- Seidler, J.; McGovern, S.L.; Doman, T.N.; Shoichet, B.K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- McGovern, S.L.; Caselli, E.; Grigorieff, N.; Shoichet, B.K. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J. Med. Chem. 2002, 45, 1712–1722. [Google Scholar] [CrossRef]
- Bajorath, J. Evolution of assay interference concepts in drug discovery. Expert Opin. Drug Discov. 2021, 16, 719–721. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhang, Y.; Ruan, Q.; Tang, S.; Sun, Z.; Zeng, R. Gains from the separation of conjoined twins. Plast. Reconstr. Surg. 2008, 122, 87e–88e. [Google Scholar] [CrossRef]
- Reker, D.; Bernardes, G.J.L.; Rodrigues, T. Computational advances in combating colloidal aggregation in drug discovery. Nat. Chem. 2019, 11, 402–418. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zhong, H.; Wang, K.; Li, N.; Chen, L. Gains from no real PAINS: Where ‘Fair Trial Strategy’ stands in the development of multi-target ligands. Acta Pharm. Sin. B 2021, 11, 3417–3432. [Google Scholar] [CrossRef] [PubMed]
- Dahlin, J.L.; Auld, D.S.; Rothenaigner, I.; Haney, S.; Sexton, J.Z.; Nissink, J.W.M.; Walsh, J.; Lee, J.A.; Strelow, J.M.; Willard, F.S.; et al. Nuisance compounds in cellular assays. Cell Chem. Biol. 2021, 28, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Muruato, A.E.; Zhang, X.; Lokugamage, K.G.; Fontes-Garfias, C.R.; Zou, J.; Liu, J.; Ren, P.; Balakrishnan, M.; Cihlar, T.; et al. A nanoluciferase SARS-CoV-2 for rapid neutralization testing and screening of anti-infective drugs for COVID-19. Nat. Commun. 2020, 11, 5214. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 293T/ACE2 Cells | HT1080/ACE2 Cells | A549/ACE2 Cells | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) a | CC50 (µM) a | SI | IC50 (nM) a | CC50 (µM) a | SI | IC50 (nM) a | CC50 (µM) a | SI | |
| NBCoV1 | 51 ± 17 | 38.5 ± 1 | 755 | 32.3 ± 4.6 | 89 ± 2 | 2755 | 63.6 ± 4.6 | 86 ± 8.7 | 1352 |
| NBCoV2 | 22.8 ± 0.8 | 37.5 ± 2 | 1630 | 25.3 ± 0.6 | >100 | >4000 | 58 ± 1.7 | >100 | >1724 |
| NBCoV3 | 60.1 ± 8.5 | 45 ± 0.5 | 750 | 64 ± 18 | >100 | >1563 | 120 ± 5 | >100 | >833 |
| NBCoV4 | 26 ± 1 | 40.7 ± 2.3 | 1565 | 47.7 ± 16 | >100 | >2096 | 73 ± 4.1 | >100 | >1370 |
| NBCoV5 | 1205 ± 240 | 35 ± 2 | 29 | 1050 ± 252 | >100 | >95 | >2000 | >100 | N/A b |
| NBCoV6 | 185 ± 5.8 | 40 ± 2.6 | 216 | 245 ± 5 | >100 | >408 | 613 ± 72 | >100 | >163 |
| NBCoV7 | 298 ± 60 | 45 ± 0.4 | 151 | 290 ± 57 | >100 | >345 | 416 ± 25 | >100 | >240 |
| NBCoV8 | 65.8 ± 6.2 | 38.7 ± 1.2 | 586 | 94 ± 9 | >100 | >1063 | 254 ± 27 | >100 | >394 |
| NBCoV9 | 342 ± 46 | 33.7 ± 2.5 | 98 | 365 ± 73 | >100 | >274 | 596 ± 42 | >100 | >168 |
| NBCoV15 | >2000 | 64 ± 14 | N/A b | >2000 | >100 | N/A b | N/A b | N/A b | N/A b |
| NBCoV17 | >2000 | >100 | N/A b | >2000 | >100 | N/A b | N/A b | N/A b | N/A b |
| NBCoV28 | >2000 | >100 | N/A b | >2000 | 92 ± 3 | N/A b | N/A b | N/A b | N/A b |
| NBCoV34 | >2000 | 83 ± 4 | N/A b | >2000 | ~100 | N/A b | N/A b | N/A b | N/A b |
| Compound | 293T/ACE2 Cells | HT1080/ACE2 Cells | A549/ACE2 Cells | |||
|---|---|---|---|---|---|---|
| IC50 (nM) a | SI | IC50 (nM) a | SI | IC50 (nM) a | SI | |
| NBCoV1 | 17 ± 2.6 | 2265 | 39 ± 8.2 | 2282 | 98 ± 4 | 878 |
| NBCoV2 | 13.8 ± 0.2 | 2717 | 19.3 ± 1.1 | >5181 | 111 ± 9 | >901 |
| NBCoV3 | 17.8 ± 4 | 2528 | 25.7 ± 0.6 | >3891 | 157 ± 12.5 | >637 |
| NBCoV4 | 80 ± 2 | 509 | 54 ± 3.5 | >1852 | 100 ± 13 | >1000 |
| NBCoV5 | 1853 ± 179 | 19 | 1760 ± 330 | >57 | >2000 | N/A b |
| NBCoV6 | 175 ± 7 | 229 | 133 ± 9.5 | >752 | 867 ± 62 | >115 |
| NBCoV7 | 111 ± 1.7 | 405 | 52 ± 8.9 | >1923 | 271 ± 76 | >369 |
| NBCoV8 | 128 ± 6.7 | 302 | 225 ± 26 | >444 | 700 ± 70 | >143 |
| NBCoV9 | 217 ± 22 | 155 | 246 ± 10 | >407 | 350 ± 15 | >286 |
| Compound | HuH-7 Cells | MRC-5 Cells | ||||
|---|---|---|---|---|---|---|
| IC50 (nM) a | CC50 (µM) a | SI | IC50 (nM) a | CC50 (µM) a | SI | |
| NBCoV1 | 95 ± 22 | ~100 | 1053 | 76.5 ± 0.3 | 69 ± 1 | 902 |
| NBCoV2 | 112 ± 7 | 80 ± 1 | 714 | 77. ± 0.5 | 63 ± 2.8 | 818 |
| NBCoV3 | 158 ± 14 | 92 ± 2 | 582 | 123 ± 17 | >50 | >407 |
| NBCoV4 | 131 ± 6 | 80 ± 6 | 611 | 80 ± 18 | 63.5 ± 3.5 | 794 |
| NBCoV5 | >2000 | 76 ± 2 | N/A b | >2000 | 72.5 ± 1 | N/A b |
| NBCoV6 | 569 ± 31 | 71 ± 1 | 125 | 945 ± 77 | 71 ± 2.8 | 75 |
| NBCoV7 | 933 ± 153 | N/Ab | N/A b | 927 ± 63.5 | 68.8 ± 1 | 74 |
| NBCoV8 | 509 ± 43 | 83 ± 2 | 163 | 559 ± 66 | 67.5 ± 3.5 | 121 |
| NBCoV9 | 214 ± 23 | 55 ± 9 | 257 | 232 ± 17 | 42.5 ± 3.5 | 183 |
| Compound | 293T-ACE2 Cells/ A-MLV |
|---|---|
| IC50 (nM) a | |
| NBCoV1 | 1397 ± 12 |
| NBCoV2 | 783 ± 76 |
| NBCoV3 | 960 ± 57 |
| NBCoV4 | 1623 ± 115 |
| NBCoV5 | >2000 |
| NBCoV6 | >2000 |
| NBCoV7 | >2000 |
| NBCoV8 | >2000 |
| NBCoV9 | 1627 ± 118 |
| Compound | SARS-CoV-2 (US_WA-1/2020) | NL4-3ΔEnv-NanoLuc/SARS-CoV-2 |
|---|---|---|
| Vero E6 Cells | 293T/ACE2, HT1080/ACE2 and A549/ACE2 Cells | |
| IC100 (µM) a | IC100 (µM) b | |
| NBCov1 | 1.25 | 0.25–1 |
| NBCoV2 | 1.25 | 0.3–1 |
| NBCoV3 | 2.5 | 0.5–1.2 |
| NBCoV4 | 2.5 | 0.3–1 |
| NBCoV5 | >10 | >2 |
| NBCoV6 | >10 | 1–3 |
| NBCoV7 | 5 | 1–3 |
| NBCoV8 | 5 | 1–2.5 |
| NBCoV9 | 2.5 | 2–3 |
| Compound → | NBCoV1 IC50 (nM) a | NBCoV2 IC50 (nM) a | NBCoV3 IC50 (nM) a | NBCoV4 IC50 (nM) a | NBCoV5 IC50 (nM) a |
|---|---|---|---|---|---|
| SARS-CoV-2 ↓ | |||||
| WT | 51 ± 17 | 22.8 ± 0.8 | 60.1 ± 8.5 | 26 ± 1 | 1205 ± 240 |
| B.1.1.7 UK (Alpha) | |||||
| N501Y | 52.5 ± 0.2 | 49 ± 0.5 | 44.5 ± 0.5 | 51 ± 0.4 | >2000 |
| ∆69–70 | 48 ± 3 | 55 ± 0.5 | 69 ± 7 | 53 ± 0.5 | >2000 |
| P681H | 35 ± 0.5 | 51 ± 0.3 | 55 ± 0.3 | 44 ± 0.4 | 1720 ± 215 |
| N501Y/∆69–70 | 46 ± 0.5 | 66 ± 1 | 94 ± 16 | 53 ± 11 | >2000 |
| N501Y/∆69–70/P681H | 57 ± 0.5 | 79 ± 14 | 232 ± 17 | 158 ± 31 | >2000 |
| B.1.351 RSA (Beta) | |||||
| E484K | 42 ± 0.7 | 45 ± 0.2 | 80 ± 10.6 | 45 ± 0.4 | >2000 |
| D614G | 56 ± 0.6 | 61 ± 0.3 | 145 ± 28 | 58 ± 0.4 | 1730 ± 62.5 |
| N501Y/D614G | 57 ± 2 | 59 ± 13 | 104 ± 1 | 77 ± 29 | >2000 |
| E484K/N501Y | 32 ± 0.5 | 36 ± 0.4 | 42 ± 1 | 51 ± 5 | >2000 |
| E484K/D614G | 33 ± 0.5 | 35 ± 0.2 | 44 ± 0.8 | 44 ± 3 | 1846 ± 124 |
| E484K/N501Y/D614G | 45 ± 2 | 43 ± 0.5 | 101 ± 2 | 43 ± 2.5 | >2000 |
| B.1.617.2 India (Delta) | |||||
| D950N | 47.7 ± 15 | 88.7 ± 6 | N/A b | 207 ± 29 | >2000 |
| D614G/P681R | 49 ± 13 | 65 ± 18 | N/A b | 148 ± 30 | >2000 |
| D614G/D950N | 33 ± 10 | 26 ± 2 | N/A b | 77.5 ± 16.5 | >2000 |
| D614G/P681R/D950N | 58 ± 7 | 53 ± 12 | N/A b | 239 ± 7 | >2000 |
| Assay Performed | In Vitro ADMET | Inhibitor | |
|---|---|---|---|
| NBCoV1 | |||
| Solubility (µM) | Phosphate buffer, pH7.4 | 4.72 | |
| Caco-2 permeability (mean Papp, × 10−6 cm/s) | NBCoV1 | A-to-B | 16.9 |
| B-to-A | 20.4 | ||
| Efflux Ratio | 1.21 | ||
| NBCoV1 + 1 µM valspodar | A-to-B | 20.5 | |
| B-to-A | 23.6 | ||
| Efflux Ration | 1.15 | ||
| P-gp Substrate classification | - | Negative | |
| Metabolic Stability (human liver microsomes) | Clint (mL/min/mg protein) | 0.0124 | |
| Half-life (min) | 112 | ||
| Protein binding (human plasma) | % bound | >99.5 | |
| Cytochrome P450 inhibition, IC50 (µM) | CYP1A2 (Phenacetin) | 7.40 | |
| CYP2B6 (Bupropion) | 3.19 | ||
| CYP2C8 (Amodiaquine) | 2.08 | ||
| CYP2C9 (Diclofenac) | 5.01 | ||
| CYP2C19 (S-Mephenytoin) | 7.31 | ||
| CYP2D6 (Bufuralol) | >10 | ||
| CYP3A (Midazolam) | >10 | ||
| CYP3A (Testosterone) | >10 | ||
| Parameters a | NBCoV1 | NBCoV2 | UNITS | ||
|---|---|---|---|---|---|
| PO | IV | PO | IV | ||
| Dose | 10 | 5 | 10 | 5 | mg/kg |
| t1/2 | 11.32 | 3.57 | 3.96 | 3.52 | h |
| Tmax | 2 | 0.25 | 4 | 2 | h |
| Cmax | 1499.76 | 7815.76 | 30.12 | 2219.45 | ng/mL |
| C0 | - | 7407.25 | - | 1710.87 | ng/mL |
| AUC0-t | 12,023.00 | 37,515.91 | 318.78 | 18,074.45 | ng/mL ∗ h |
| MRT0-inf_obs | 14.34 | 4.02 | 7.28 | 5.09 | h |
| CLobs | - | 0.00013 | - | 0.00027 | (mg/kg)/(ng/mL)/h |
| Vss_obs | - | 0.00053 | - | 0.00140 | (mg/kg)/(ng/mL) |
| Vz/F_obs | 0.01078 | - | 0.17593 | - | (mg/kg)/(ng/mL) |
| CL/F_obs | 0.00066 | - | 0.03082 | - | (mg/kg)/(ng/mL)/h |
| F% | 20.1 | - | 0.9 | - | 100 ∗ AUC(PO)/AUC(IV) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curreli, F.; Ahmed, S.; Victor, S.M.B.; Drelich, A.; Panda, S.S.; Altieri, A.; Kurkin, A.V.; Tseng, C.-T.K.; Hillyer, C.D.; Debnath, A.K. Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays. Viruses 2022, 14, 69. https://doi.org/10.3390/v14010069
Curreli F, Ahmed S, Victor SMB, Drelich A, Panda SS, Altieri A, Kurkin AV, Tseng C-TK, Hillyer CD, Debnath AK. Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays. Viruses. 2022; 14(1):69. https://doi.org/10.3390/v14010069
Chicago/Turabian StyleCurreli, Francesca, Shahad Ahmed, Sofia M. B. Victor, Aleksandra Drelich, Siva S. Panda, Andrea Altieri, Alexander V. Kurkin, Chien-Te K. Tseng, Christopher D. Hillyer, and Asim K. Debnath. 2022. "Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays" Viruses 14, no. 1: 69. https://doi.org/10.3390/v14010069
APA StyleCurreli, F., Ahmed, S., Victor, S. M. B., Drelich, A., Panda, S. S., Altieri, A., Kurkin, A. V., Tseng, C.-T. K., Hillyer, C. D., & Debnath, A. K. (2022). Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays. Viruses, 14(1), 69. https://doi.org/10.3390/v14010069