
Evaluating the Impact of Anthropogenic Factors on the Dissemination of Contemporary Cosmopolitan, Arctic, and Arctic-like Rabies Viruses

 , ,
, , 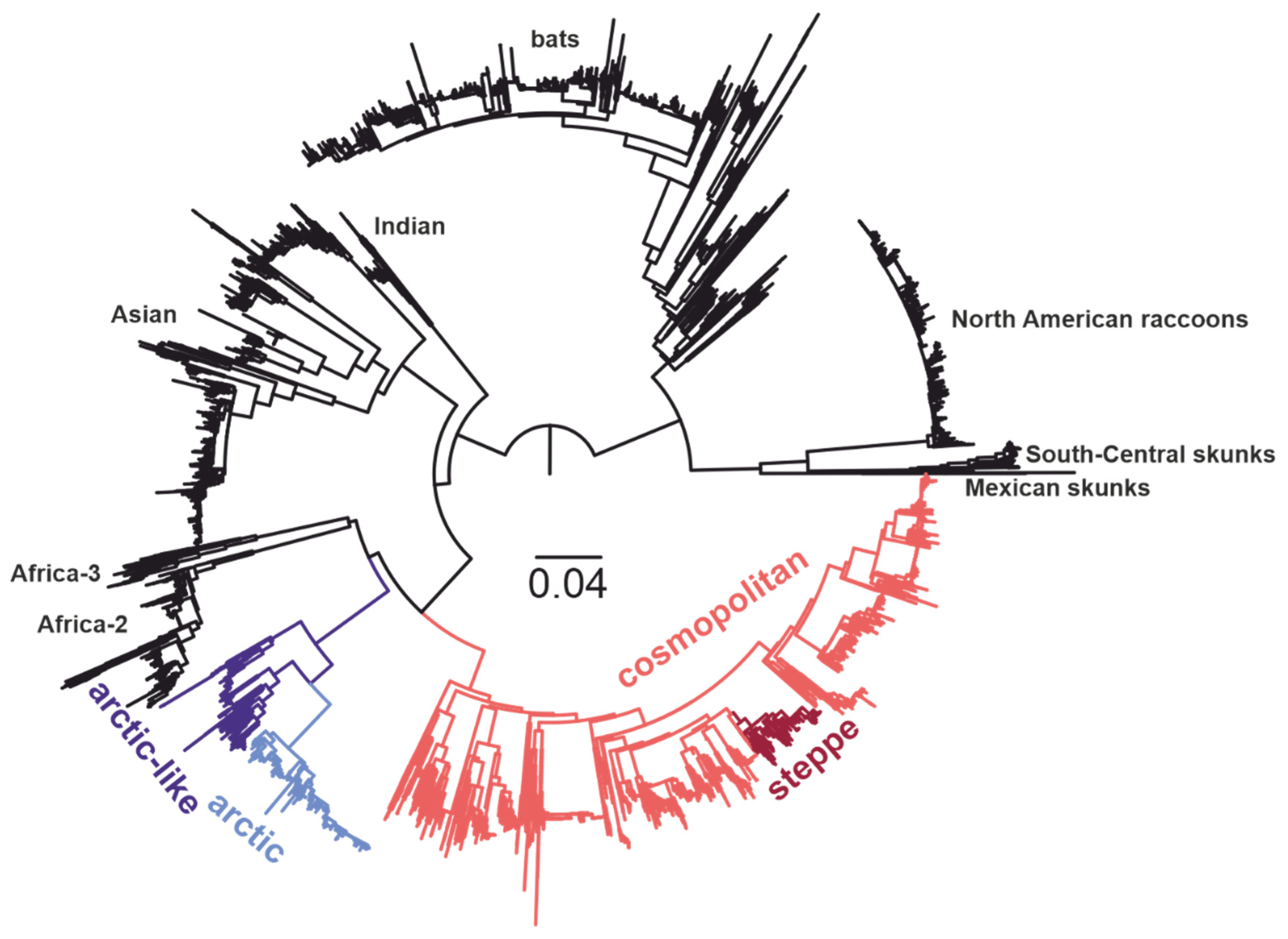{kind=link}
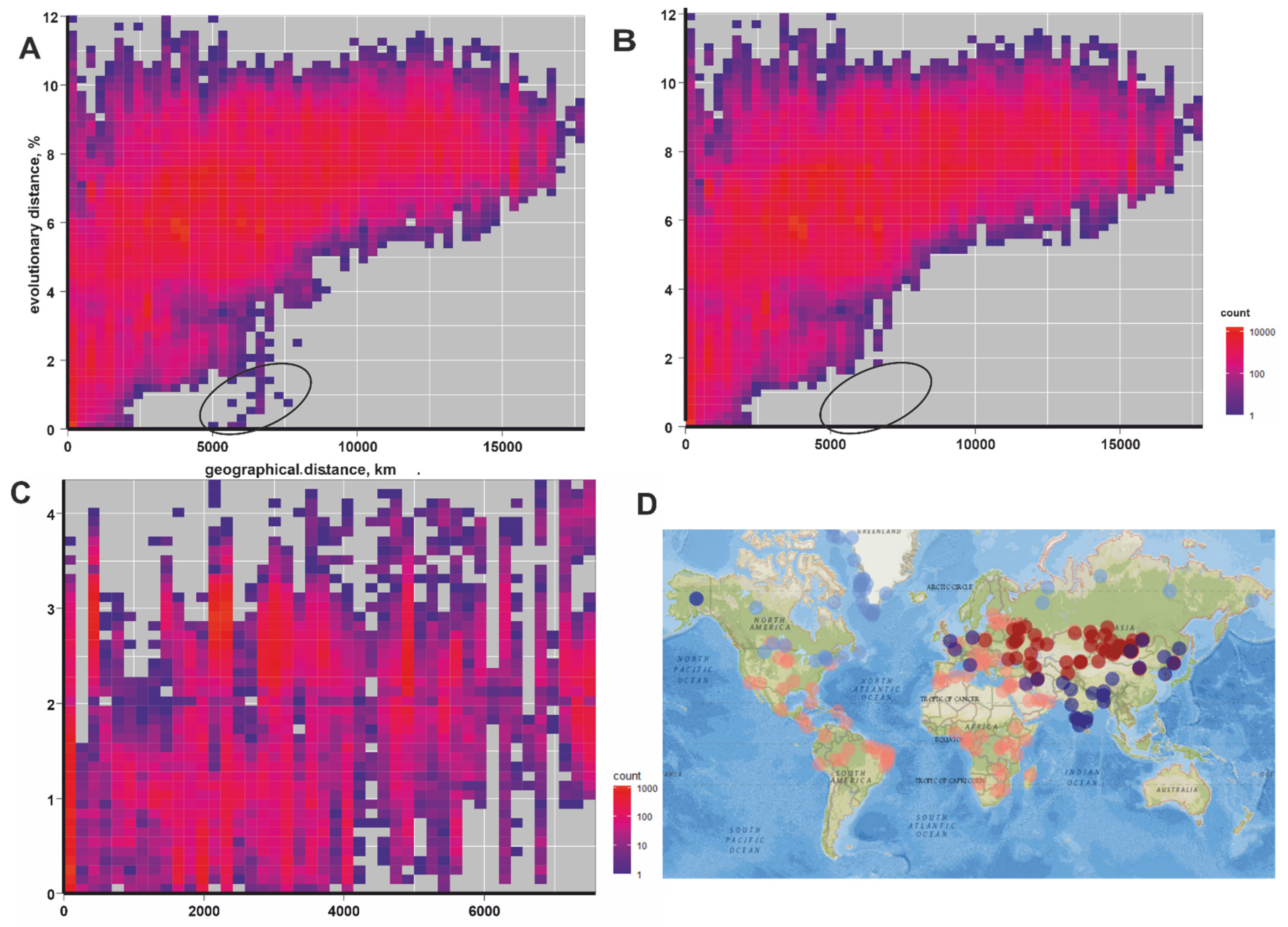{kind=link}
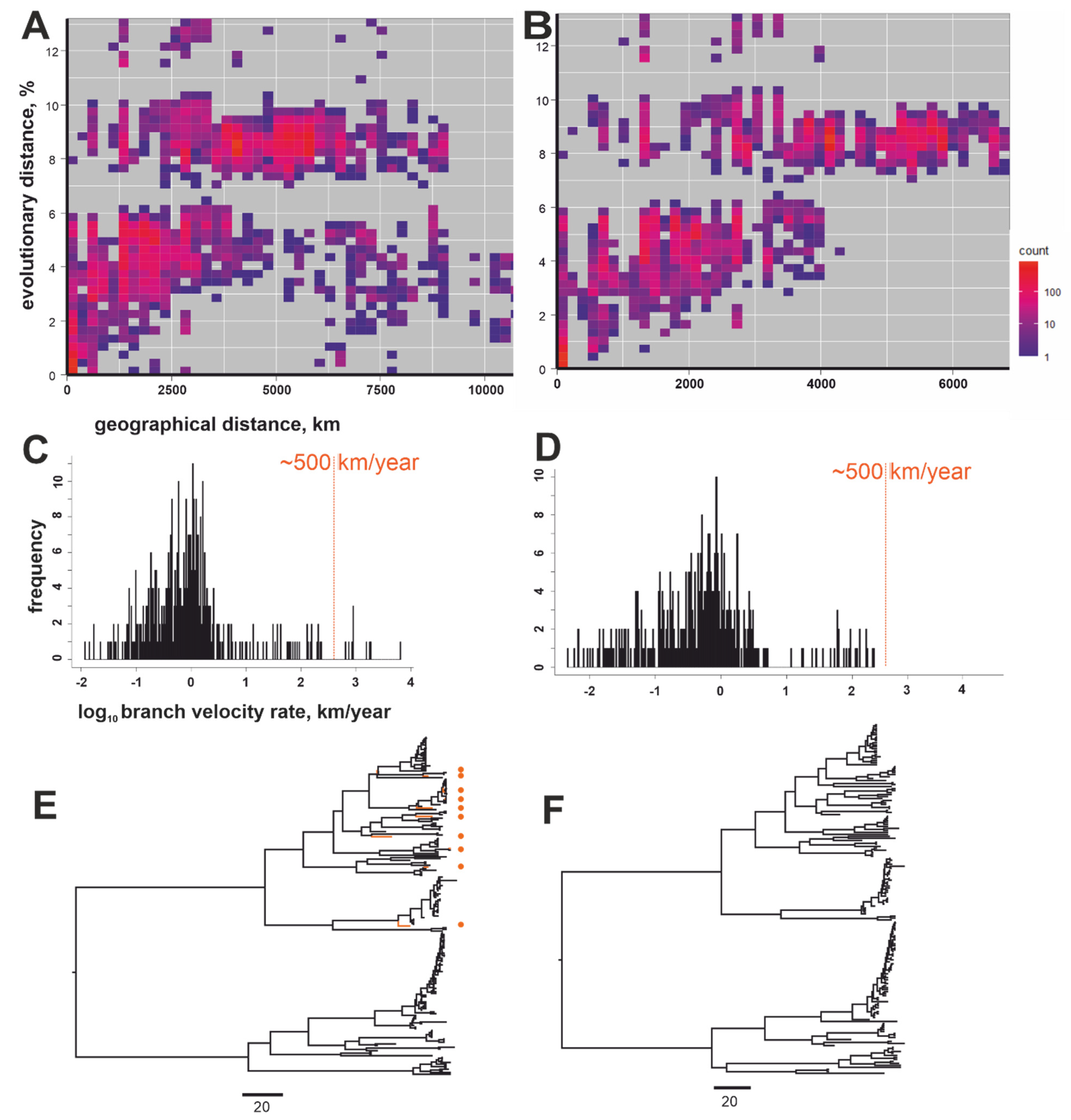{kind=link}
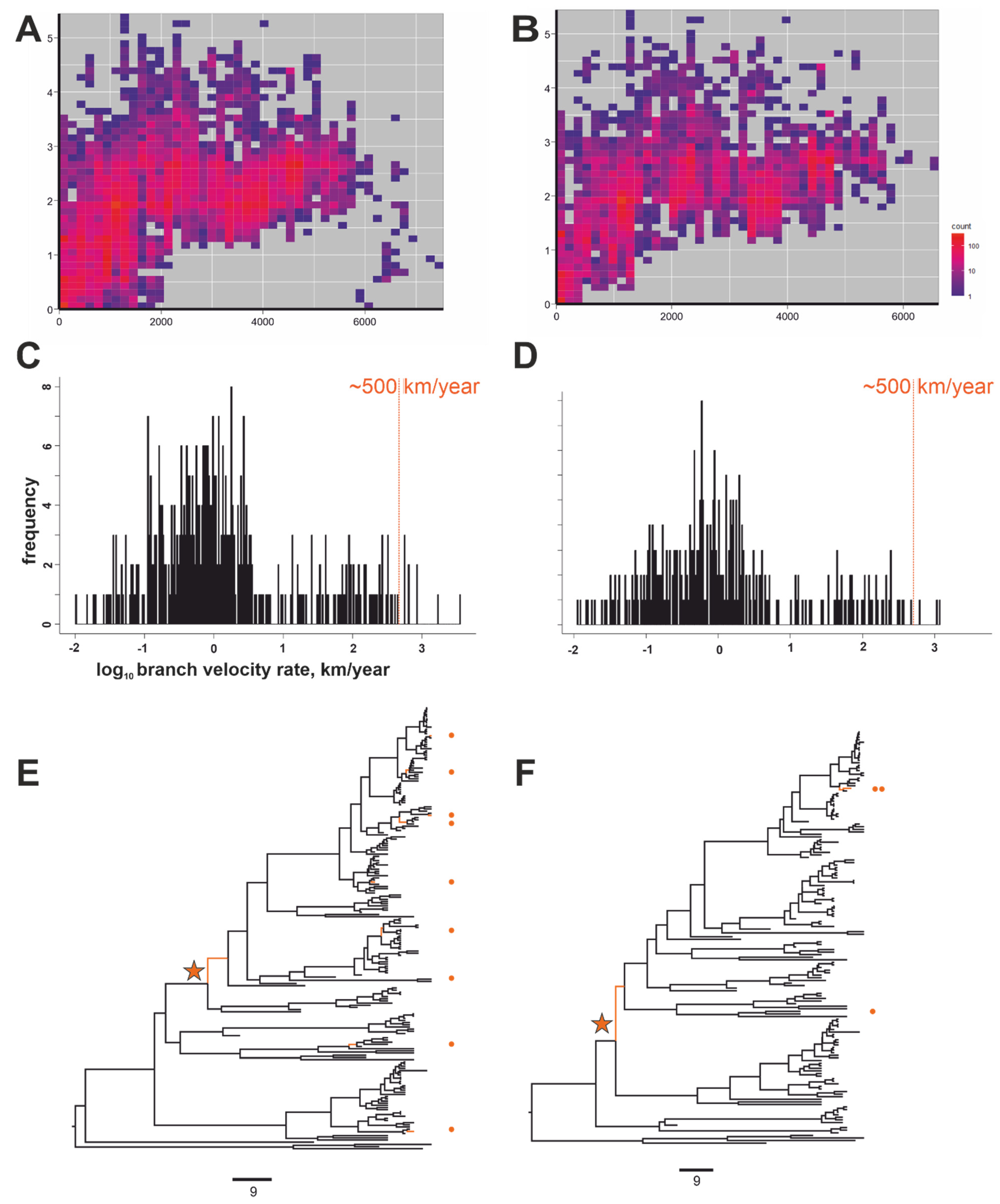{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hayman, D.T.S.; Fooks, A.R.; Marston, D.; Garcia-R, J.C. The Global Phylogeography of Lyssaviruses—Challenging the ’Out of Africa’ Hypothesis. PLoS Negl. Trop. Dis. 2016, 10, e0005266. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Expert Consultation on Rabies: Third Report; WHO Technical Report Series 1012; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.; Bourhy, H. Large-Scale Phylogenomic Analysis Reveals the Complex Evolutionary History of Rabies Virus in Multiple Carnivore Hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef] [PubMed]
- Bourhy, H.; Reynes, J.-M.; Dunham, E.J.; Dacheux, L.; Larrous, F.; Huong, V.T.Q.; Xu, G.; Yan, J.; Miranda, M.E.G.; Holmes, E. The origin and phylogeography of dog rabies virus. J. Gen. Virol. 2008, 89, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.; Freuling, C.M.; Müller, T.; Pfaff, F.; Bodenhofer, U.; Höper, D.; Fischer, M.; Marston, D.A.; Fooks, A.R.; Mettenleiter, T.C.; et al. Defining objective clusters for rabies virus sequences using affinity propagation clustering. PLoS Negl. Trop. Dis. 2018, 12, e0006182. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Villa, A.; Mauldin, M.R.; Shi, M.; Escobar, L.E.; Gallardo-Romero, N.F.; Damon, I.; Olson, V.A.; Streicker, D.G.; Emerson, G. The history of rabies in the Western Hemisphere. Antivir. Res. 2017, 146, 221–232. [Google Scholar] [CrossRef]
- Mey, C.; Metlin, A.; Duong, V.; Ong, S.; In, S.; Horwood, P.F.; Reynes, J.-M.; Bourhy, H.; Tarantola, A.; Buchy, P. Evidence of two distinct phylogenetic lineages of dog rabies virus circulating in Cambodia. Infect. Genet. Evol. 2016, 38, 55–61. [Google Scholar] [CrossRef]
- Shchelkanov, M.Y.; Deviatkin, A.; Ananiev, V.Y.; Frolov, E.V.; Dombrovskaya, I.E.; Dedkov, V.G.; Ardashev, A.V.; Kolomeets, S.A.; Korotkova, I.P.; Lyubchenko, E.N.; et al. Isolation and complete genome sequencing of rabies virus strain isolated from a brown bear (Ursus arctos) that attacked a human in Primorsky krai (November 2014). Probl. Virol. 2016, 61, 180–186. [Google Scholar] [CrossRef]
- Boland, T.A.; McGuone, D.; Jindal, J.; Rocha, M.; Cumming, M.; Rupprecht, C.E.; Barbosa, T.F.S.; Oliveira, R.D.N.; Chu, C.J.; Cole, A.J.; et al. Phylogenetic and epidemiologic evidence of multiyear incubation in human rabies. Ann. Neurol. 2013, 75, 155–160. [Google Scholar] [CrossRef]
- Turcitu, M.; Barboi, G.; Vuta, V.; Mihai, I.; Boncea, D.; Dumitrescu, F.; Codreanu, M.; Johnson, N.; Fooks, A.; Müller, T.; et al. Molecular epidemiology of rabies virus in Romania provides evidence for a high degree of heterogeneity and virus diversity. Virus Res. 2010, 150, 28–33. [Google Scholar] [CrossRef]
- Anderson, R.; Jackson, H.C.; May, R.M.; Smith, A.M. Population dynamics of fox rabies in Europe. Nat. Cell Biol. 1981, 289, 765–771. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Botvinkin, A.D.; McElhinney, L.; Smith, J.S.; Orciari, L.A.; Hughes, G.J.; Fooks, A.R.; Rupprecht, C.E. Molecular epidemiology of terrestrial rabies in the former soviet Union. J. Wildl. Dis. 2004, 40, 617–631. [Google Scholar] [CrossRef][Green Version]
- Fuglei, E.; Tarroux, A. Arctic fox dispersal from Svalbard to Canada: One female’s long run across sea ice. Polar Res. 2019, 38, 3512. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Falardeau, E.; Flynn, A.; Whitney, H.; Marshall, H.D. Relationships between fox populations and rabies virus spread in northern Canada. PLoS ONE 2021, 16, e0246508. [Google Scholar] [CrossRef]
- Joly, K.; Gurarie, E.; Sorum, M.S.; Kaczensky, P.; Cameron, M.D.; Jakes, A.F.; Borg, B.L.; Nandintsetseg, D.; Hopcraft, J.G.C.; Buuveibaatar, B.; et al. Longest terrestrial migrations and movements around the world. Sci. Rep. 2019, 9, 15333. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Lukashev, A.N.; Poleshchuk, E.; Dedkov, V.G.; Tkachev, S.; Sidorov, G.N.; Karganova, G.G.; Galkina, I.V.; Shchelkanov, M.Y.; Shipulin, G. The phylodynamics of the rabies virus in the Russian Federation. PLoS ONE 2017, 12, e0171855. [Google Scholar] [CrossRef]
- Mansfield, K.; Racloz, V.; McElhinney, L.; Marston, D.; Johnson, N.; Rønsholt, L.; Christensen, L.; Neuvonen, E.; Botvinkin, A.; Rupprecht, C.; et al. Molecular epidemiological study of Arctic rabies virus isolates from Greenland and comparison with isolates from throughout the Arctic and Baltic regions. Virus Res. 2006, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hanke, D.; Freuling, C.M.; Fischer, S.; Hueffer, K.; Hundertmark, K.; Nadin-Davis, S.; Marston, D.; Fooks, A.R.; Bøtner, A.; Mettenleiter, T.C.; et al. Spatio-temporal Analysis of the Genetic Diversity of Arctic Rabies Viruses and Their Reservoir Hosts in Greenland. PLoS Negl. Trop. Dis. 2016, 10, e0004779. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, I.V.; Hughes, G.J.; Botvinkin, A.D.; Gribencha, S.G.; Rupprecht, C.E. Arctic and Arctic-like rabies viruses: Distribution, phylogeny and evolutionary history. Epidemiol. Infect. 2008, 136, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Nadin-Davis, S.A.; Turner, G.; Paul, J.P.V.; Madhusudana, S.N.; Wandeler, A.I. Emergence of Arctic-like Rabies Lineage in India. Emerg. Infect. Dis. 2007, 13, 111–116. [Google Scholar] [CrossRef]
- Freuling, C.M.; Hampson, K.; Selhorst, T.; Schröder, R.; Meslin, F.X.; Mettenleiter, T.C.; Müller, T. The elimination of fox rabies from Europe: Determinants of success and lessons for the future. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120142. [Google Scholar] [CrossRef]
- Johnson, N.; Un, H.; Fooks, A.R.; Freuling, C.; Müller, T.; Aylan, O.; Vos, A. Rabies epidemiology and control in Turkey: Past and present. Epidemiol. Infect. 2009, 138, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Fèvre, E.M.; Bronsvoort, M.; Hamilton, K.A.; Cleaveland, S. Animal movements and the spread of infectious diseases. Trends Microbiol. 2006, 14, 125–131. [Google Scholar] [CrossRef]
- Hutchison, D.W.; Templeton, A.R. Correlation of Pairwise Genetic and Geographic Distance Measures: Inferring the Relative Influences of Gene Flow and Drift on the Distribution of Genetic Variability. Evolution 1999, 53, 1898. [Google Scholar] [CrossRef]
- Deviatkin, A.A.; Kholodilov, I.S.; Belova, O.A.; Bugmyrin, S.V.; Bespyatova, L.A.; Ivannikova, A.Y.; Vakulenko, Y.A.; Lukashev, A.N.; Karganova, G.G. Baltic Group Tick-Borne Encephalitis Virus Phylogeography: Systemic Inconsistency Pattern between Genetic and Geographic Distances. Microorganisms 2020, 8, 1589. [Google Scholar] [CrossRef]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Lemey, P. Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 2011, 1, 423–429. [Google Scholar] [CrossRef]
- Talbi, C.; Lemey, P.; Suchard, M.A.; Abdelatif, E.; Elharrak, M.; Jalal, N.; Faouzi, A.; Echevarría, J.E.; Morón, S.V.; Rambaut, A.; et al. Phylodynamics and Human-Mediated Dispersal of a Zoonotic Virus. PLoS Pathog. 2010, 6, e1001166. [Google Scholar] [CrossRef]
- Dellicour, S.; Rose, R.; Faria, N.R.; Vieira, L.F.P.; Bourhy, H.; Gilbert, M.; Lemey, P.; Pybus, O. Using Viral Gene Sequences to Compare and Explain the Heterogeneous Spatial Dynamics of Virus Epidemics. Mol. Biol. Evol. 2017, 34, 2563–2571. [Google Scholar] [CrossRef]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data—Reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2011, 61, 170–173. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed]
- FigTree v1.4.4. Available online: https://github.com/rambaut/figtree/releases (accessed on 28 December 2021).
- Vaiente, M.A.; Scotch, M. Going back to the roots: Evaluating Bayesian phylogeographic models with discrete trait uncertainty. Infect. Genet. Evol. 2020, 85, 104501. [Google Scholar] [CrossRef] [PubMed]
- McElhinney, L.M.; Marston, D.; Freuling, C.M.; Cragg, W.; Stankov, S.; Lalošević, D.; Müller, T.; Fooks, A.R. Molecular diversity and evolutionary history of rabies virus strains circulating in the Balkans. J. Gen. Virol. 2011, 92, 2171–2180. [Google Scholar] [CrossRef][Green Version]
- Talbi, C.; Holmes, E.; De Benedictis, P.; Faye, O.; Nakouné, E.; Gamatié, D.; Diarra, A.; Elmamy, B.O.; Sow, A.; Adjogoua, E.V.; et al. Evolutionary history and dynamics of dog rabies virus in western and central Africa. J. Gen. Virol. 2009, 90, 783–791. [Google Scholar] [CrossRef]
- Guo, Z.; Tao, X.; Yin, C.; Han, N.; Yu, J.; Li, H.; Liu, H.; Fang, W.; Adams, J.; Wang, J.; et al. National Borders Effectively Halt the Spread of Rabies: The Current Rabies Epidemic in China Is Dislocated from Cases in Neighboring Countries. PLoS Negl. Trop. Dis. 2013, 7, e2039. [Google Scholar] [CrossRef]
- Biek, R.; Henderson, J.C.; Waller, L.A.; Rupprecht, C.E.; Real, L.A. A high-resolution genetic signature of demographic and spatial expansion in epizootic rabies virus. Proc. Natl. Acad. Sci. USA 2007, 104, 7993–7998. [Google Scholar] [CrossRef]
- Wang, W.; Ma, J.; Nie, J.; Li, J.; Cao, S.; Wang, L.; Yu, C.; Huang, W.; Li, Y.; Yu, Y.; et al. Antigenic variations of recent street rabies virus. Emerg. Microbes Infect. 2019, 8, 1584–1592. [Google Scholar] [CrossRef]
- Wang, L.; Wu, X.; Bao, J.; Song, C.; Du, J. Phylodynamic and transmission pattern of rabies virus in China and its neighboring countries. Arch. Virol. 2019, 164, 2119–2129. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Villa, A.; Reeder, S.A.; Orciari, L.A.; Yager, P.A.; Franka, R.; Blanton, J.D.; Zuckero, L.; Hunt, P.; Oertli, E.H.; Robinson, L.E.; et al. Enzootic Rabies Elimination from Dogs and Reemergence in Wild Terrestrial Carnivores, United States. Emerg. Infect. Dis. 2008, 14, 1849–1854. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, S.R.; Winkler, W.G. Descriptive epidemiology from an epizootic of raccoon rabies in the middle Atlantic states, 1982–1983. Am. J. Epidemiol. 1987, 126, 429–437. [Google Scholar] [CrossRef]
- Horton, D.; McElhinney, L.M.; Freuling, C.M.; Marston, D.; Banyard, A.C.; Goharrriz, H.; Wise, E.; Breed, A.; Saturday, G.; Kolodziejek, J.; et al. Complex Epidemiology of a Zoonotic Disease in a Culturally Diverse Region: Phylogeography of Rabies Virus in the Middle East. PLoS Negl. Trop. Dis. 2015, 9, e0003569. [Google Scholar] [CrossRef]
- Carnieli, P.; Oliveira, R.D.N.; Macedo, C.I.; Castilho, J.G. Phylogeography of rabies virus isolated from dogs in Brazil between 1985 and 2006. Arch. Virol. 2011, 156, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Brunker, K.; Marston, D.; Horton, D.L.; Cleaveland, S.; Fooks, A.R.; Kazwala, R.; Ngeleja, C.; Lembo, T.; Sambo, M.; Mtema, Z.J.; et al. Elucidating the phylodynamics of endemic rabies virus in eastern Africa using whole-genome sequencing. Virus Evol. 2015, 1, vev011. [Google Scholar] [CrossRef]
- Dibia, I.N.; Sumiarto, B.; Susetya, H.; Putra, A.A.G.; Scott-Orr, H.; Mahardika, G.N. Phylogeography of the current rabies viruses in Indonesia. J. Veter-Sci. 2015, 16, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Vakulenko, Y.; Deviatkin, A.; Lukashev, A. The Effect of Sample Bias and Experimental Artefacts on the Statistical Phylogenetic Analysis of Picornaviruses. Viruses 2019, 11, 1032. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deviatkin, A.A.; Vakulenko, Y.A.; Dashian, M.A.; Lukashev, A.N. Evaluating the Impact of Anthropogenic Factors on the Dissemination of Contemporary Cosmopolitan, Arctic, and Arctic-like Rabies Viruses. Viruses 2022, 14, 66. https://doi.org/10.3390/v14010066
Deviatkin AA, Vakulenko YA, Dashian MA, Lukashev AN. Evaluating the Impact of Anthropogenic Factors on the Dissemination of Contemporary Cosmopolitan, Arctic, and Arctic-like Rabies Viruses. Viruses. 2022; 14(1):66. https://doi.org/10.3390/v14010066
Chicago/Turabian StyleDeviatkin, Andrei A., Yulia A. Vakulenko, Mariia A. Dashian, and Alexander N. Lukashev. 2022. "Evaluating the Impact of Anthropogenic Factors on the Dissemination of Contemporary Cosmopolitan, Arctic, and Arctic-like Rabies Viruses" Viruses 14, no. 1: 66. https://doi.org/10.3390/v14010066
APA StyleDeviatkin, A. A., Vakulenko, Y. A., Dashian, M. A., & Lukashev, A. N. (2022). Evaluating the Impact of Anthropogenic Factors on the Dissemination of Contemporary Cosmopolitan, Arctic, and Arctic-like Rabies Viruses. Viruses, 14(1), 66. https://doi.org/10.3390/v14010066