
Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions

Abstract
:1. Introduction
2. Human Cytomegalovirus Replication Cycle
Overview of Nuclear Egress of HCMV
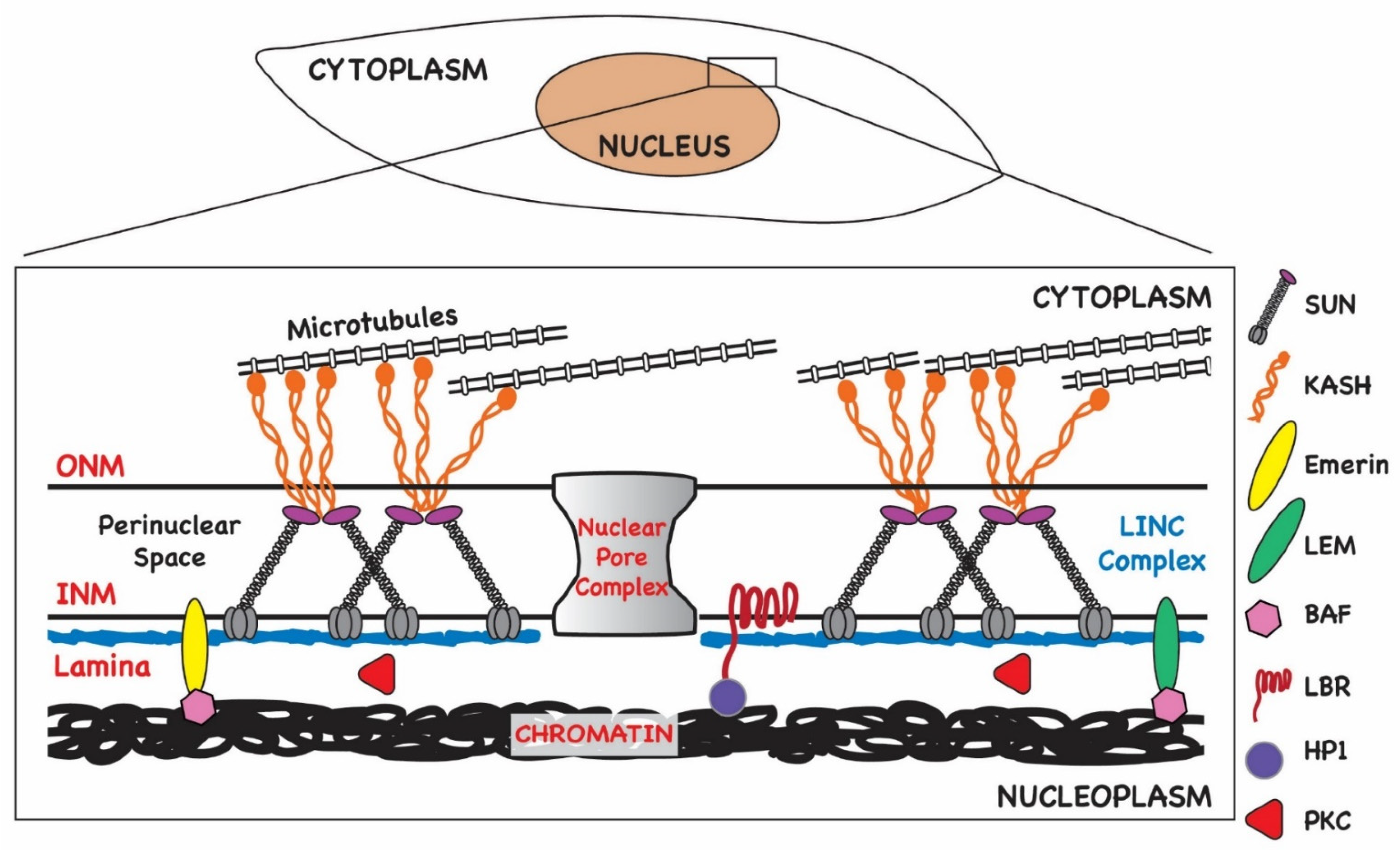
3. Interphase Nuclear Architecture
4. Herpesviruses Circumvent Nuclear Barriers
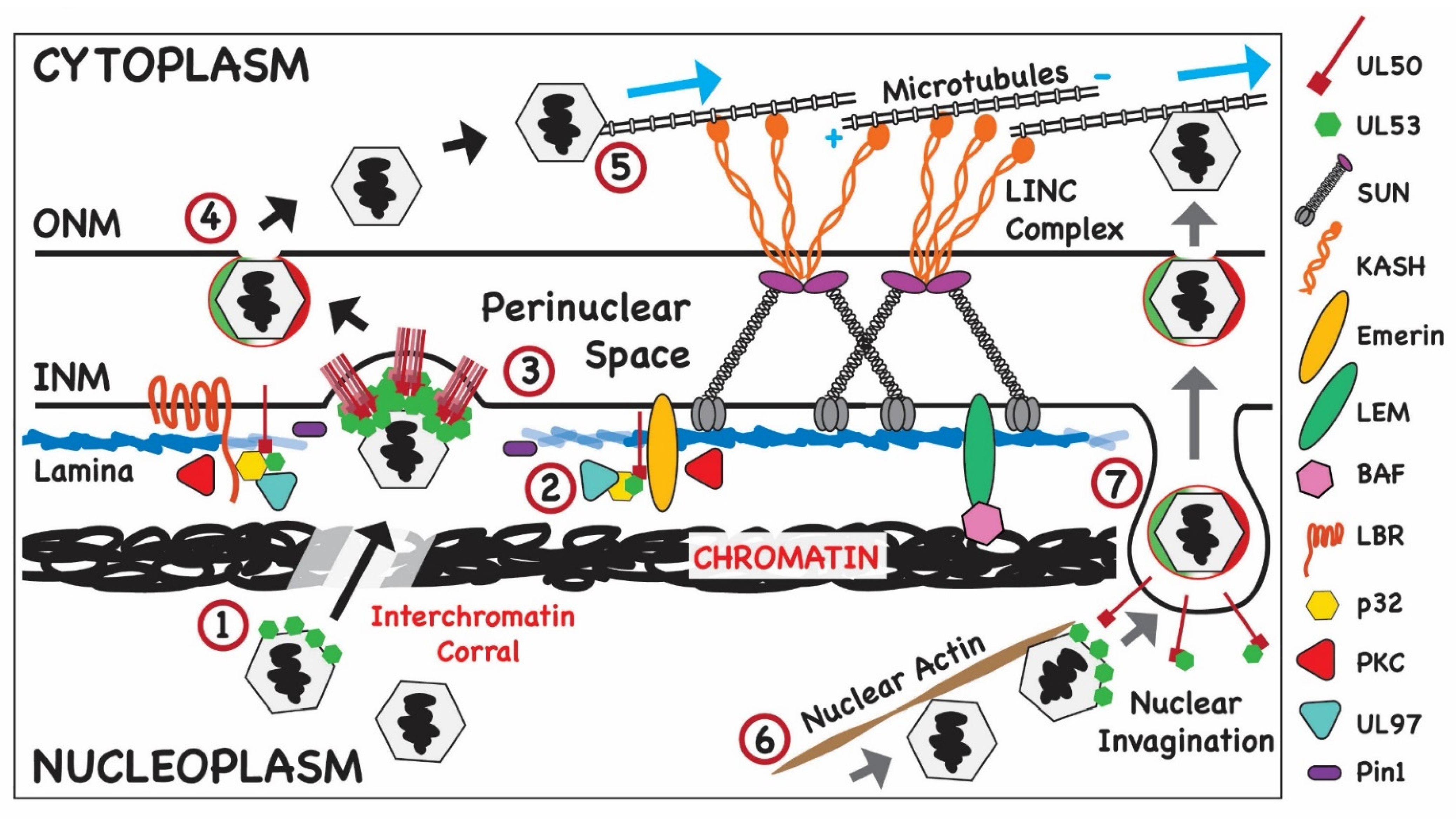
4.1. Nuclear Egress: Budding of Newly Assembled HCMV Capsids into the PNS
Role of Virus-Encoded Proteins
4.2. Role of Cellular Proteins in Nuclear Egress of HCMV: Modification of Cellular Barriers to Capsid Budding into the PNS
4.2.1. Chromatin as a Barrier to Nuclear Egress
4.2.2. Modifications of Nuclear Lamins and HCMV Nuclear Egress
4.2.3. Nuclear Actin and Nuclear Egress
4.2.4. ESCRT-III and Nuclear Egress
4.3. Nuclear Egress: Transit of Newly Assembled HCMV Capsids from the PNS through the ONM to the Cytoplasm
4.3.1. Role of Viral Proteins
4.3.2. Role of Cellular Proteins
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kicuntod, J.; Alkhashrom, S.; Häge, S.; Diewald, B.; Müller, R.; Hahn, F.; Lischka, P.; Sticht, H.; Eichler, J.; Marschall, M. Properties of Oligomeric Interaction of the Cytomegalovirus Core Nuclear Egress Complex (NEC) and Its Sensitivity to an NEC Inhibitory Small Molecule. Viruses 2021, 13, 462. [Google Scholar] [CrossRef]
- Monier, K.; Armas, J.C.G.; Etteldorf, S.; Ghazal, P.; Sullivan, K. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2000, 2, 661–665. [Google Scholar] [CrossRef]
- Aho, V.; Mäntylä, E.; Ekman, A.; Hakanen, S.; Mattola, S.; Chen, J.-H.; Weinhardt, V.; Ruokolainen, V.; Sodeik, B.; Larabell, C.; et al. Quantitative Microscopy Reveals Stepwise Alteration of Chromatin Structure during Herpesvirus Infection. Viruses 2019, 11, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchkovich, N.J.; Maguire, T.G.; Alwine, J.C. Role of the Endoplasmic Reticulum Chaperone BiP, SUN Domain Proteins, and Dynein in Altering Nuclear Morphology during Human Cytomegalovirus Infection. J. Virol. 2010, 84, 7005–7017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aho, V.; Myllys, M.; Ruokolainen, V.; Hakanen, S.; Mäntylä, E.; Virtanen, J.; Hukkanen, V.; Kühn, T.; Timonen, J.; Mattila, K.; et al. Chromatin organization regulates viral egress dynamics. Sci. Rep. 2017, 7, 3692. [Google Scholar] [CrossRef] [PubMed]
- Alwine, J.C. The Human Cytomegalovirus Assembly Compartment: A Masterpiece of Viral Manipulation of Cellular Processes That Facilitates Assembly and Egress. PLoS Pathog. 2012, 8, e1002878. [Google Scholar] [CrossRef] [Green Version]
- Villinger, C.; Neusser, G.; Kranz, C.; Walther, P.; Mertens, T. 3D Analysis of HCMV Induced-Nuclear Membrane Structures by FIB/SEM Tomography: Insight into an Unprecedented Membrane Morphology. Viruses 2015, 7, 5686–5704. [Google Scholar] [CrossRef]
- Procter, D.J.; Furey, C.; Garza-Gongora, A.G.; Kosak, S.T.; Walsh, D. Cytoplasmic control of intranuclear polarity by human cytomegalovirus. Nat. Cell Biol. 2020, 587, 109–114. [Google Scholar] [CrossRef]
- Sanchez, V.; Clark, C.L.; Yen, J.Y.; Dwarakanath, R.; Spector, D.H. Viable Human Cytomegalovirus Recombinant Virus with an Internal Deletion of the IE2 86 Gene Affects Late Stages of Viral Replication. J. Virol. 2002, 76, 2973–2989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, V.; McElroy, A.K.; Yen, J.; Tamrakar, S.; Clark, C.L.; Schwartz, R.A.; Spector, D.H. Cyclin-Dependent Kinase Activity Is Required at Early Times for Accurate Processing and Accumulation of the Human Cytomegalovirus UL122-123 and UL37 Immediate-Early Transcripts and at Later Times for Virus Production. J. Virol. 2004, 78, 11219–11232. [Google Scholar] [CrossRef] [Green Version]
- Salvant, B.S.; Fortunato, E.A.; Spector, D.H. Cell Cycle Dysregulation by Human Cytomegalovirus: Influence of the Cell Cycle Phase at the Time of Infection and Effects on Cyclin Transcription. J. Virol. 1998, 72, 3729–3741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, V.; Greis, K.D.; Sztul, E.; Britt, W.J. Accumulation of Virion Tegument and Envelope Proteins in a Stable Cytoplasmic Compartment during Human Cytomegalovirus Replication: Characterization of a Potential Site of Virus Assembly. J. Virol. 2000, 74, 975–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Vasanji, A.; Pellett, P.E. Three-Dimensional Structure of the Human Cytomegalovirus Cytoplasmic Virion Assembly Complex Includes a Reoriented Secretory Apparatus. J. Virol. 2007, 81, 11861–11869. [Google Scholar] [CrossRef] [Green Version]
- Hook, L.; Grey, F.; Grabski, R.; Tirabassi, R.; Doyle, T.; Hancock, M.; Landais, I.; Jeng, S.; McWeeney, S.; Britt, W.; et al. Cytomegalovirus miRNAs Target Secretory Pathway Genes to Facilitate Formation of the Virion Assembly Compartment and Reduce Cytokine Secretion. Cell Host Microbe 2014, 15, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Rebmann, G.M.; Grabski, R.; Sanchez, V.; Britt, W.J. Phosphorylation of Golgi Peripheral Membrane Protein Grasp65 Is an Integral Step in the Formation of the Human Cytomegalovirus Cytoplasmic Assembly Compartment. mBio 2016, 7, e01554-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Procter, D.J.; Banerjee, A.; Nukui, M.; Kruse, K.; Gaponenko, V.; Murphy, E.A.; Komarova, Y.; Walsh, D. The HCMV Assembly Compartment Is a Dynamic Golgi-Derived MTOC that Controls Nuclear Rotation and Virus Spread. Dev. Cell 2018, 45, 83–100.e7. [Google Scholar] [CrossRef] [Green Version]
- Indran, S.V.; Ballestas, M.E.; Britt, W.J. Bicaudal D1-Dependent Trafficking of Human Cytomegalovirus Tegument Protein pp150 in Virus-Infected Cells. J. Virol. 2010, 84, 3162–3177. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, A.D.; Sembongi, H.; Su, W.-M.; Abreu, S.; Reggiori, F.; Carman, G.M.; Siniossoglou, S. Lipid partitioning at the nuclear envelope controls membrane biogenesis. Mol. Biol. Cell 2015, 26, 3641–3657. [Google Scholar] [CrossRef]
- Malhas, A.; Goulbourne, C.; Vaux, D. The nucleoplasmic reticulum: Form and function. Trends Cell Biol. 2011, 21, 362–373. [Google Scholar] [CrossRef]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Rose, K.M.; Spada, S.J.; Hirsch, V.M.; Bouamr, F. When in Need of an ESCRT: The Nature of Virus Assembly Sites Suggests Mechanistic Parallels between Nuclear Virus Egress and Retroviral Budding. Viruses 2021, 13, 1138. [Google Scholar] [CrossRef] [PubMed]
- Vietri, M.; Stenmark, H.; Campsteijn, C. Closing a gap in the nuclear envelope. Curr. Opin. Cell Biol. 2016, 40, 90–97. [Google Scholar] [CrossRef]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef] [Green Version]
- Lin, D.H.; Hoelz, A. The Structure of the Nuclear Pore Complex (An Update). Annu. Rev. Biochem. 2019, 88, 725–783. [Google Scholar] [CrossRef] [Green Version]
- Starr, D.A.; Fridolfsson, H.N. Interactions Between Nuclei and the Cytoskeleton Are Mediated by SUN-KASH Nuclear-Envelope Bridges. Annu. Rev. Cell Dev. Biol. 2010, 26, 421–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, S.G.; Zhang, Q.; Prasad, N.; Li, Y.; Chamala, S.; Kuchibhotla, R.; Kc, B.; Aggarwal, V.; Shrestha, S.; Jones, A.L.; et al. The mammalian LINC complex regulates genome transcriptional responses to substrate rigidity. Sci. Rep. 2016, 6, 38063. [Google Scholar] [CrossRef]
- Gundersen, G.G.; Worman, H.J. Nuclear Positioning. Cell 2013, 152, 1376–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maninová, M.; Klímová, Z.; Parsons, J.T.; Weber, M.J.; Iwanicki, M.P.; Vomastek, T. The Reorientation of Cell Nucleus Promotes the Establishment of Front–Rear Polarity in Migrating Fibroblasts. J. Mol. Biol. 2013, 425, 2039–2055. [Google Scholar] [CrossRef]
- Razafsky, D.; Wirtz, D.; Hodzic, D. Nuclear Envelope in Nuclear Positioning and Cell Migration. Adv. Exp. Med. Biol. 2014, 773, 471–490. [Google Scholar] [CrossRef] [Green Version]
- Burke, B. Chain reaction: LINC complexes and nuclear positioning. F1000Research 2019, 8, 136. [Google Scholar] [CrossRef]
- Burla, R.; La Torre, M.; Maccaroni, K.; Verni, F.; Giunta, S.; Saggio, I. Interplay of the nuclear envelope with chromatin in physiology and pathology. Nucleus 2020, 11, 205–218. [Google Scholar] [CrossRef]
- Uhler, C.; Shivashankar, G.V. Regulation of genome organization and gene expression by nuclear mechanotransduction. Nat. Rev. Mol. Cell Biol. 2017, 18, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Genet. 2011, 9, 382–394. [Google Scholar] [CrossRef]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The Interacting UL31 and UL34 Gene Products of Pseudorabies Virus Are Involved in Egress from the Host-Cell Nucleus and Represent Components of Primary Enveloped but Not Mature Virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milbradt, J.; Kraut, A.; Hutterer, C.; Sonntag, E.; Schmeiser, C.; Ferro, M.; Wagner, S.; Lenac, T.; Claus, C.; Pinkert, S.; et al. Proteomic Analysis of the Multimeric Nuclear Egress Complex of Human Cytomegalovirus. Mol. Cell. Proteom. 2014, 13, 2132–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milbradt, J.; Auerochs, S.; Sticht, H.; Marschall, M. Cytomegaloviral proteins that associate with the nuclear lamina: Components of a postulated nuclear egress complex. J. Gen. Virol. 2009, 90, 579–590. [Google Scholar] [CrossRef]
- Lee, C.-P.; Liu, P.-T.; Kung, H.-N.; Su, M.-T.; Chua, H.-H.; Chang, Y.-H.; Chang, C.-W.; Tsai, C.-H.; Liu, F.-T.; Chen, M.-R. The ESCRT Machinery Is Recruited by the Viral BFRF1 Protein to the Nucleus-Associated Membrane for the Maturation of Epstein-Barr Virus. PLoS Pathog. 2012, 8, e1002904. [Google Scholar] [CrossRef] [Green Version]
- Gonnella, R.; Farina, A.; Santarelli, R.; Raffa, S.; Feederle, R.; Bei, R.; Granato, M.; Modesti, A.; Frati, L.; Delecluse, H.-J.; et al. Characterization and Intracellular Localization of the Epstein-Barr Virus Protein BFLF2: Interactions with BFRF1 and with the Nuclear Lamina. J. Virol. 2005, 79, 3713–3727. [Google Scholar] [CrossRef] [Green Version]
- Farina, A.; Feederle, R.; Raffa, S.; Gonnella, R.; Santarelli, R.; Frati, L.; Angeloni, A.; Torrisi, M.R.; Faggioni, A.; Delecluse, H.-J. BFRF1 of Epstein-Barr Virus Is Essential for Efficient Primary Viral Envelopment and Egress. J. Virol. 2005, 79, 3703–3712. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Granzow, H.; Fuchs, W.; Keil, G.M.; Finke, S.; Mettenleiter, T.C. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 7241–7246. [Google Scholar] [CrossRef] [Green Version]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun. 2014, 5, 4131. [Google Scholar] [CrossRef]
- Draganova, E.B.; Zhang, J.; Zhou, Z.H.; E Heldwein, E. Structural basis for capsid recruitment and coat formation during HSV-1 nuclear egress. eLife 2020, 9, e56627. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.; Vasishtan, D.; Bäuerlein, F.J.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagenaar, F.; Pol, J.M.A.; Peeters, B.; Gielkens, A.L.J.; De Wind, N.; Kimman, T.G. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 1995, 76, 1851–1859. [Google Scholar] [CrossRef]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus Recruitment of Cellular Kinases to Dissolve the Nuclear Lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Kamil, J.P.; Coughlin, M.; Reim, N.I.; Coen, D.M. Human Cytomegalovirus UL50 and UL53 Recruit Viral Protein Kinase UL97, Not Protein Kinase C, for Disruption of Nuclear Lamina and Nuclear Egress in Infected Cells. J. Virol. 2014, 88, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Kuan, M.I.; O’Dowd, J.M.; Chughtai, K.; Hayman, I.; Brown, C.J.; Fortunato, E.A. Human Cytomegalovirus nuclear egress and secondary envelopment are negatively affected in the absence of cellular p53. Virology 2016, 497, 279–293. [Google Scholar] [CrossRef]
- I Kuan, M.; O’Dowd, J.M.; Fortunato, E.A. The absence of p53 during Human Cytomegalovirus infection leads to decreased UL53 expression, disrupting UL50 localization to the inner nuclear membrane, and thereby inhibiting capsid nuclear egress. Virology 2016, 497, 262–278. [Google Scholar] [CrossRef]
- Monte, P.D.; Pignatelli, S.; Zini, N.; Maraldi, N.M.; Perret, E.; Prevost, M.C.; Landini, M.P. Analysis of intracellular and intraviral localization of the human cytomegalovirus UL53 protein. J. Gen. Virol. 2002, 83, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Schmeiser, C.; Borst, E.; Sticht, H.; Marschall, M.; Milbradt, J. The cytomegalovirus egress proteins pUL50 and pUL53 are translocated to the nuclear envelope through two distinct modes of nuclear import. J. Gen. Virol. 2013, 94, 2056–2069. [Google Scholar] [CrossRef]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [Green Version]
- Leigh, K.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.; Hogle, J.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, Y.A.; Häge, S.; Alkhashrom, S.; Höllriegl, T.; Weigert, S.; Dolles, S.; Hof, K.; Walzer, S.A.; Egerer-Sieber, C.; Conrad, M.; et al. High-resolution crystal structures of two prototypical β- and γ-herpesviral nuclear egress complexes unravel the determinants of subfamily specificity. J. Biol. Chem. 2020, 295, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [Green Version]
- Borst, E.M.; Bauerfeind, R.; Binz, A.; Stephan, T.M.; Neuber, S.; Wagner, K.; Steinbrück, L.; Sodeik, B.; Roviš, T.L.; Jonjić, S.; et al. The Essential Human Cytomegalovirus Proteins pUL77 and pUL93 are Structural Components Necessary for Viral Genome Encapsidation. J. Virol. 2016, 90, 5860–5875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köppen-Rung, P.; Dittmer, A.; Bogner, E. Intracellular Distribution of Capsid-Associated pUL77 of Human Cytomegalovirus and Interactions with Packaging Proteins and pUL93. J. Virol. 2016, 90, 5876–5885. [Google Scholar] [CrossRef] [Green Version]
- Hamirally, S.; Kamil, J.P.; Ndassa-Colday, Y.M.; Lin, A.J.; Jahng, W.J.; Baek, M.-C.; Noton, S.; Silva, L.A.; Simpson-Holley, M.; Knipe, D.M.; et al. Viral Mimicry of Cdc2/Cyclin-Dependent Kinase 1 Mediates Disruption of Nuclear Lamina during Human Cytomegalovirus Nuclear Egress. PLoS Pathog. 2009, 5, e1000275. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Bender, B.J.; Kamil, J.; Lye, M.F.; Pesola, J.M.; Reim, N.I.; Hogle, J.; Coen, D.M. Human Cytomegalovirus UL97 Phosphorylates the Viral Nuclear Egress Complex. J. Virol. 2014, 89, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; He, Y.S.; Kim, Y.; Chu, L.; Ohmstede, C.; Biron, K.K.; Coen, D.M. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 1997, 71, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Sonntag, E.; Milbradt, J.; Svrlanska, A.; Strojan, H.; Häge, S.; Kraut, A.; Hesse, A.-M.; Amin, B.; Sonnewald, U.; Couté, Y.; et al. Protein kinases responsible for the phosphorylation of the nuclear egress core complex of human cytomegalovirus. J. Gen. Virol. 2017, 98, 2569–2581. [Google Scholar] [CrossRef]
- Milbradt, J.; Auerochs, S.; Marschall, M. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 2007, 88, 2642–2650. [Google Scholar] [CrossRef]
- Marschall, M.; Marzi, A.; aus dem Siepen, P.; Jochmann, R.; Kalmer, M.; Auerochs, S.; Lischka, P.; Leis, M.; Stamminger, T. Cellular p32 Recruits Cytomegalovirus Kinase pUL97 to Redistribute the Nuclear Lamina. J. Biol. Chem. 2005, 280, 33357–33367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milbradt, J.; Webel, R.; Auerochs, S.; Sticht, H.; Marschall, M. Novel Mode of Phosphorylation-triggered Reorganization of the Nuclear Lamina during Nuclear Egress of Human Cytomegalovirus. J. Biol. Chem. 2010, 285, 13979–13989. [Google Scholar] [CrossRef] [Green Version]
- Milbradt, J.; Hutterer, C.; Bahsi, H.; Wagner, S.; Sonntag, E.; Horn, A.H.C.; Kaufer, B.B.; Mori, Y.; Sticht, H.; Fossen, T.; et al. The Prolyl Isomerase Pin1 Promotes the Herpesvirus-Induced Phosphorylation-Dependent Disassembly of the Nuclear Lamina Required for Nucleocytoplasmic Egress. PLoS Pathog. 2016, 12, e1005825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Liu, X.-J.; Yao, Y.; Jiang, X.; Wang, X.-Z.; Yang, H.; Sun, J.-Y.; Miao, Y.; Wang, W.; Huang, Z.-L.; et al. WDR5 Facilitates Human Cytomegalovirus Replication by Promoting Capsid Nuclear Egress. J. Virol. 2018, 92, e00207-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, A.R.; Lawler, J.; Coen, D.M. A Role for Nuclear F-Actin Induction in Human Cytomegalovirus Nuclear Egress. mBio 2016, 7, e01254-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkie, A.R.; Sharma, M.; Pesola, J.M.; Ericsson, M.; Fernandez, R.; Coen, D.M. A Role for Myosin Va in Human Cytomegalovirus Nuclear Egress. J. Virol. 2018, 92, e01849-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arii, J.; Takeshima, K.; Maruzuru, Y.; Koyanagi, N.; Nakayama, Y.; Kato, A.; Mori, Y.; Kawaguchi, Y. Role of the arginine cluster in the disordered domain of Herpes Simplex Virus 1 UL34 for the recruitment of ESCRT-III for viral primary envelopment. J. Virol. 2021. [Google Scholar] [CrossRef]
- Wright, C.C.; Wisner, T.W.; Hannah, B.P.; Eisenberg, R.J.; Cohen, G.H.; Johnson, D.C. Fusion between Perinuclear Virions and the Outer Nuclear Membrane Requires the Fusogenic Activity of Herpes Simplex Virus gB. J. Virol. 2009, 83, 11847–11856. [Google Scholar] [CrossRef] [Green Version]
- Maric, M.; Shao, J.; Ryan, R.J.; Wong, C.-S.; Gonzalez-Alegre, P.; Roller, R.J. A Functional Role for TorsinA in Herpes Simplex Virus 1 Nuclear Egress. J. Virol. 2011, 85, 9667–9679. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Hellberg, T.; Granzow, H.; Franzke, K.; Gonzalez, B.D.; Goodchild, R.E.; Mettenleiter, T.C. Integrity of the Linker of Nucleoskeleton and Cytoskeleton Is Required for Efficient Herpesvirus Nuclear Egress. J. Virol. 2017, 91, e00330-17. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sam, M.D.; Evans, B.T.; Coen, D.M.; Hogle, J.M. Biochemical, Biophysical, and Mutational Analyses of Subunit Interactions of the Human Cytomegalovirus Nuclear Egress Complex. J. Virol. 2009, 83, 2996–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigalke, J.M.; E Heldwein, E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Marschall, M.; Häge, S.; Conrad, M.; Alkhashrom, S.; Kicuntod, J.; Schweininger, J.; Kriegel, M.; Lösing, J.; Tillmanns, J.; Neipel, F.; et al. Nuclear Egress Complexes of HCMV and Other Herpesviruses: Solving the Puzzle of Sequence Coevolution, Conserved Structures and Subfamily-Spanning Binding Properties. Viruses 2020, 12, 683. [Google Scholar] [CrossRef]
- Arii, J.; Takeshima, K.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Roles of the Interhexamer Contact Site for Hexagonal Lattice Formation of the Herpes Simplex Virus 1 Nuclear Egress Complex in Viral Primary Envelopment and Replication. J. Virol. 2019, 93, e00498-19. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.J.; Bjerke, S.L.; Haugo, A.C.; Hanson, S. Analysis of a Charge Cluster Mutation of Herpes Simplex Virus Type 1 UL34 and Its Extragenic Suppressor Suggests a Novel Interaction between pUL34 and pUL31 That Is Necessary for Membrane Curvature around Capsids. J. Virol. 2010, 84, 3921–3934. [Google Scholar] [CrossRef] [Green Version]
- Bjerke, S.L.; Cowan, J.M.; Kerr, J.K.; Reynolds, A.E.; Baines, J.D.; Roller, R.J. Effects of Charged Cluster Mutations on the Function of Herpes Simplex Virus Type 1 U L 34 Protein. J. Virol. 2003, 77, 7601–7610. [Google Scholar] [CrossRef] [Green Version]
- Milbradt, J.; Sonntag, E.; Wagner, S.; Strojan, H.; Wangen, C.; Rovis, T.L.; Lisnic, B.; Jonjic, S.; Sticht, H.; Britt, W.J.; et al. Human Cytomegalovirus Nuclear Capsids Associate with the Core Nuclear Egress Complex and the Viral Protein Kinase pUL97. Viruses 2018, 10, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcomb, W.W.; Homa, F.L.; Brown, J.C. Herpes Simplex Virus Capsid Structure: DNA Packaging Protein UL25 Is Located on the External Surface of the Capsid near the Vertices. J. Virol. 2006, 80, 6286–6294. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.F.; Cockrell, S.K.; Copeland, A.M.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Labeling and Localization of the Herpes Simplex Virus Capsid Protein UL25 and Its Interaction with the Two Triplexes Closest to the Penton. J. Mol. Biol. 2010, 397, 575–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Keil, G.M.; Mettenleiter, T.C. The Capsid-Associated UL25 Protein of the Alphaherpesvirus Pseudorabies Virus Is Nonessential for Cleavage and Encapsidation of Genomic DNA but Is Required for Nuclear Egress of Capsids. J. Virol. 2006, 80, 6235–6246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Karger, A.; Mettenleiter, T.C. Identification, Subviral Localization, and Functional Characterization of the Pseudorabies Virus UL17 Protein. J. Virol. 2005, 79, 13442–13453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Wills, E.; Lim, H.Y.; Zhou, Z.H.; Baines, J.D. Association of Herpes Simplex Virus pUL31 with Capsid Vertices and Components of the Capsid Vertex-Specific Complex. J. Virol. 2014, 88, 3815–3825. [Google Scholar] [CrossRef] [Green Version]
- Takeshima, K.; Arii, J.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Identification of the Capsid Binding Site in the Herpes Simplex Virus 1 Nuclear Egress Complex and Its Role in Viral Primary Envelopment and Replication. J. Virol. 2019, 93, e01290-19. [Google Scholar] [CrossRef]
- McNab, A.R.; Desai, P.; Person, S.; Roof, L.L.; Thomsen, D.R.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 1998, 72, 1060–1070. [Google Scholar] [CrossRef] [Green Version]
- Trus, B.L.; Newcomb, W.W.; Cheng, N.; Cardone, G.; Marekov, L.; Homa, F.L.; Brown, J.C.; Steven, A.C. Allosteric Signaling and a Nuclear Exit Strategy: Binding of UL25/UL17 Heterodimers to DNA-Filled HSV-1 Capsids. Mol. Cell 2007, 26, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Toropova, K.; Huffman, J.B.; Homa, F.L.; Conway, J. The Herpes Simplex Virus 1 UL17 Protein Is the Second Constituent of the Capsid Vertex-Specific Component Required for DNA Packaging and Retention. J. Virol. 2011, 85, 7513–7522. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 2001, 82, 2363–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckman, B.J.; Roller, R.J. Herpes Simplex Virus Type 1 Primary Envelopment: UL34 Protein Modification and the US3-UL34 Catalytic Relationship. J. Virol. 2004, 78, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.J.; Baines, J.D. Herpesvirus Nuclear Egress. Adv. Anat. Embryol. Cell Biol. 2017, 223, 143–169. [Google Scholar] [CrossRef] [PubMed]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-Dependent Kinase-Like Function Is Shared by the Beta- and Gamma- Subset of the Conserved Herpesvirus Protein Kinases. PLoS Pathog. 2010, 6, e1001092. [Google Scholar] [CrossRef] [PubMed]
- Jacob, T.; van den Broeke, C.; Favoreel, H.W. Viral Serine/Threonine Protein Kinases. J. Virol. 2011, 85, 1158–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Yamamoto, M.; Ohno, T.; Tanaka, M.; Sata, T.; Nishiyama, Y.; Kawaguchi, Y. Herpes Simplex Virus 1-Encoded Protein Kinase UL13 Phosphorylates Viral Us3 Protein Kinase and Regulates Nuclear Localization of Viral Envelopment Factors UL34 and UL31. J. Virol. 2006, 80, 1476–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muradov, J.H.; Finnen, R.L.; Gulak, M.A.; Hay, T.J.M.; Banfield, B.W. pUL21 regulation of pUs3 kinase activity influences the nature of nuclear envelope deformation by the HSV-2 nuclear egress complex. PLoS Pathog. 2021, 17, e1009679. [Google Scholar] [CrossRef] [PubMed]
- Wysocka, J.; Swigut, T.; Milne, T.; Dou, Y.; Zhang, X.; Burlingame, A.L.; Roeder, R.G.; Brivanlou, A.H.; Allis, C.D. WDR5 Associates with Histone H3 Methylated at K4 and Is Essential for H3 K4 Methylation and Vertebrate Development. Cell 2005, 121, 859–872. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.-Z.; Tsai, Y.-P.; Yang, M.-H.; Huang, C.-H.; Chang, S.-Y.; Chang, C.-C.; Teng, S.-C.; Wu, K.-J. Interplay between HDAC3 and WDR5 Is Essential for Hypoxia-Induced Epithelial-Mesenchymal Transition. Mol. Cell 2011, 43, 811–822. [Google Scholar] [CrossRef]
- Wang, P.; Dreger, M.; Madrazo, E.; Williams, C.J.; Samaniego, R.; Hodson, N.W.; Monroy, F.; Baena, E.; Sánchez-Mateos, P.; Hurlstone, A.; et al. WDR5 modulates cell motility and morphology and controls nuclear changes induced by a 3D environment. Proc. Natl. Acad. Sci. USA 2018, 115, 8581–8586. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Yang, F.W.; Lindert, M.T.; Isermann, P.; Schepens, J.; Maas, R.J.A.; Venkataraman, C.; Lammerding, J.; Madzvamuse, A.; Hendriks, W.; et al. Cell migration through three-dimensional confining pores: Speed accelerations by deformation and recoil of the nucleus. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20180225. [Google Scholar] [CrossRef]
- Mazumder, A.; Roopa, T.; Basu, A.; Mahadevan, L.; Shivashankar, G. Dynamics of Chromatin Decondensation Reveals the Structural Integrity of a Mechanically Prestressed Nucleus. Biophys. J. 2008, 95, 3028–3035. [Google Scholar] [CrossRef] [Green Version]
- Nava, M.; Miroshnikova, Y.A.; Biggs, L.; Whitefield, D.B.; Metge, F.; Boucas, J.; Vihinen, H.; Jokitalo, E.; Li, X.; Arcos, J.M.G.; et al. Heterochromatin-Driven Nuclear Softening Protects the Genome against Mechanical Stress-Induced Damage. Cell 2020, 181, 800–817.e22. [Google Scholar] [CrossRef]
- Bosse, J.B.; Hogue, I.B.; Feric, M.; Thiberge, S.Y.; Sodeik, B.; Brangwynne, C.P.; Enquist, L.W. Remodeling nuclear architecture allows efficient transport of herpesvirus capsids by diffusion. Proc. Natl. Acad. Sci. USA 2015, 112, E5725–E5733. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Ikegami, K. Nuclear lamin phosphorylation: An emerging role in gene regulation and pathogenesis of laminopathies. Nucleus 2020, 11, 299–314. [Google Scholar] [CrossRef]
- Machowska, M.; Piekarowicz, K.; Rzepecki, R. Regulation of lamin properties and functions: Does phosphorylation do it all? Open Biol. 2015, 5, 150094. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Coen, N.M. Comparison of Effects of Inhibitors of Viral and Cellular Protein Kinases on Human Cytomegalovirus Disruption of Nuclear Lamina and Nuclear Egress. J. Virol. 2014, 88, 10982–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forest, T.; Barnard, S.; Baines, J.D. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 2005, 7, 429–431. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Holley, M.; Colgrove, R.C.; Nalepa, G.; Harper, J.W.; Knipe, D.M. Identification and Functional Evaluation of Cellular and Viral Factors Involved in the Alteration of Nuclear Architecture during Herpes Simplex Virus 1 Infection. J. Virol. 2005, 79, 12840–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feierbach, B.; Piccinotti, S.; Bisher, M.; Denk, W.; Enquist, L.W. Alpha-Herpesvirus Infection Induces the Formation of Nuclear Actin Filaments. PLoS Pathog. 2006, 2, e85. [Google Scholar] [CrossRef]
- Bosse, J.B.; Virding, S.; Thiberge, S.; Scherer, J.; Wodrich, H.; Ruzsics, Z.; Koszinowski, U.H.; Enquist, L.W. Nuclear Herpesvirus Capsid Motility Is Not Dependent on F-Actin. mBio 2014, 5, e01909-14. [Google Scholar] [CrossRef] [Green Version]
- Olmos, Y.; Hodgson, L.; Mantell, J.M.; Verkade, P.; Carlton, J.G. ESCRT-III controls nuclear envelope reformation. Nat. Cell Biol. 2015, 522, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.-R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Duménil, A.-M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Penfield, L.; Shankar, R.; Szentgyörgyi, E.; Laffitte, A.; Mauro, M.S.; Audhya, A.; Müller-Reichert, T.; Bahmanyar, S. Regulated lipid synthesis and LEM2/CHMP7 jointly control nuclear envelope closure. J. Cell Biol. 2020, 219, e201908179. [Google Scholar] [CrossRef] [Green Version]
- Tandon, R.; AuCoin, D.P.; Mocarski, E.S. Human Cytomegalovirus Exploits ESCRT Machinery in the Process of Virion Maturation. J. Virol. 2009, 83, 10797–10807. [Google Scholar] [CrossRef] [Green Version]
- Streck, N.T.; Carmichael, J.C.; Buchkovich, N.J. Nonenvelopment Role for the ESCRT-III Complex during Human Cytomegalovirus Infection. J. Virol. 2018, 92, e02096-17. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.M.; Yates, C.; Minson, T. Herpes Simplex Virus Type 1 Cytoplasmic Envelopment Requires Functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulz, K.S.; Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Glycoproteins gB and gH Are Required for Syncytium Formation but Not for Herpesvirus-Induced Nuclear Envelope Breakdown. J. Virol. 2013, 87, 9733–9741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dultz, E.; Ellenberg, J. Live imaging of single nuclear pores reveals unique assembly kinetics and mechanism in interphase. J. Cell Biol. 2010, 191, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, S.; Ellenberg, J. Mechanisms of nuclear pore complex assembly—Two different ways of building one molecular machine. FEBS Lett. 2018, 592, 475–488. [Google Scholar] [CrossRef]
- Hölper, J.E.; Klupp, B.G.; Luxton, G.W.G.; Franzke, K.; Mettenleiter, T.C. Function of Torsin AAA+ ATPases in Pseudorabies Virus Nuclear Egress. Cells 2020, 9, 738. [Google Scholar] [CrossRef] [Green Version]
- Laudermilch, E.; Tsai, P.-L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting Torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971. [Google Scholar] [CrossRef] [PubMed]
- Rampello, A.J.; Laudermilch, E.; Vishnoi, N.; Prophet, S.M.; Shao, L.; Zhao, C.; Lusk, C.P.; Schlieker, C. Torsin ATPase deficiency leads to defects in nuclear pore biogenesis and sequestration of MLF2. J. Cell Biol. 2020, 219, e201910185. [Google Scholar] [CrossRef] [PubMed]
- Sosa, B.A.; Rothballer, A.; Kutay, U.; Schwartz, T.U. LINC Complexes Form by Binding of Three KASH Peptides to Domain Interfaces of Trimeric SUN Proteins. Cell 2012, 149, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurusaran, M.; Davies, O.R. A molecular mechanism for LINC complex branching by structurally diverse SUN-KASH 6:6 assemblies. eLife 2021, 10, e60175. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Role during Infection | Citation | |
|---|---|---|
| VIRAL NUCLEAR EGRESS COMPLEX | ||
| pUL50/53 | Core NEC, forms hexameric ring structure that can deform membranes and perform membrane scission | [36,49,50,51,52,53,54] |
| pUL93/pUL77 | Capsid vertex components 1/2, potential adaptor proteins binding to core NEC, required for encapsidation | [55,56] |
| pUL97 | Conserved herpesvirus protein kinase, phosphorylates lamins and NEC components, possibly host proteins; disrupts lamin polymerization | [46,57,58,59] |
| NEC-DIRECTED TRANSIT THROUGH INM | ||
| Lamins | Phosphorylated by viral and possibly cellular kinases; focal disruption of lamin network required for egress | [46,57,60,61,62,63,64] |
| WDR5 | Required for efficient egress by unknown mechanism; may remodel chromatin and regulate nuclear architecture | [65] |
| Protein kinase C | Host kinase that phosphorylates lamina and disrupts lamin polymerization | [45,61] |
| p32 | Recruited to INM by interactions with NEC; binds lamin B receptor; recruits UL97 to NEC | [62] |
| Nuclear actin/Myosin Va | Required for efficient transit of capsids to INM | [66,67] |
| ESCRT-III | Required for efficient egress in α-herpesviruses; repairs nuclear membrane ruptures | [21,23,68] |
| p53 | Regulates expression of core NEC protein UL53 | [47,48] |
| Emerin | Polarized expression in HCMV-infected cells; interacts with NEC; reduced expression disrupts AC formation | [8,35] |
| TRANSIT FROM THE PNS THOUGH THE ONM | ||
| Viral gB | In α-herpesvirus infected cells, reported to be required for membrane fusion at ONM | [69] |
| Torsin A | ATPase; may regulate LINC complex; deletion causes accumulation of capsids in PNS in α-herpesvirus infected cells | [70] |
| LINC COMPLEX | Sun2 levels downregulated by infection; Sun1 localization polarized, levels may be downregulated; reduction in expression correlated with dilation of PNS; expression of Sun dominant-negative proteins reduces virus yield | [4,8,71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, V.; Britt, W. Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions. Viruses 2022, 14, 15. https://doi.org/10.3390/v14010015
Sanchez V, Britt W. Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions. Viruses. 2022; 14(1):15. https://doi.org/10.3390/v14010015
Chicago/Turabian StyleSanchez, Veronica, and William Britt. 2022. "Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions" Viruses 14, no. 1: 15. https://doi.org/10.3390/v14010015
APA StyleSanchez, V., & Britt, W. (2022). Human Cytomegalovirus Egress: Overcoming Barriers and Co-Opting Cellular Functions. Viruses, 14(1), 15. https://doi.org/10.3390/v14010015