
Adenovirus Co-Opts Neutrophilic Inflammation to Enhance Transduction of Epithelial Cells

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cell Culture and Reagents
2.3. Adenovirus Transduction
2.4. PMN Isolation and Co-Culture
2.5. Western Blot
2.6. Cell Surface Biotinylation
2.7. Statistical Analyses
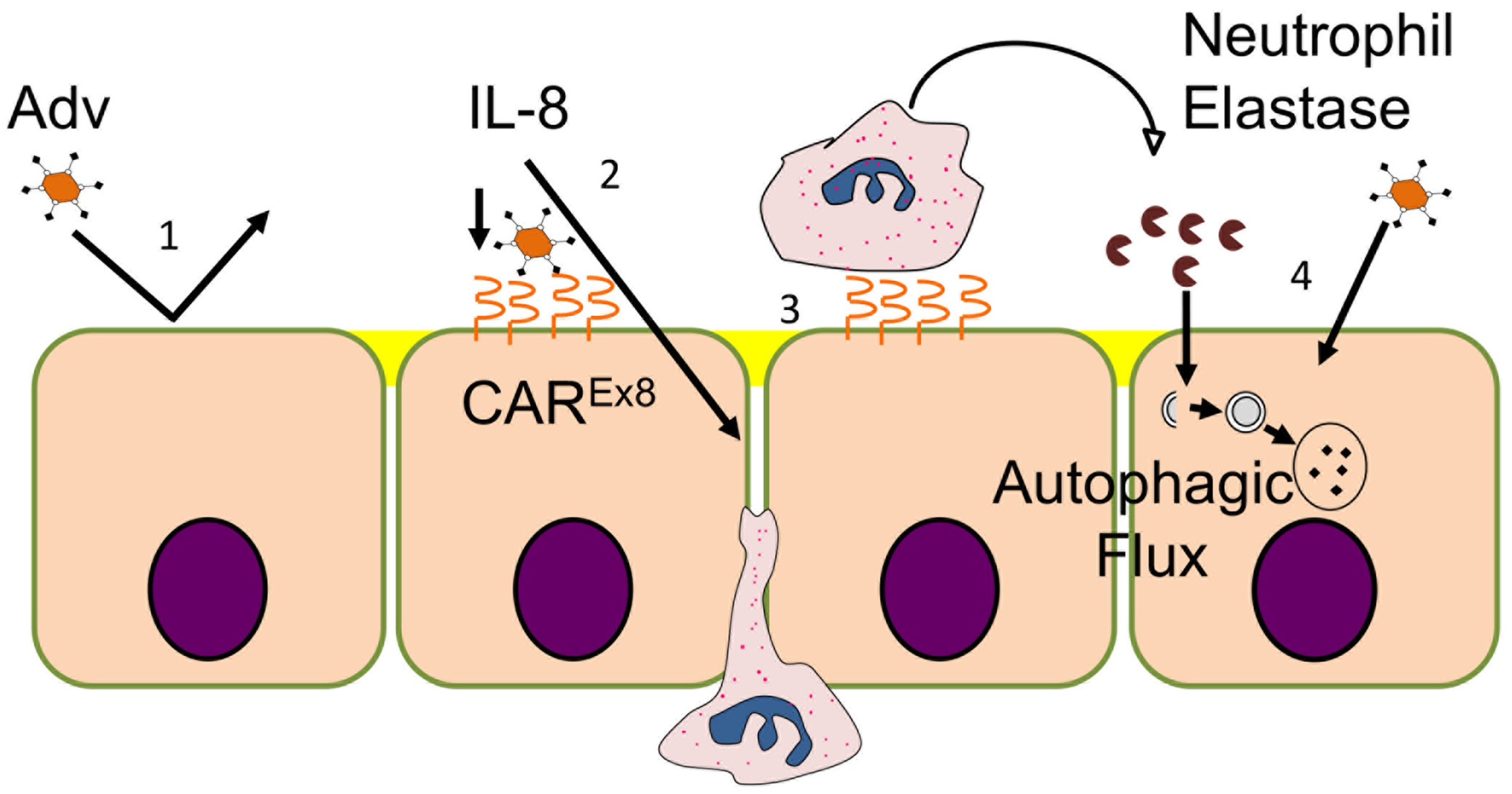
3. Results
3.1. Donor Demography
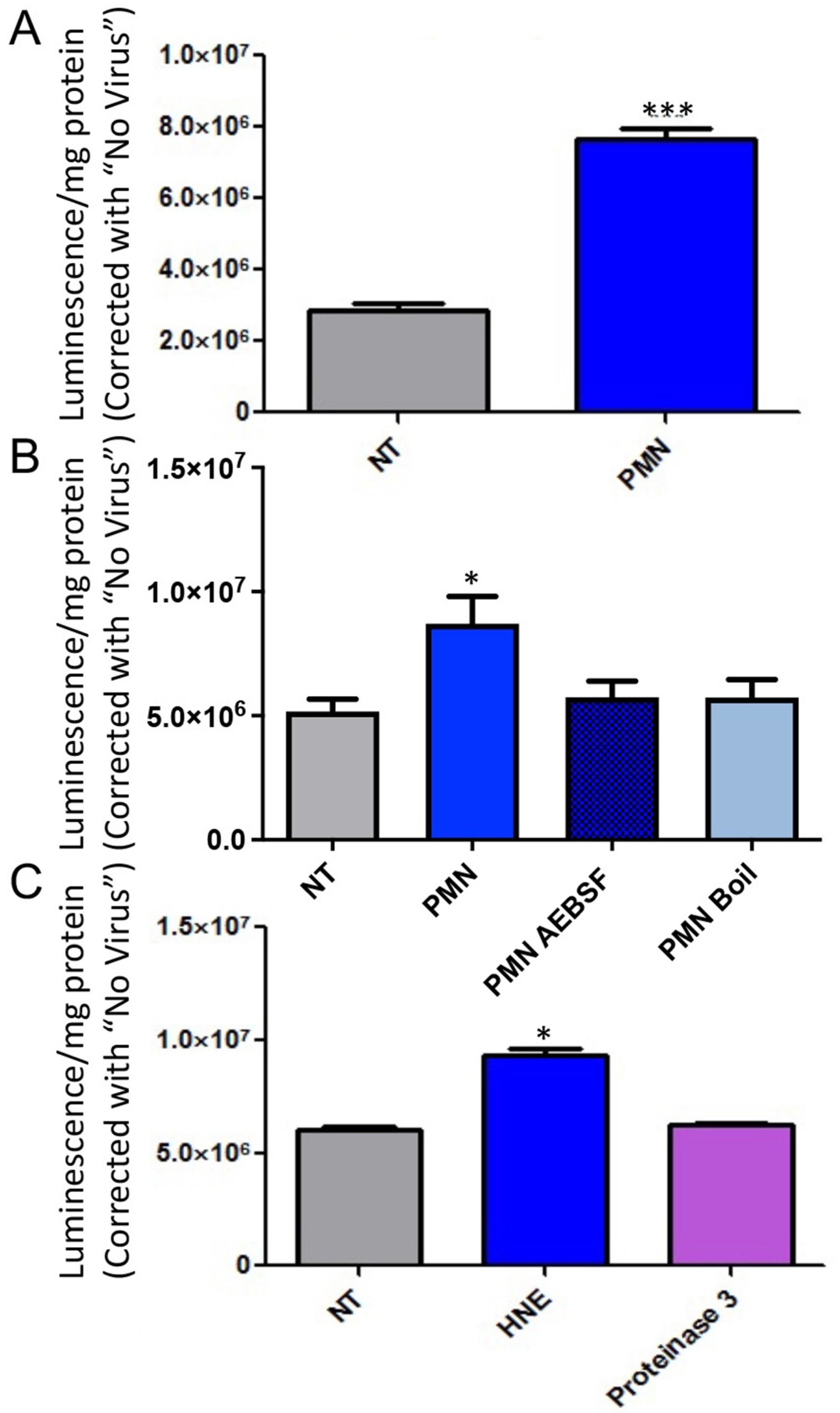
3.2. Serine Protease Contributes to PMN Mediated Enhancement of Epithelial HAdV Transduction
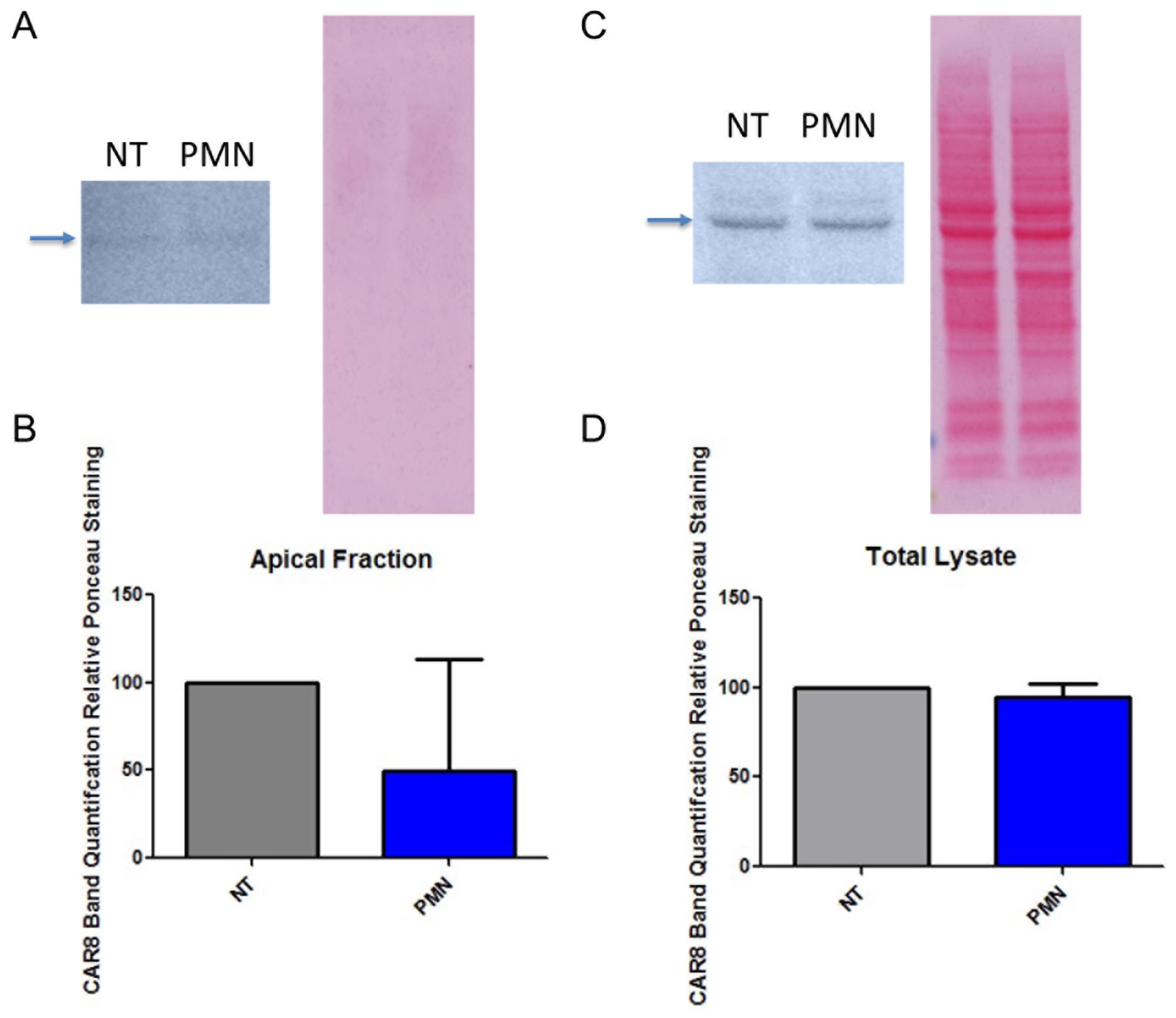
3.3. PMN Exposure Does Not Increase Epithelial CAREx8 Expression
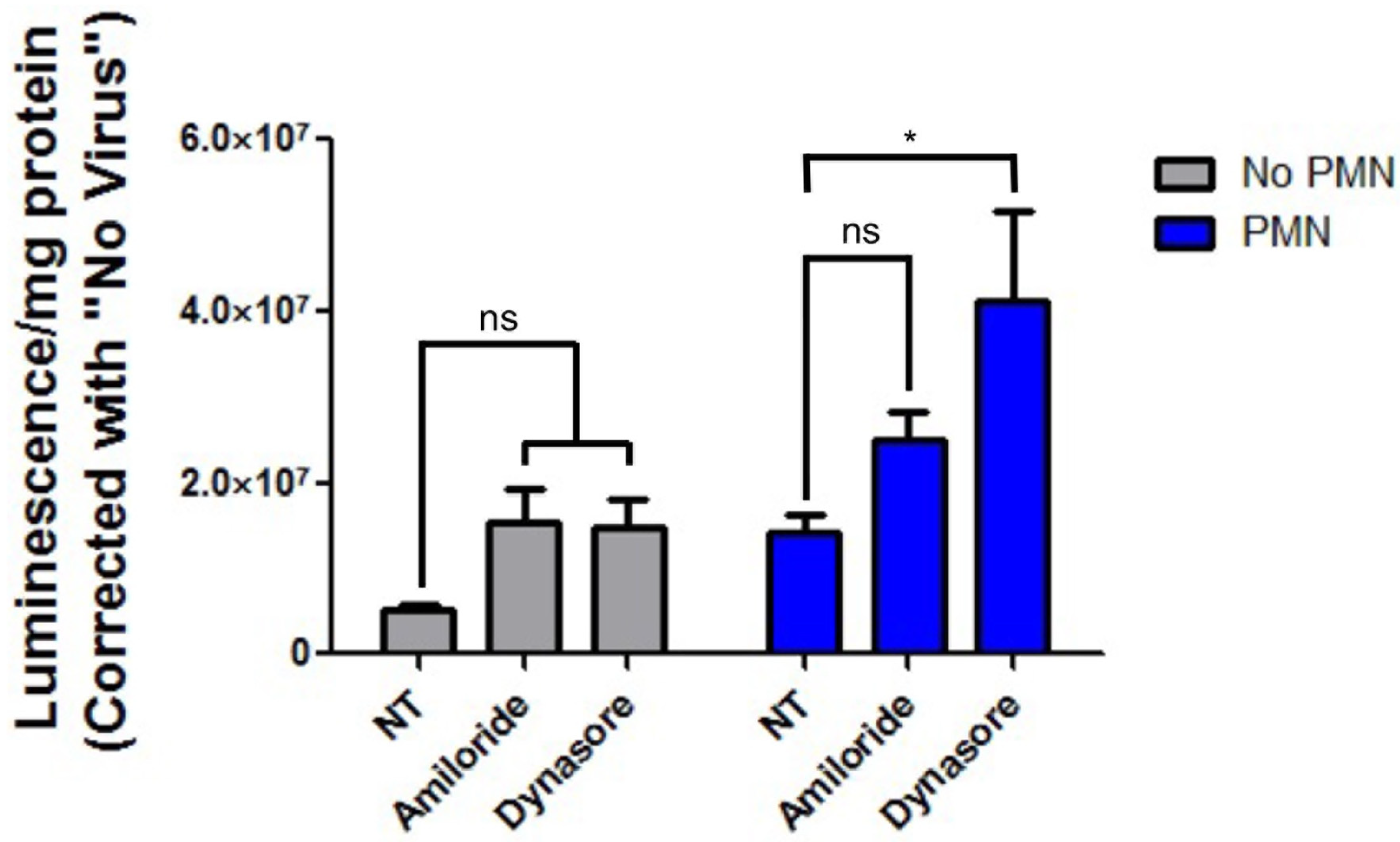
3.4. Inhibition of Epithelial Endocytosis Does Not Decrease PMN-Mediated Enhancement of HAdV5 Transduction
3.5. HNE Activates Autophagic Flux in MDCK-CAREx8 Epithelia
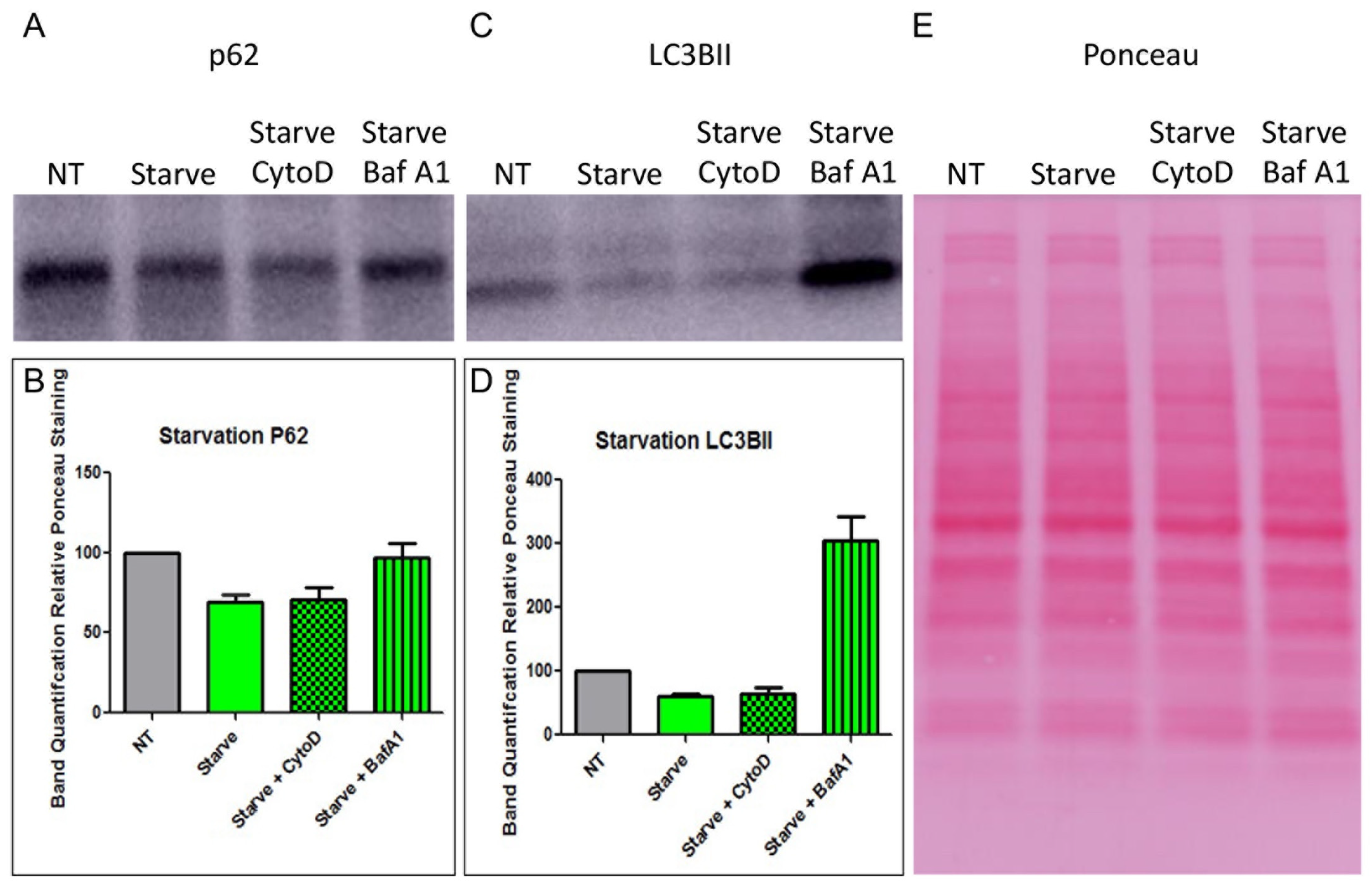
3.6. Starvation, HNE, and Whole PMN-Mediated Enhancement of Epithelial HAdV5 Transduction in MDCK-CAREx8 Cells Is Differentially Affected by Cytochalasin D and Bafilomcyin A1
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar]
- Lion, T. Adenovirus infections in immunocompetent and immunocompromised patients. Clin. Microbiol. Rev. 2014, 27, 441–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florescu, D.F.; Hoffman, J.A.; on behalf of the AST Infectious Diseases Community of Practice. Adenovirus in solid organ transplantation. Am. J. Transpl. 2013, 13 (Suppl. S4), 206–211. [Google Scholar] [CrossRef]
- Florescu, D.F.; Schaenman, J.M.; on behalf of the AST Infectious Diseases Community of Practice. Adenovirus in solid organ transplant recipients: Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transpl. 2019, 33, e13527. [Google Scholar] [CrossRef]
- Vora, S.B.; Brothers, A.W.; Englund, J.A. Renal Toxicity in Pediatric Patients Receiving Cidofovir for the Treatment of Adenovirus Infection. J. Pediatr. Infect. Dis. Soc. 2017, 6, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.; Kuhnel, F. Oncolytic viruses: Adenoviruses. Virus Genes 2017, 53, 700–706. [Google Scholar] [CrossRef]
- Shaw, A.R.; Suzuki, M. Recent advances in oncolytic adenovirus therapies for cancer. Curr. Opin. Virol. 2016, 21, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zhao, S.; Ou, J.; Zhang, J.; Lan, W.; Guan, W.; Wu, X.; Yan, Y.; Zhao, W.; Wu, J.; et al. COVID-19: Coronavirus Vaccine Development Updates. Front. Immunol. 2020, 11, 602256. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; Liu, X.; Klionsky, D.J. SnapShot: Selective autophagy. Cell 2013, 152, 368. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Montespan, C.; Marvin, S.A.; Austin, S.; Burrage, A.M.; Roger, B.; Rayne, F.; Faure, M.; Campell, E.M.; Schneider, C.; Reimer, R.; et al. Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog. 2017, 13, e1006217. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Carlin, C.R. Host cell autophagy modulates early stages of adenovirus infections in airway epithelial cells. J. Virol. 2013, 87, 2307–2319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef] [PubMed]
- Bewley, M.C.; Springer, K.; Zhang, Y.-B.; Freimuth, P.; Flanagan, J.M. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 1999, 286, 1579–1583. [Google Scholar] [CrossRef] [Green Version]
- Excoffon, K.J.D.A.; Gansemer, N.D.; Mobily, M.E.; Karp, P.H.; Parekh, K.R.; Zabner, J. Isoform-specific regulation and localization of the coxsackie and adenovirus receptor in human airway epithelia. PLoS ONE 2010, 5, e9909. [Google Scholar] [CrossRef]
- Kotha, P.L.N.; Sharma, P.; Kolawole, A.; Yan, R.; AlGhamri, M.S.; Brockman, T.L.; Gomez-Cambronero, J.; Excoffon, K.J.D.A. Adenovirus entry from the apical surface of polarized epithelia is facilitated by the host innate immune response. PLoS Pathog. 2015, 11, e1004696. [Google Scholar] [CrossRef]
- Lutschg, V.; Boucke, K.; Hemmi, S.; Greber, U.F. Chemotactic antiviral cytokines promote infectious apical entry of human adenovirus into polarized epithelial cells. Nat. Commun. 2011, 2, 391. [Google Scholar] [CrossRef] [Green Version]
- Witherden, D.A.; Verdino, P.; Rieder, S.E.; Garijo, O.; Mills, R.E.; Teyton, L.; Fischer, W.H.; Wilson, I.A.; Havran, W.L. The junctional adhesion molecule JAML is a costimulatory receptor for epithelial gammadelta T cell activation. Science 2010, 329, 1205–1210. [Google Scholar] [CrossRef] [Green Version]
- Zen, K.; Liu, Y.; McCall, I.C.; Wu, T.; Lee, W.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol. Biol. Cell 2005, 16, 2694–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Kolawole, A.; Wiltshire, S.M.; Frondorf, K.; Excoffon, K.J.D.A. Accessibility of the coxsackievirus and adenovirus receptor and its importance in adenovirus gene transduction efficiency. J. Gen. Virol. 2012, 93 Pt 1, 155–158. [Google Scholar] [CrossRef]
- Readler, J.M.; AlKahlout, A.S.; Sharma, P.; Excoffon, K.J. Isoform specific editing of the coxsackievirus and adenovirus receptor. Virology 2019, 536, 20–26. [Google Scholar] [CrossRef]
- Sharma, P.; Kolawole, A.; Core, S.; Kajon, A.E.; Excoffon, K.J.D.A. Sidestream smoke exposure increases the susceptibility of airway epithelia to adenoviral infection. PLoS ONE 2012, 7, e49930. [Google Scholar] [CrossRef]
- Golden, J.W.; Schiff, L.A. Neutrophil elastase, an acid-independent serine protease, facilitates reovirus uncoating and infection in U937 promonocyte cells. Virol. J. 2005, 2, 48. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.C.; Lin, H.-C.; Liu, C.-Y.; Wang, C.-H.; Hwang, T.; Huang, T.-T.; Lin, C.-H.; Kuo, H.-P. Neutrophil elastase induces IL-8 synthesis by lung epithelial cells via the mitogen-activated protein kinase pathway. J. Biomed. Sci. 2004, 11, 49–58. [Google Scholar] [CrossRef]
- Bergin, D.A.; Greene, C.M.; Sterchi, E.E.; Kenna, C.; Geraghty, P.; Belaaouaj, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J. Biol. Chem. 2008, 283, 31736–31744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiorpelidou, E.; Foster, B.; Farrell, J.; Ogese, M.O.; Faulkner, L.; Goldring, C.E.; Park, B.K.; Naisbitt, D.J. IL-8 release from human neutrophils cultured with pro-haptenic chemical sensitizers. Chem. Res. Toxicol. 2012, 25, 2054–2056. [Google Scholar] [CrossRef] [PubMed]
- Luisoni, S.; Suomalainen, M.; Boucke, K.; Tanner, L.B.; Wenk, M.R.; Guan, X.L.; Grzybek, M.; Coskun, U.; Greber, U.F. Co-option of Membrane Wounding Enables Virus Penetration into Cells. Cell Host Microbe 2015, 18, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Koivusalo, M.; Welch, C.; Hayashi, H.; Scott, C.C.; Kim, M.; Alexander, T.; Touret, N.; Grinstein, H.S. Amiloride inhibits macropinocytosis by lowering submembranous pH and preventing Rac1 and Cdc42 signaling. J. Cell Biol. 2010, 188, 547–563. [Google Scholar] [CrossRef] [Green Version]
- Macia, E.; Ehrlich, M.; Massol, R.; Boucrot, E.; Brunner, C.; Kirchhausen, T. Dynasore, a cell-permeable inhibitor of dynamin. Dev. Cell 2006, 10, 839–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preta, G.; Cronin, J.G.; Sheldon, I.M. Dynasore—Not just a dynamin inhibitor. Cell Commun. Signal. 2015, 13, 24. [Google Scholar] [CrossRef] [Green Version]
- Hou, H.H.; Cheng, S.L.; Chung, K.P.; Kuo, M.Y.P.; Yeh, C.C.; Chang, B.E.; Lu, H.H.; Wang, H.C.; Yu, C.J. Elastase induces lung epithelial cell autophagy through placental growth factor: A new insight of emphysema pathogenesis. Autophagy 2014, 10, 1509–1521. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Arozena, A.A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Takanezawa, Y.; Nakamura, R.; Sone, Y.; Uraguchi, S.; Kobayashi, K.; Tomoda, H.; Kiyono, M. Variation in the activity of distinct cytochalasins as autophagy inhibitiors in human lung A549 cells. Biochem. Biophys. Res. Commun. 2017, 494, 641–647. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Deng, Y.; Herbert, J.; Robinson, E.; Ren, L.; Smyth, R.L.; Smith, C.M. Neutrophil-Airway Epithelial Interactions Result in Increased Epithelial Damage and Viral Clearance during Respiratory Syncytial Virus Infection. J. Virol. 2020, 94, e02161-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotter, M.J.; Zaiss, A.K.; Muruve, D.A. Neutrophils interact with adenovirus vectors via Fc receptors and complement receptor 1. J. Virol. 2005, 79, 14622–14631. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xu, S.; Wang, N.; Ma, Q.; Peng, P.; Yu, Y.; Zhang, L.; Ying, Z.; Wang, H. Dynasore Suppresses mTORC1 Activity and Induces Autophagy to Regulate the Clearance of Protein Aggregates in Neurodegenerative Diseases. Neurotox. Res. 2019, 36, 108–116. [Google Scholar] [CrossRef]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.; Henry, C.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry, C.M.; Sullivan, G.P.; Clancy, D.M.; Afonina, I.S.; Kulms, D.; Martin, S.J. Neutrophil-Derived Proteases Escalate Inflammation through Activation of IL-36 Family Cytokines. Cell Rep. 2016, 14, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thierry, A.R.; Roch, B. Neutrophil Extracellular Traps and By-Products Play a Key Role in COVID-19: Pathogenesis, Risk Factors, and Therapy. J. Clin. Med. 2020, 9, 2942. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Ota, C.; Kitaoka, S.; Chiba, Y.; Takayanagi, M.; Kitamura, T.; Yamamoto, K.; Fujie, H.; Mikami, H.; Uematsu, M.; et al. Elevated serum levels of neutrophil elastase in patients with influenza virus-associated encephalopathy. J. Neurol. Sci. 2015, 349, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Stupack, D.; Bokoch, G.M.; Nemerow, G.R. Adenovirus endocytosis requires actin cytoskeleton reorganization mediated by Rho family GTPases. J. Virol. 1998, 72, 8806–8812. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Readler, J.M.; Burke, M.R.; Sharma, P.; Excoffon, K.J.D.A.; Kolawole, A.O. Adenovirus Co-Opts Neutrophilic Inflammation to Enhance Transduction of Epithelial Cells. Viruses 2022, 14, 13. https://doi.org/10.3390/v14010013
Readler JM, Burke MR, Sharma P, Excoffon KJDA, Kolawole AO. Adenovirus Co-Opts Neutrophilic Inflammation to Enhance Transduction of Epithelial Cells. Viruses. 2022; 14(1):13. https://doi.org/10.3390/v14010013
Chicago/Turabian StyleReadler, James M., Meghan R. Burke, Priyanka Sharma, Katherine J. D. A. Excoffon, and Abimbola O. Kolawole. 2022. "Adenovirus Co-Opts Neutrophilic Inflammation to Enhance Transduction of Epithelial Cells" Viruses 14, no. 1: 13. https://doi.org/10.3390/v14010013
APA StyleReadler, J. M., Burke, M. R., Sharma, P., Excoffon, K. J. D. A., & Kolawole, A. O. (2022). Adenovirus Co-Opts Neutrophilic Inflammation to Enhance Transduction of Epithelial Cells. Viruses, 14(1), 13. https://doi.org/10.3390/v14010013