
Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights

 ,
,  , ,
, ,  , ,
, ,
Abstract
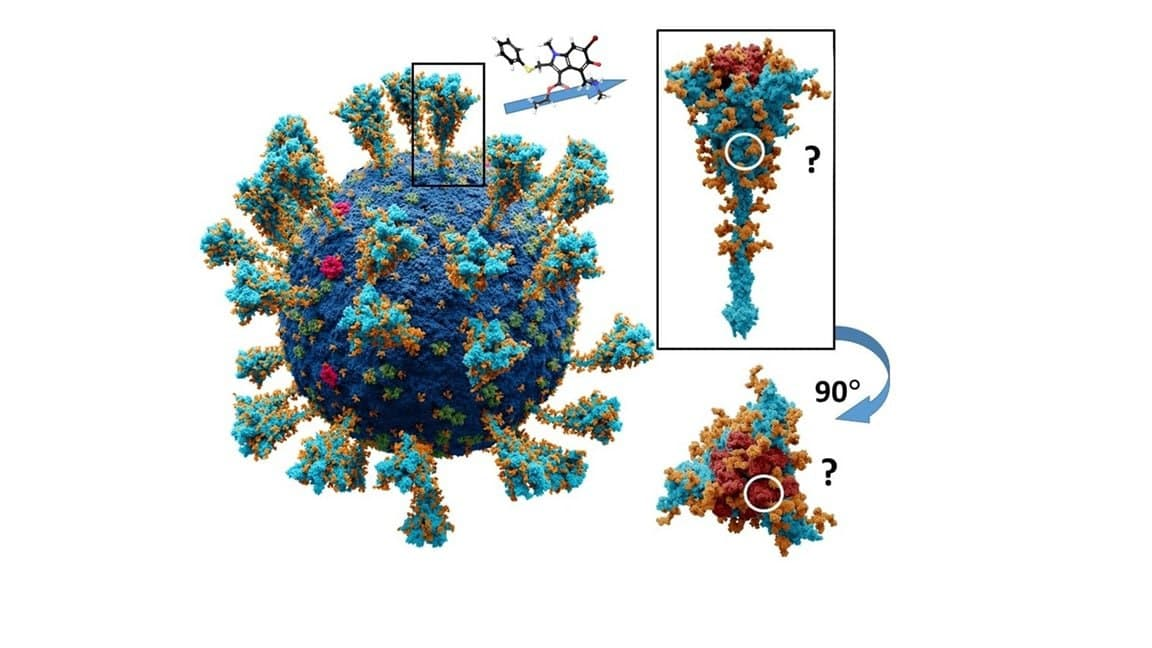
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Biological Experiments
2.1.1. Cell Cultures
2.1.2. Plasmids
2.1.3. Preparation of SARS-CoV-2 Pseudotyped Lentiviral Particles
2.1.4. Determination of the Ability of SARS-CoV-2 Pseudotyped Lentiviral Particles to Penetrate HEK293T Cells
2.1.5. Determination of Cytotoxicity of Compounds on HEK293T Cells
2.1.6. Determination of Semi-Inhibitory Concentrations of Compounds against Lenti-S SARS-CoV-2 Pseudoviruses and Calculation of Selectivity Index (SI) Values
2.2. Molecular Dynamics and Docking
2.2.1. Ligand and Protein Preparation
2.2.2. Molecular Dynamic Models
2.2.3. Population Analysis
2.2.4. Analysis of Secondary Structure Change
2.2.5. Interaction Interface Analysis
2.2.6. Molecular Docking
3. Results and Discussion
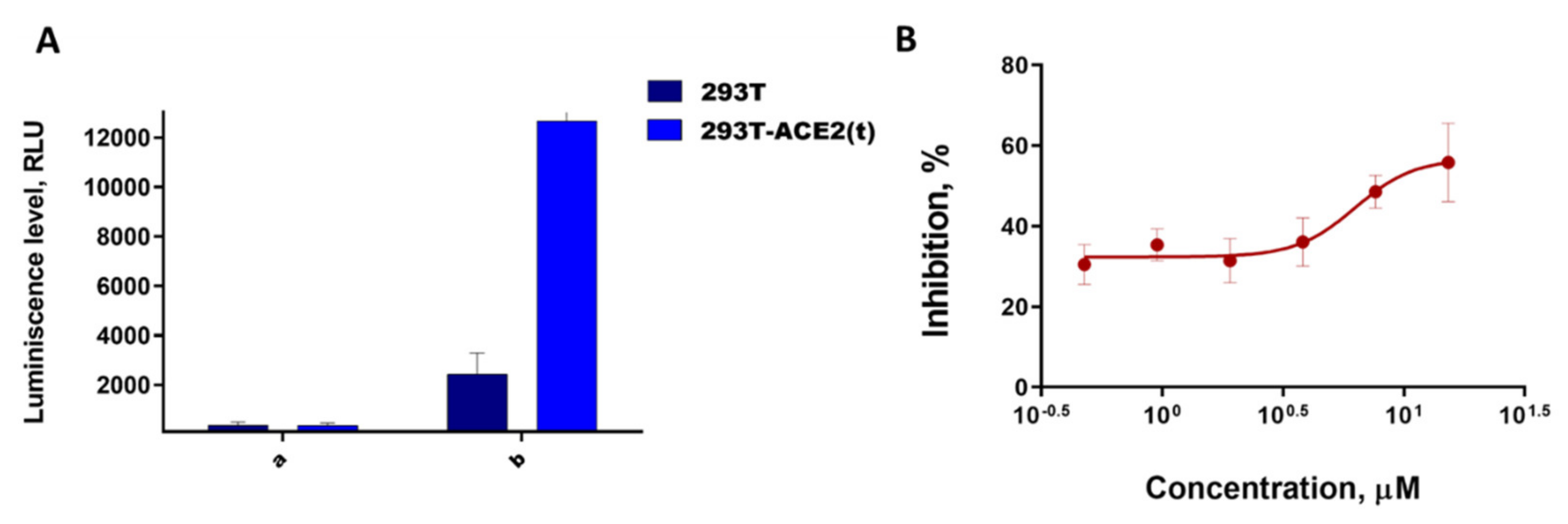
3.1. Biological Experiments
3.2. Molecular Dynamics
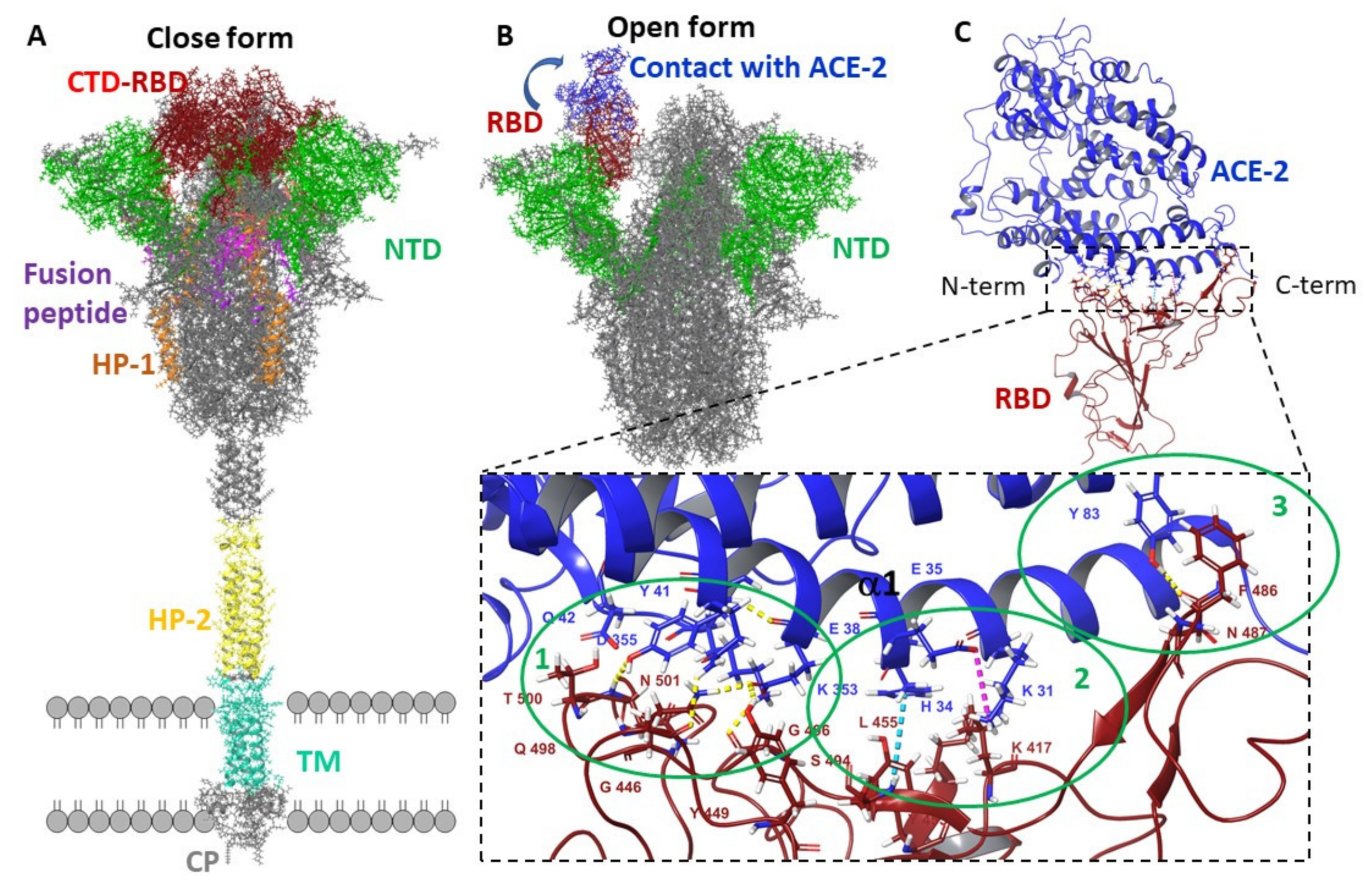
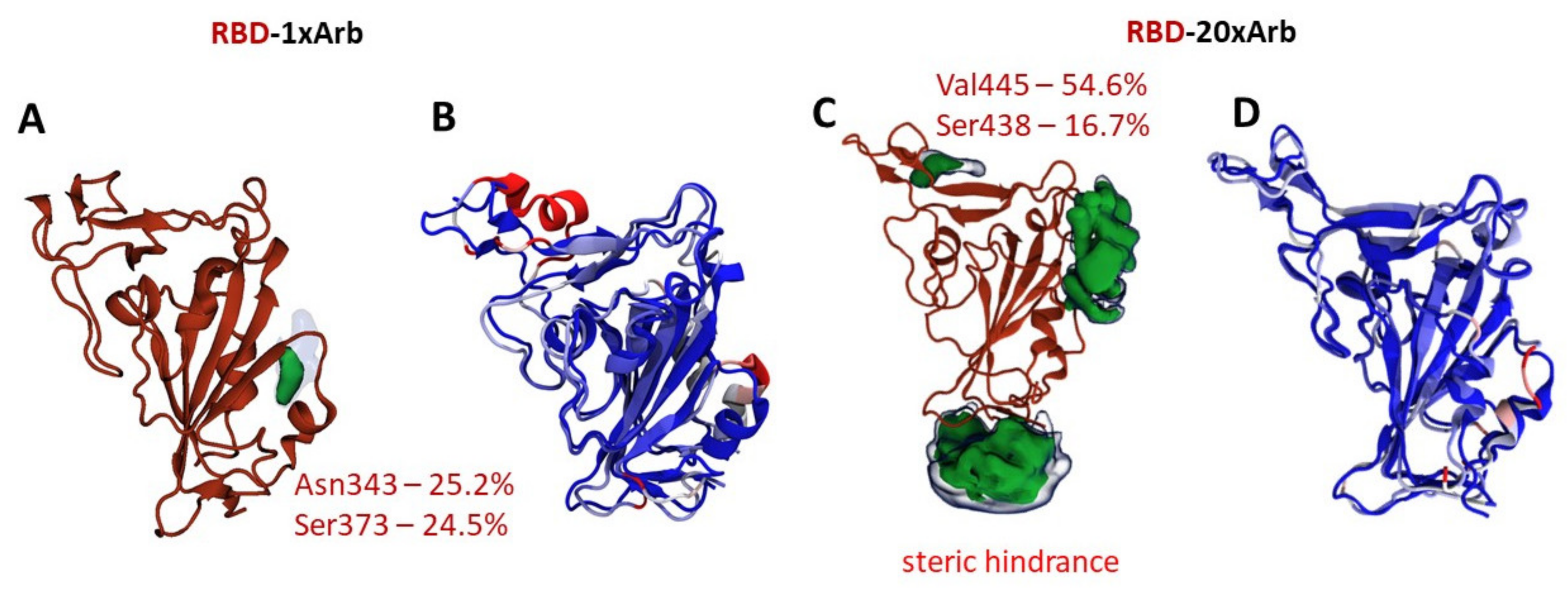
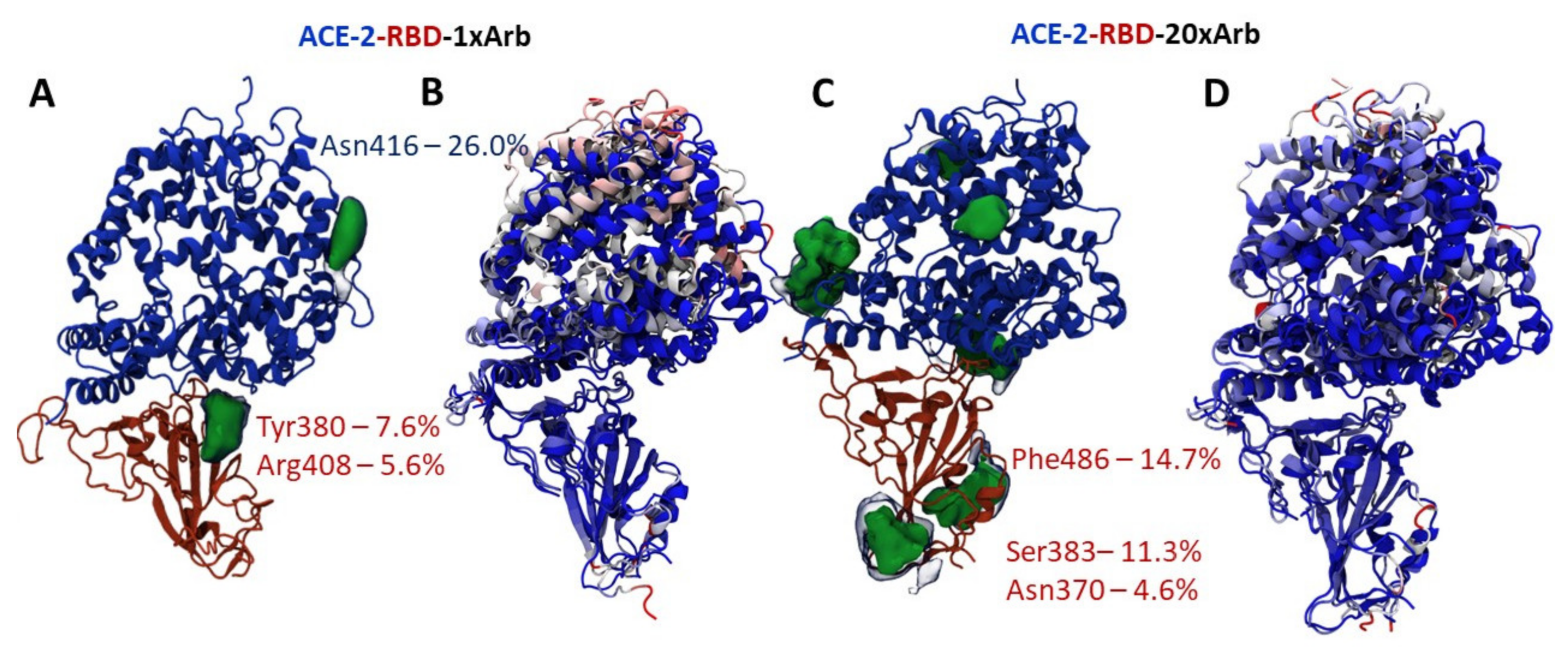
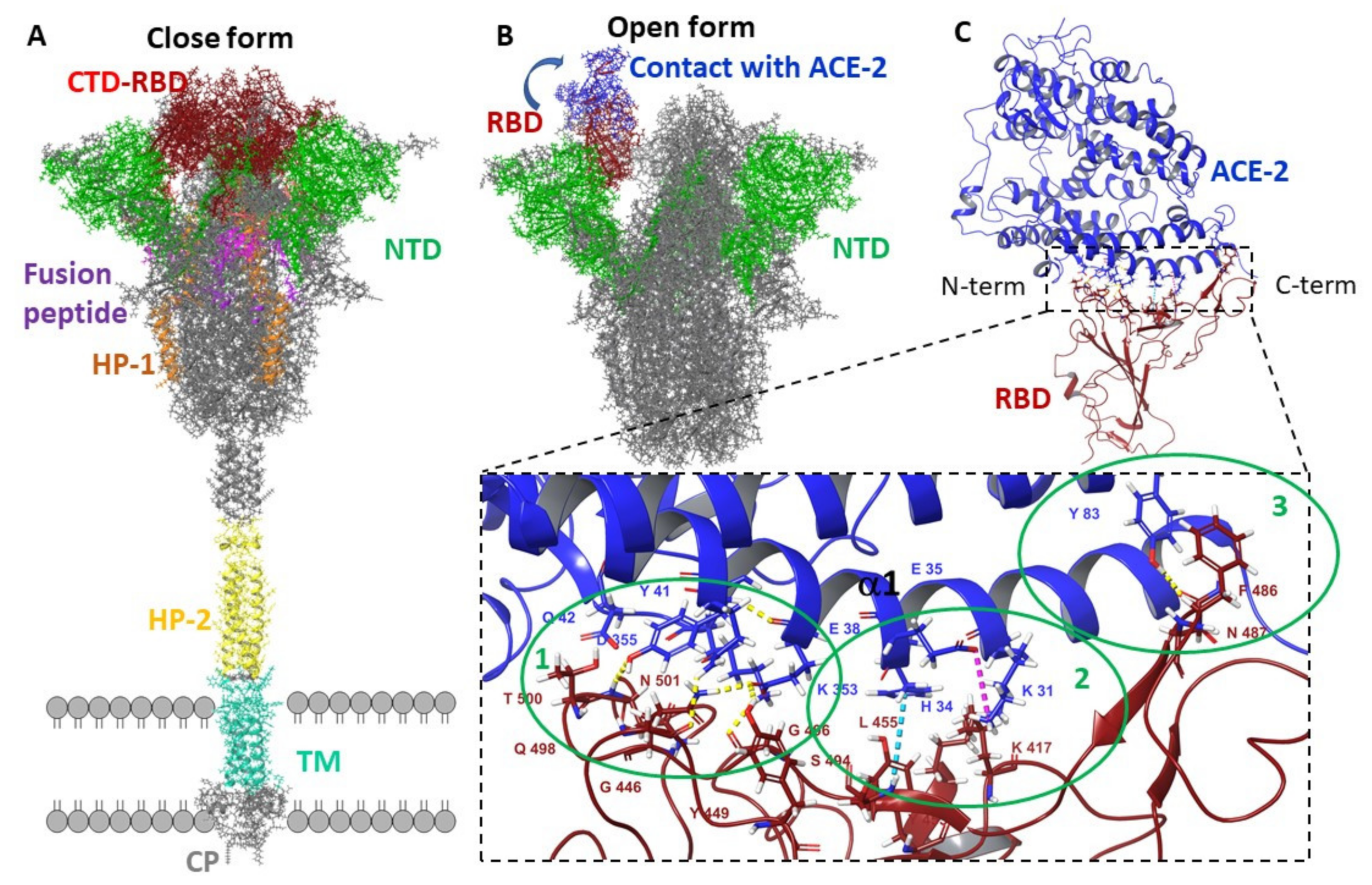
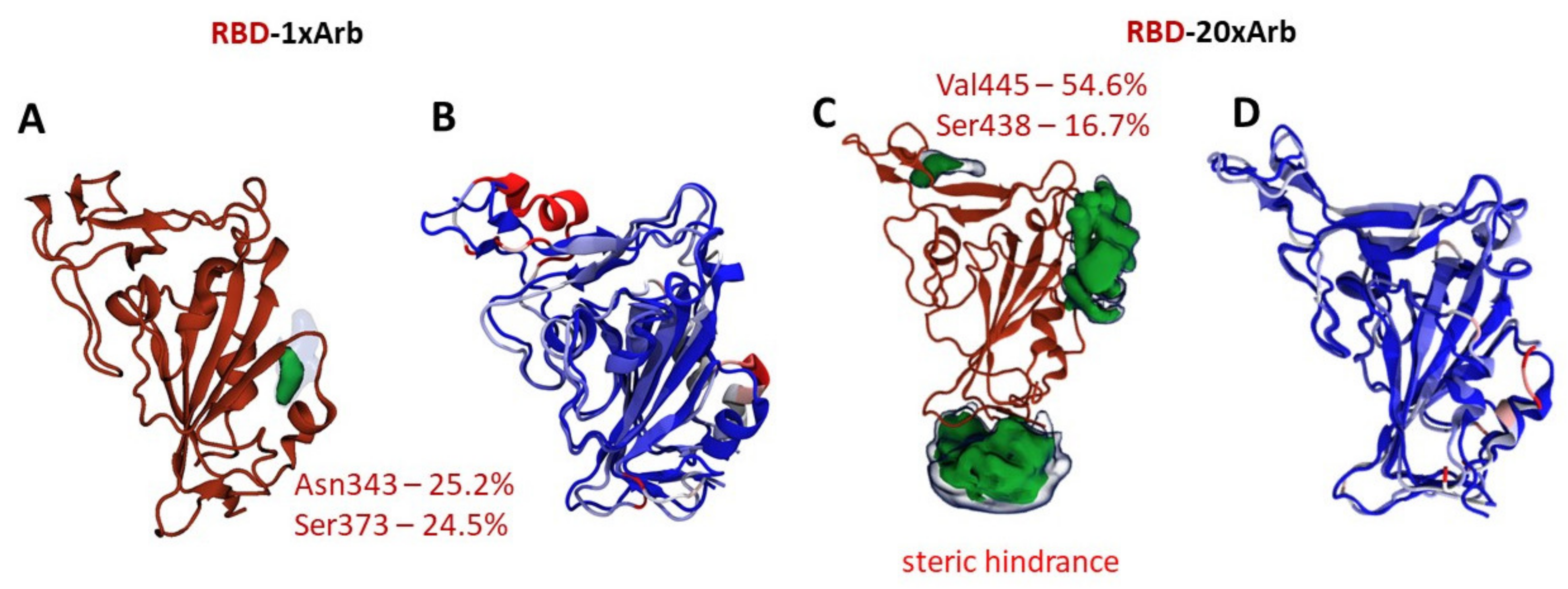
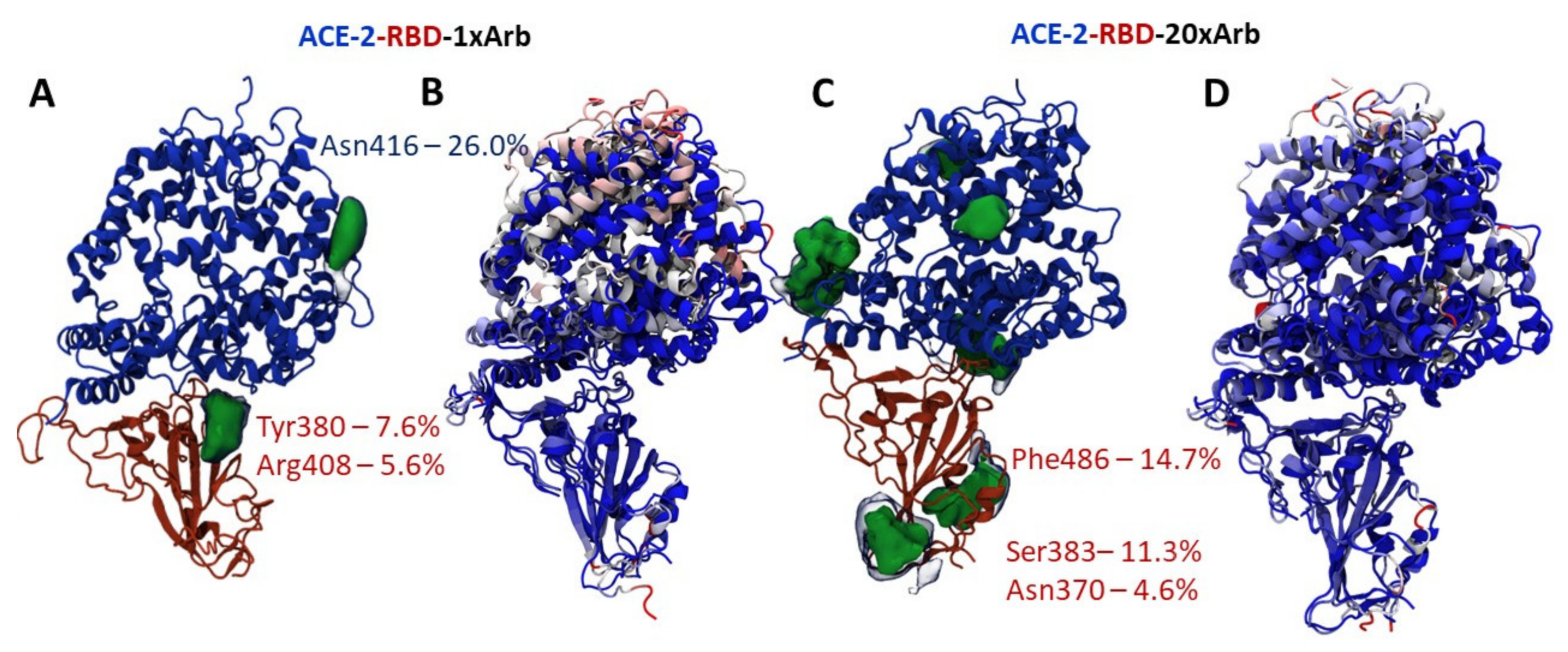
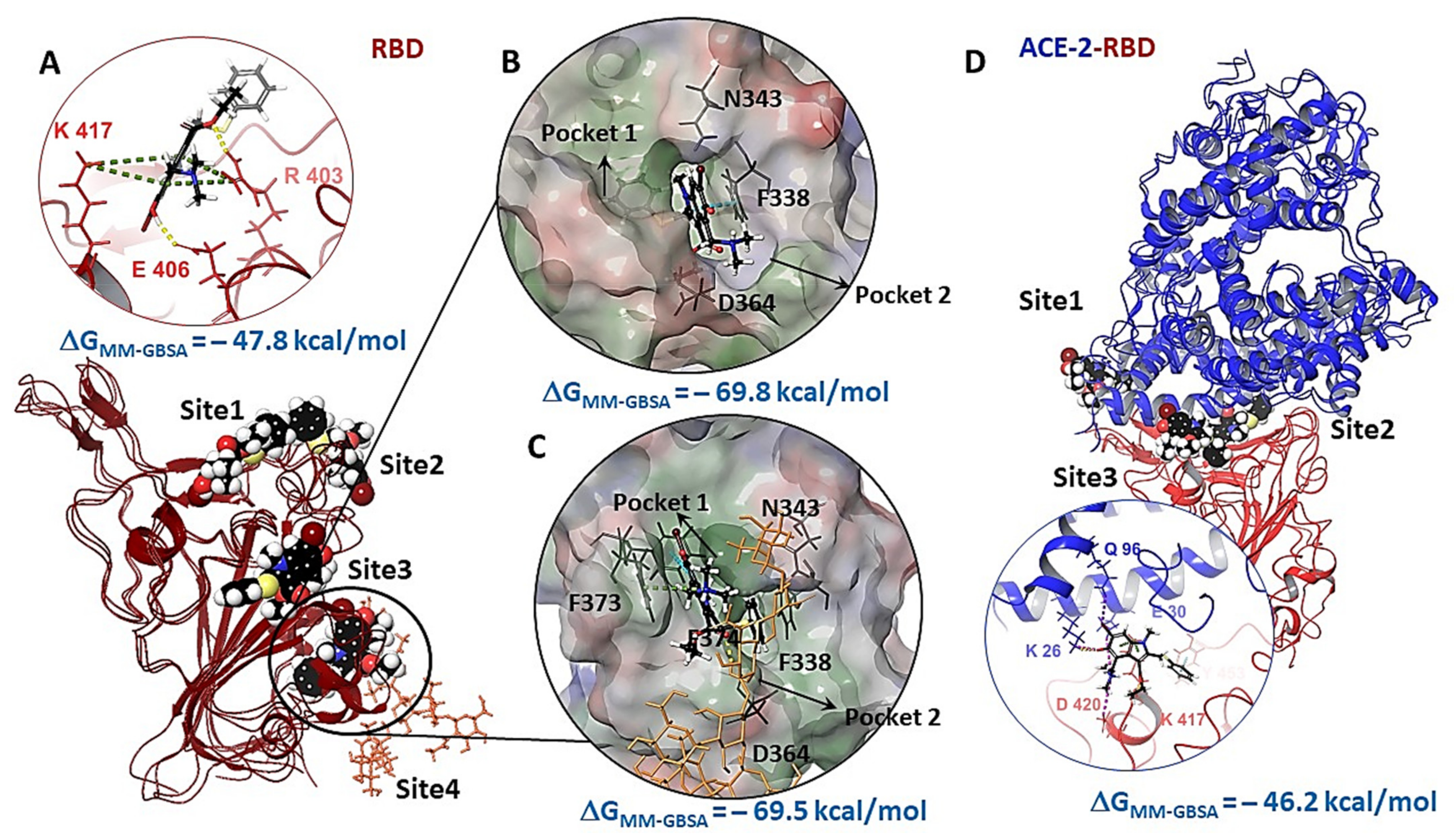
3.2.1. Molecular Dynamic of RBD-n×Arb and RBD-ACE-2-n×Arb Unglycosylated Systems
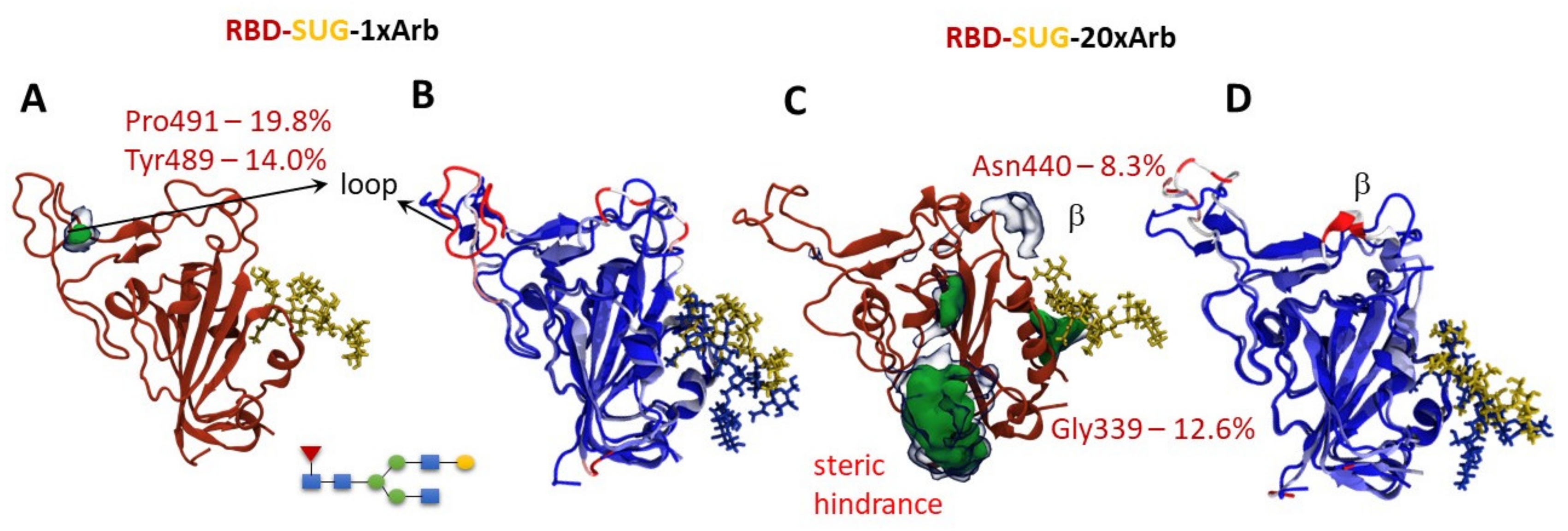
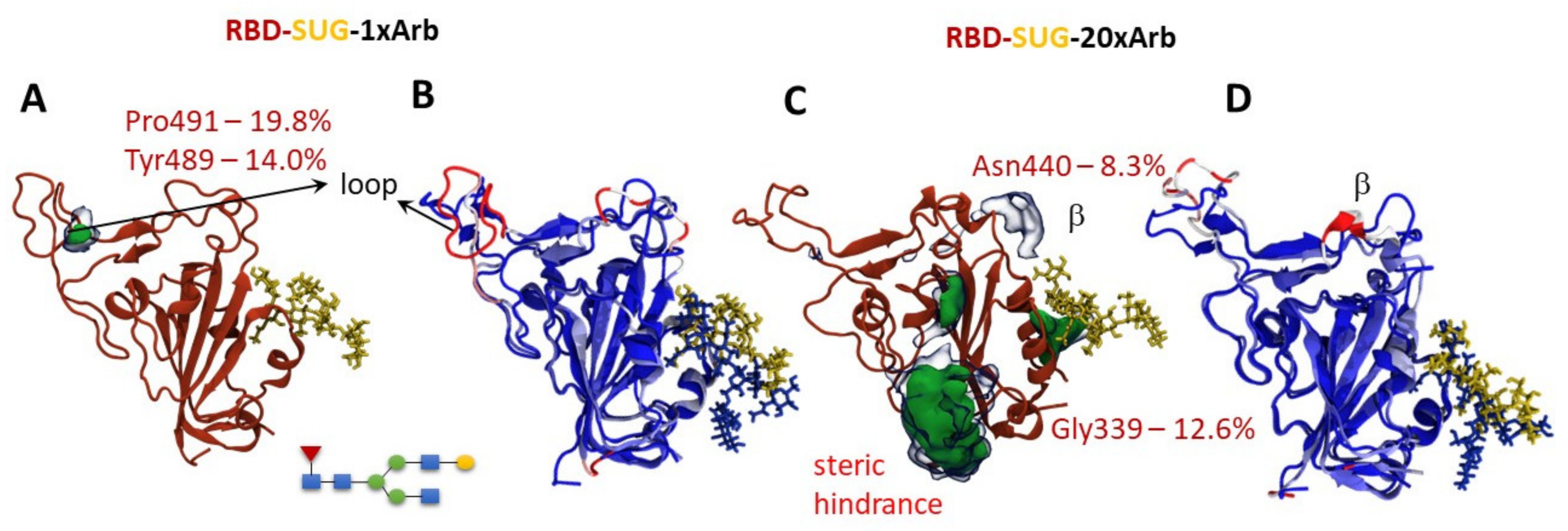
3.2.2. Molecular Dynamics of RBD-n×Arb Glycosylated Systems
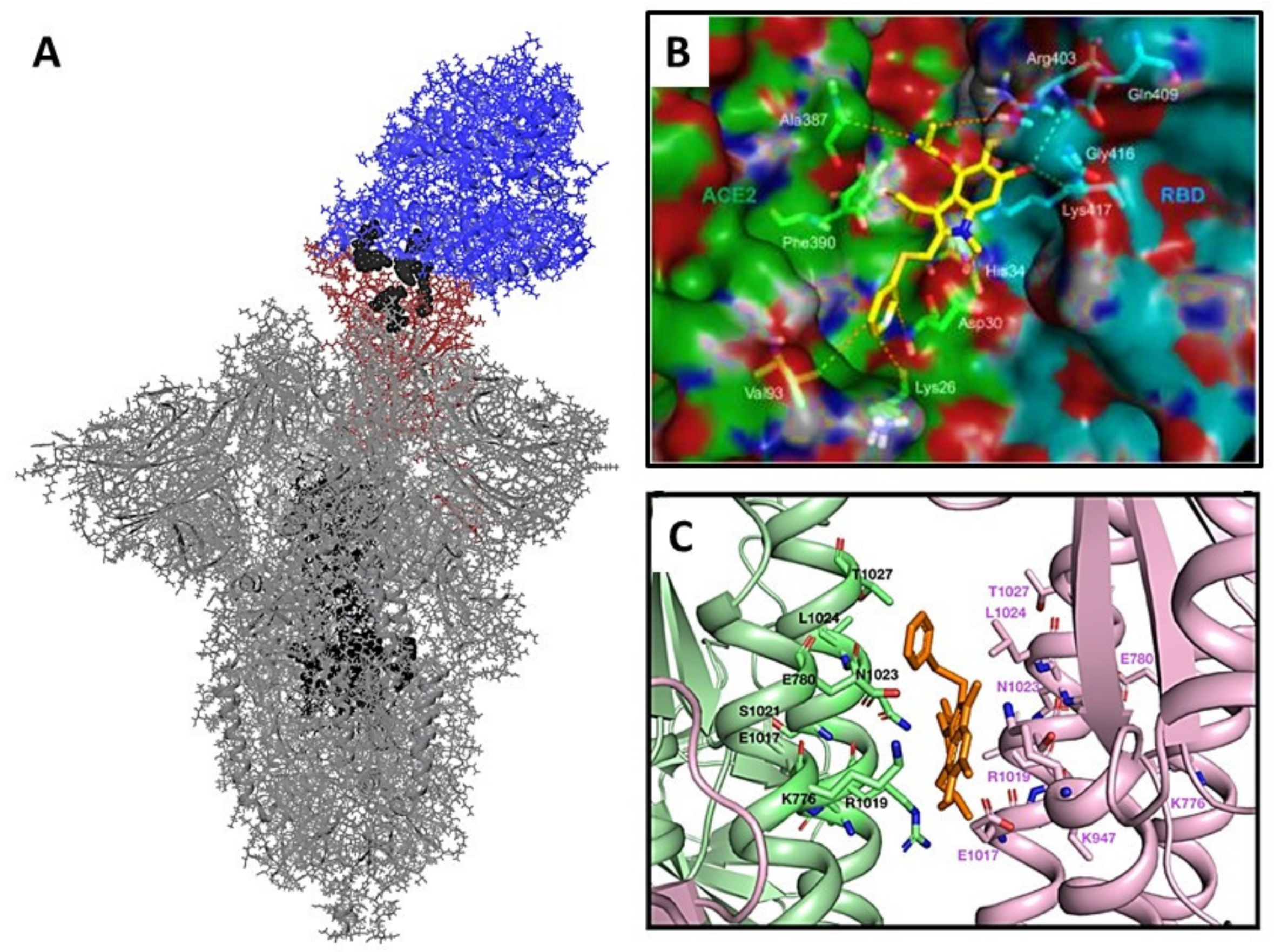
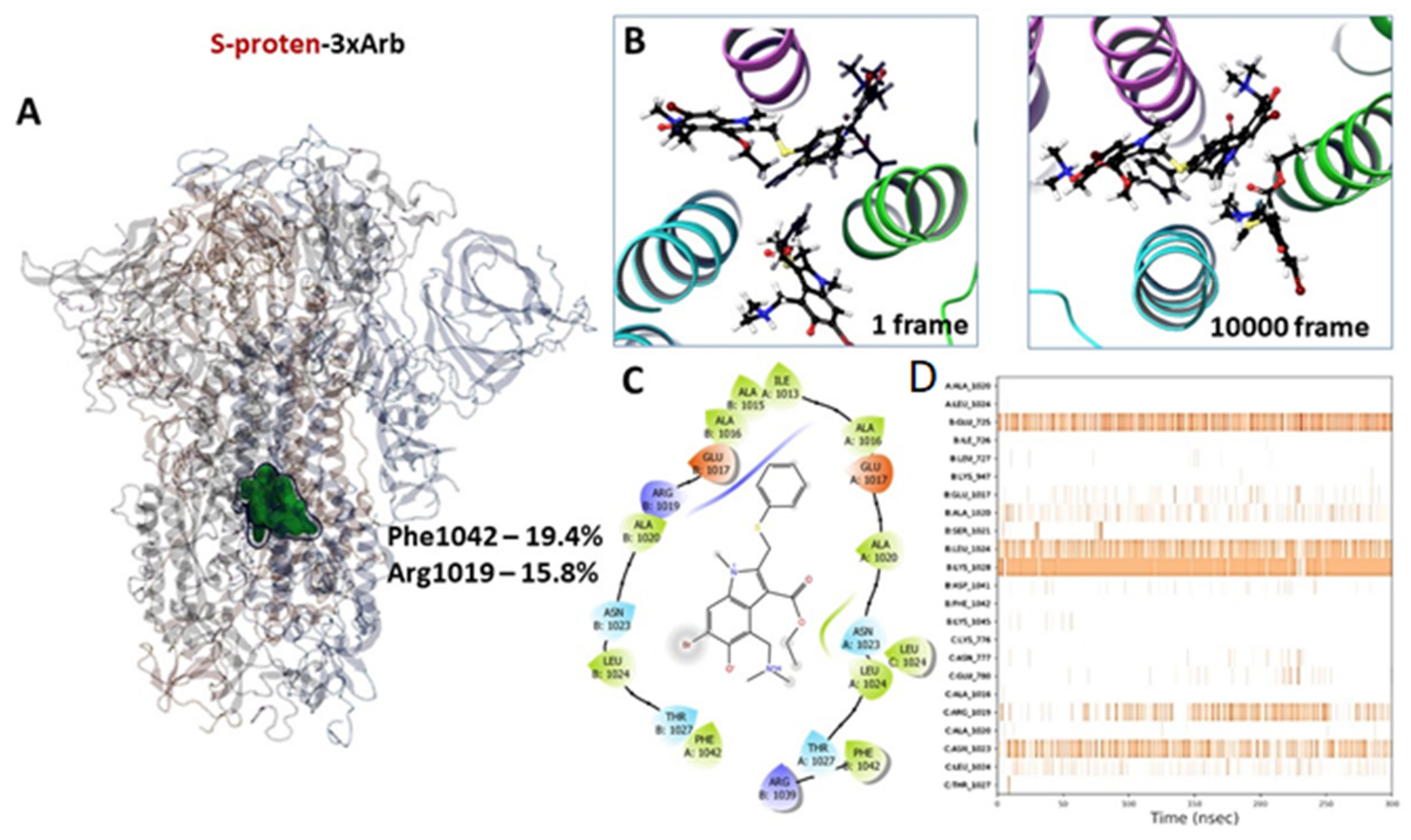
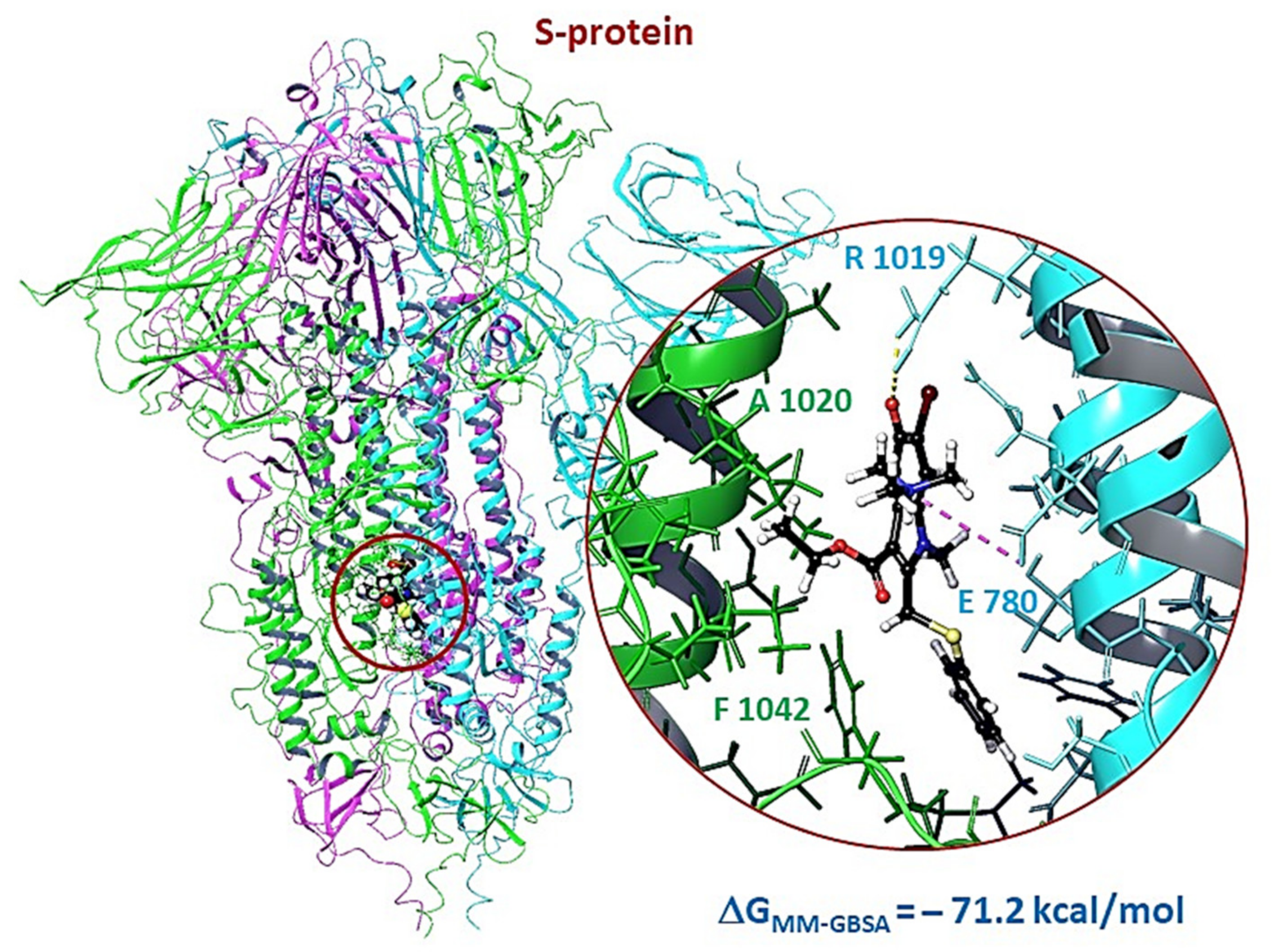
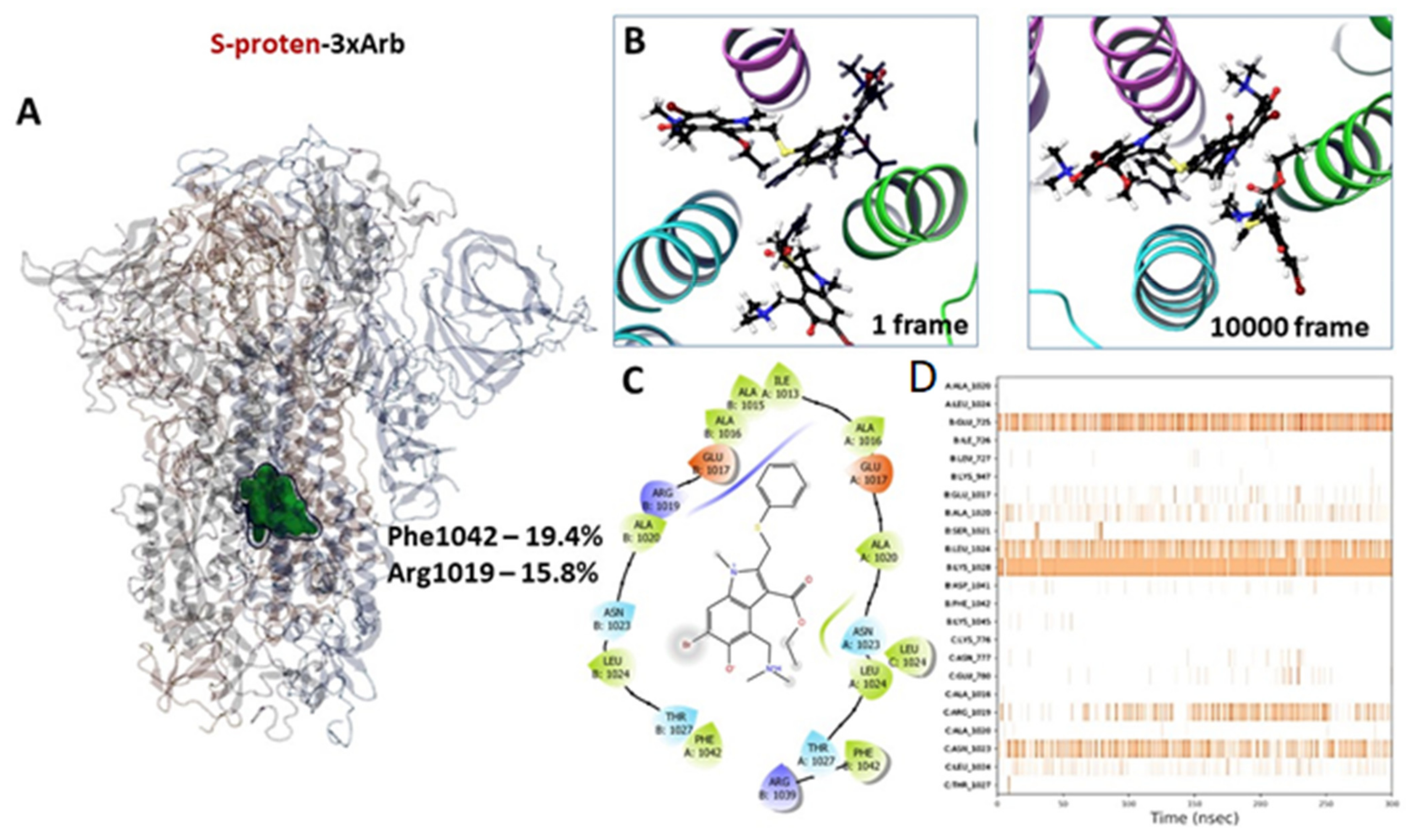
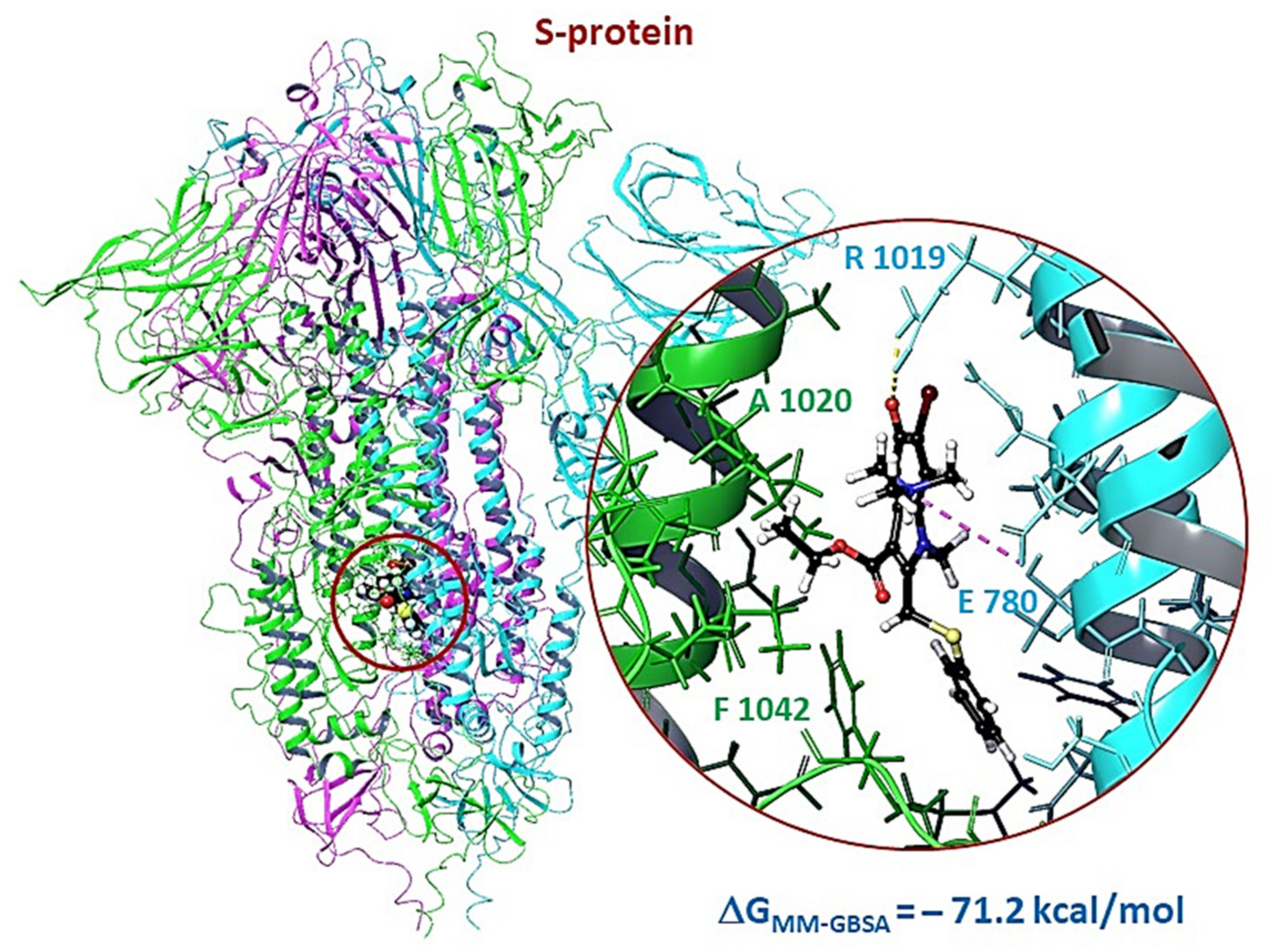
3.2.3. Molecular Dynamics of Full-Size Proteins with Three Arbidol Molecules
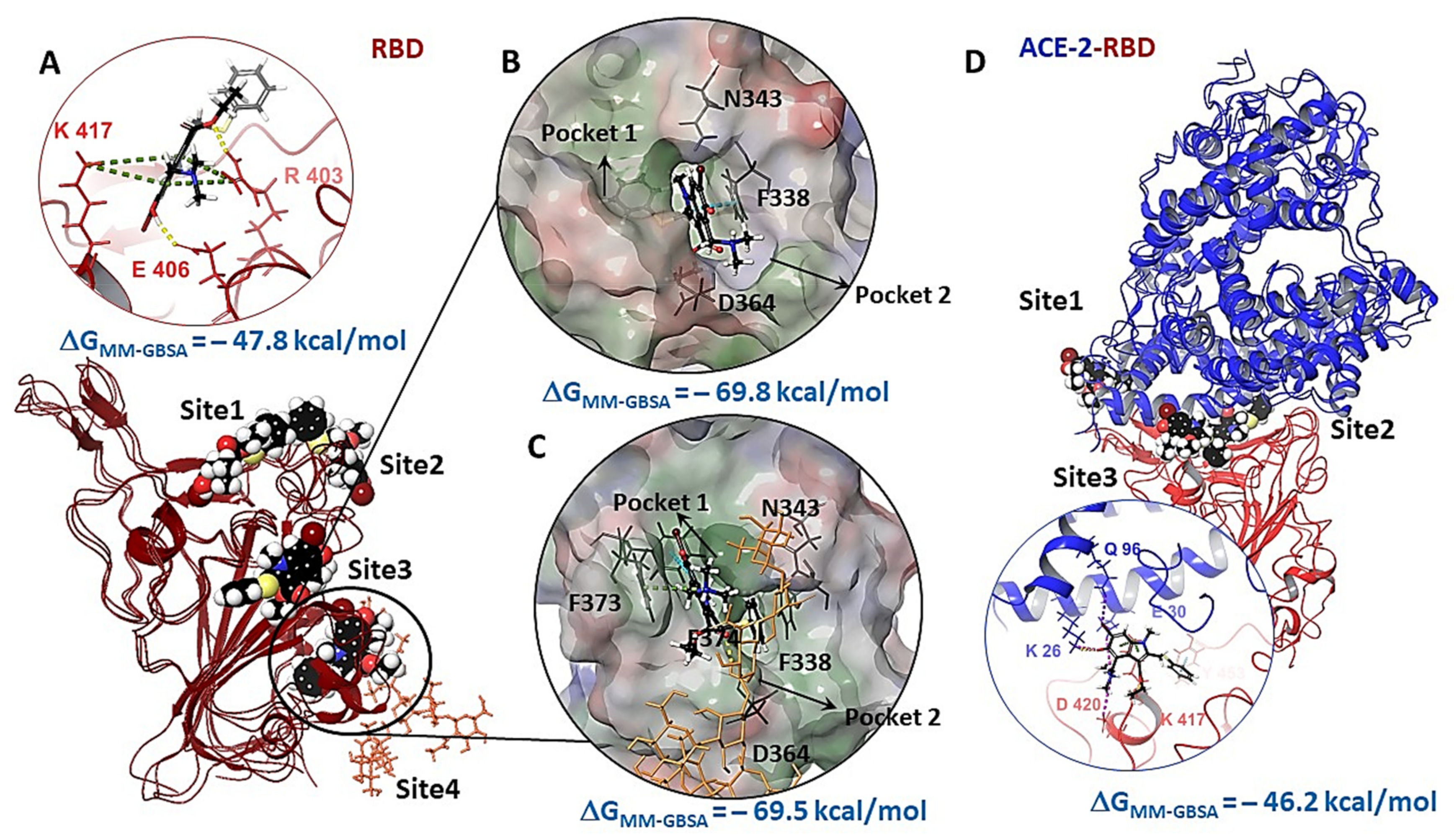
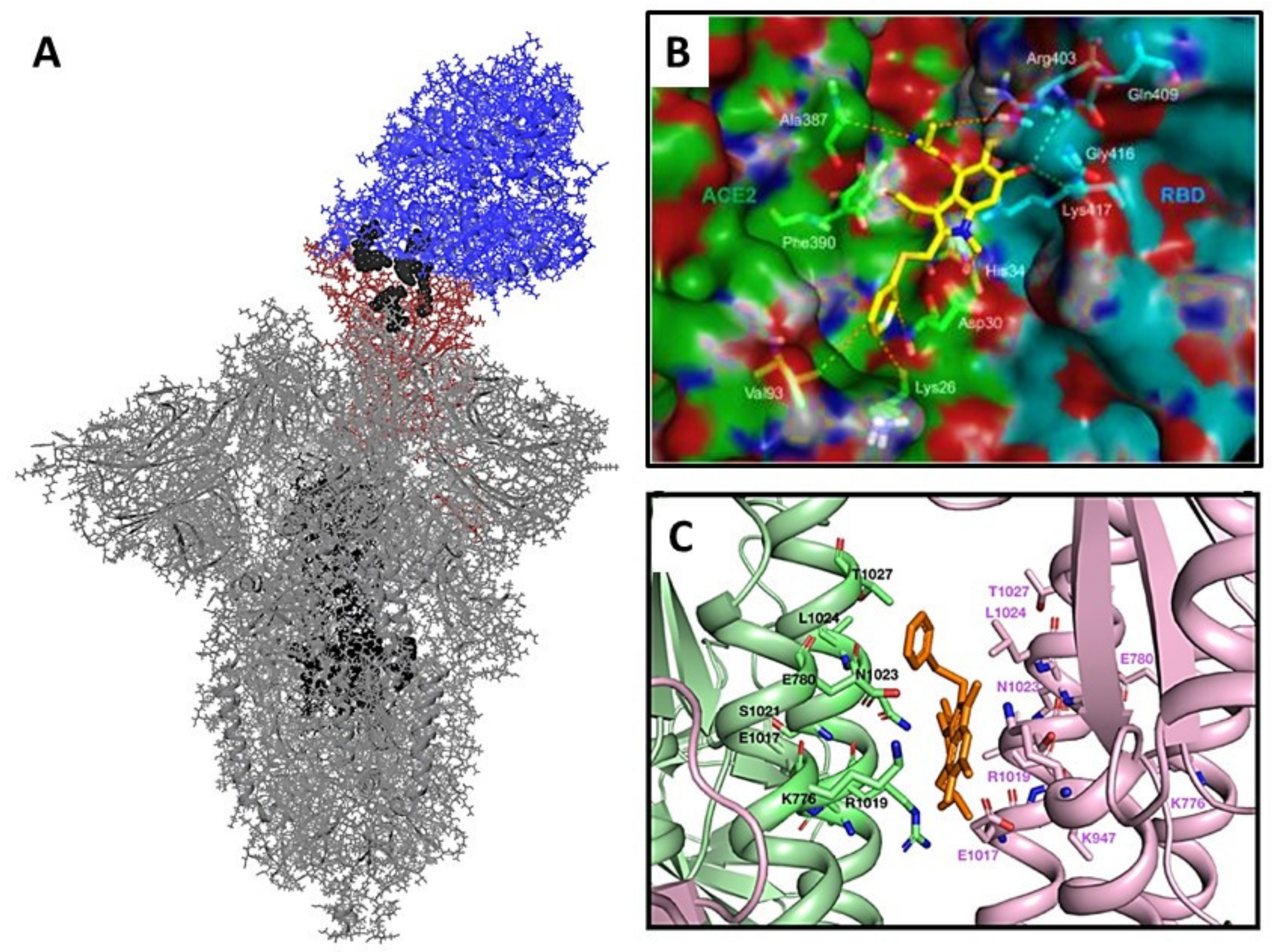
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Available online: https://www.who.int/news/item/27-04-2020-who-timeline---covid-19 (accessed on 18 March 2020).
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Pal, D. Spike protein fusion loop controls SARS-CoV-2 fusogenicity and infectivity. J. Struct. Biol. 2021, 213, 107713. [Google Scholar] [CrossRef]
- Petrosillo, N.; Viceconte, G.; Ergonul, O.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are they closely related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Mallajosyula, V.; Tato, C.M.; Tan, G.S.; Wang, T.T. SARS-CoV-2 vaccines in advanced clinical trials: Where do we stand? Adv. Drug Deliv. Rev. 2021, 172, 314–338. [Google Scholar] [CrossRef]
- WHO. DRAFT Landscape of COVID-19 Candidate Vaccines. 2020. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 28 November 2021).
- Khuroo, M.S.; Khuroo, M.; Khuroo, M.S.; Sofi, A.A.; Khuroo, N.S. COVID-19 Vaccines: A Race against Time in the Middle of Death and Devastation! J. Clin. Exp. Hepatol. 2020, 10, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Logunov, D.Y.; Dolzhikova, I.V.; Shcheblyakov, D.V.; Tukhvatulin, A.I.; Zubkova, O.V.; Dzharullaeva, A.S.; Kovyrshina, A.V.; Lubenets, N.L.; Grousova, D.M.; Erokhova, A.S.; et al. Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: An interim analysis of a randomised controlled phase 3 trial in Russia. Lancet 2021, 397, 671–681. [Google Scholar] [CrossRef]
- Kim, J.H.; Marks, F.; Clemens, J.D. Looking beyond COVID-19 vaccine phase 3 trials. Nat. Med. 2021, 27, 205–211. [Google Scholar] [CrossRef]
- Jones, I.; Roy, P. Sputnik V COVID-19 vaccine candidate appears safe and effective. Lancet 2021, 397, 642–643. [Google Scholar] [CrossRef]
- Status of COVID-19 Vaccines within WHO EUL/PQ Evaluation Process NRA. Available online: https://extranet.who.int/pqweb/sites/default/files/documents/Status_COVID_VAX_15Dec2021_0.pdf (accessed on 28 November 2021).
- Levett-Jones, T. Vaccines for preventing influenza in the elderly: A Cochrane review summary. Int. J. Nurs. Stud. 2020, 109, 103372. [Google Scholar] [CrossRef] [PubMed]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchianò, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 receptor interaction by natural and semi-synthetic steroidal agents acting on functional pockets on the receptor binding domain. Front. Chem. 2020, 8, 846. [Google Scholar] [CrossRef] [PubMed]
- Xiu, S.; Dick, A.; Ju, H.; Mirzaie, S.; Abdi, F.; Cocklin, S.; Zhan, P.; Liu, X. Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 2020, 63, 12256–12274. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef]
- Ngo, H.X.; Garneau-Tsodikova, S. What are the drugs of the future? Medchemcomm 2018, 9, 757–758. [Google Scholar] [CrossRef]
- Boriskin, Y.; Leneva, I.; Pecheur, E.-I.; Polyak, S. Arbidol: A Broad-Spectrum Antiviral Compound that Blocks Viral Fusion. Curr. Med. Chem. 2008, 15, 997–1005. [Google Scholar] [CrossRef]
- Wang, X.; Cao, R.; Zhang, H.; Liu, J.; Xu, M.; Hu, H.; Li, Y.; Zhao, L.; Li, W.; Sun, X.; et al. The anti-influenza virus drug, arbidol is an efficient inhibitor of SARS-CoV-2 in vitro. Cell Discov. 2020, 6, 28. [Google Scholar] [CrossRef]
- Cai, L.; Guo, X.; Cao, Y.; Ying, P.; Hong, L.; Zhang, Y.; Yi, G.; Fu, M. Determining available strategies for prevention and therapy: Exploring COVID-19 from the perspective of ACE2 (Review). Int. J. Mol. Med. 2021, 47, 43. [Google Scholar] [CrossRef] [PubMed]
- Leneva, I.; Kartashova, N.; Poromov, A.; Gracheva, A.; Korchevaya, E.; Glubokova, E.; Borisova, O.; Shtro, A.; Loginova, S.; Shchukina, V.; et al. Antiviral Activity of Umifenovir In Vitro against a Broad Spectrum of Coronaviruses, Including the Novel SARS-CoV-2 Virus. Viruses 2021, 13, 1665. [Google Scholar] [CrossRef]
- Padhi, A.K.; Seal, A.; Khan, J.M.; Ahamed, M.; Tripathi, T. Unraveling the mechanism of arbidol binding and inhibition of SARS-CoV-2: Insights from atomistic simulations. Eur. J. Pharmacol. 2021, 894, 173836. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N. Arbidol: A potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents 2020, 56, 105998. [Google Scholar] [CrossRef]
- Kadam, R.U.; Wilson, I.A. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. USA 2017, 114, 206–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Liu, Q.; Huang, W.; Li, X.; Wang, Y. Current status on the development of pseudoviruses for enveloped viruses. Rev. Med. Virol. 2018, 28, e1963. [Google Scholar] [CrossRef] [PubMed]
- Bentley, E.M.; Mather, S.T.; Temperton, N.J. The use of pseudotypes to study viruses, virus sero-epidemiology and vaccination. Vaccine 2015, 33, 2955–2962. [Google Scholar] [CrossRef] [PubMed]
- Sholukh, A.M.; Fiore-Gartland, A.; Ford, E.S.; Miner, M.D.; Hou, Y.J.; Tse, L.V.; Kaiser, H.; Zhu, H.; Lu, J.; Madarampalli, B.; et al. Evaluation of cell-based and surrogate SARS-CoV-2 neutralization assays. J. Clin. Microbiol. 2021, 59, e00527-21. [Google Scholar] [CrossRef]
- Tan, Y.S.; Verma, C.S. Straightforward Incorporation of Multiple Ligand Types into Molecular Dynamics Simulations for Efficient Binding Site Detection and Characterization. J. Chem. Theory Comput. 2020, 16, 6633–6644. [Google Scholar] [CrossRef] [PubMed]
- Fratev, F.; Steinbrecher, T.; Jónsdóttir, S.Ó. Prediction of Accurate Binding Modes Using Combination of Classical and Accelerated Molecular Dynamics and Free-Energy Perturbation Calculations: An Application to Toxicity Studies. ACS Omega 2018, 3, 4357–4371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarovaya, O.I.; Salakhutdinov, N.F. Mono- and sesquiterpenes as a starting platform for the development of antiviral drugs. Russ. Chem. Rev. 2021, 90, 488–510. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Putilova, V.P.; Yarovaya, O.I.; Zybkina, A.V.; Mordvinova, E.D.; Zaykovskaya, A.V.; Shcherbakov, D.N.; Orshanskaya, I.R.; Sinegubova, E.O.; Esaulkova, I.L.; et al. Synthesis and Antiviral Activity of Camphene Derivatives against Different Types of Viruses. Molecules 2021, 26, 2235. [Google Scholar] [CrossRef]
- Sokolova, A.S.; Yarovaya, O.I.; Baranova, D.V.; Galochkina, A.V.; Shtro, A.A.; Kireeva, M.V.; Borisevich, S.S.; Gatilov, Y.V.; Zarubaev, V.V.; Salakhutdinov, N.F. Quaternary ammonium salts based on (-)-borneol as effective inhibitors of influenza virus. Arch. Virol. 2021, 166, 1965–1976. [Google Scholar] [CrossRef] [PubMed]
- Borisevich, S.S.; Gureev, M.A.; Yarovaya, O.I.; Zarubaev, V.V.; Kostin, G.A.; Porozov, Y.B.; Salakhutdinov, N.F. Can molecular dynamics explain decreased pathogenicity in mutant camphecene-resistant influenza virus? J. Biomol. Struct. Dyn. 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zarubaev, V.V.; Pushkina, E.A.; Borisevich, S.S.; Galochkina, A.V.; Garshinina, A.V.; Shtro, A.A.; Egorova, A.A.; Sokolova, A.S.; Khursan, S.L.; Yarovaya, O.I.; et al. Selection of influenza virus resistant to the novel camphor-based antiviral camphecene results in loss of pathogenicity. Virology 2018, 524, 69–77. [Google Scholar] [CrossRef]
- Amani, B.; Amani, B.; Zareei, S.; Zareei, M. Efficacy and safety of arbidol (umifenovir) in patients with COVID-19: A systematic review and meta-analysis. Immun. Inflamm. Dis. 2021, 9, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Alavi Darazam, I.; Shokouhi, S.; Mardani, M.; Pourhoseingholi, M.A.; Rabiei, M.M.; Hatami, F.; Shabani, M.; Moradi, O.; Gharehbagh, F.J.; Irvani, S.S.N.; et al. Umifenovir in hospitalized moderate to severe COVID-19 patients: A randomized clinical trial. Int. Immunopharmacol. 2021, 99, 107969. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Neerukonda, S.N.; Vassell, R.; Herrup, R.; Liu, S.; Wang, T.; Takeda, K.; Yang, Y.; Lin, T.-L.; Wang, W.; Weiss, C.D. Establishment of a well-characterized SARS-CoV-2 lentiviral pseudovirus neutralization assay using 293T cells with stable expression of ACE2 and TMPRSS2. PLoS ONE 2021, 16, e0248348. [Google Scholar] [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; et al. Maraviroc (UK-427,857), a Potent, Orally Bioavailable, and Selective Small-Molecule Inhibitor of Chemokine Receptor CCR5 with Broad-Spectrum Anti-Human Immunodeficiency Virus Type 1 Activity. Antimicrob. Agents Chemother. 2005, 49, 4721–4732. [Google Scholar] [CrossRef] [Green Version]
- Blaising, J.; Polyak, S.J.; Pécheur, E.-I. Arbidol as a broad-spectrum antiviral: An update. Antivir. Res. 2014, 107, 84–94. [Google Scholar] [CrossRef]
- Woo, H.; Park, S.-J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a Fully Glycosylated Full-Length SARS-CoV-2 Spike Protein Model in a Viral Membrane. J. Phys. Chem. B 2020, 124, 7128–7137. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Praissman, J.L.; Grant, O.C.; Cai, Y.; Xiao, T.; Rosenbalm, K.E.; Aoki, K.; Kellman, B.P.; Bridger, R.; Barouch, D.H.; et al. Virus-Receptor Interactions of Glycosylated SARS-CoV-2 Spike and Human ACE2 Receptor. Cell Host Microbe 2020, 28, 586–601.e6. [Google Scholar] [CrossRef] [PubMed]
- Shuster, A.; Pechalrieu, D.; Jackson, C.B.; Abegg, D.; Choe, H.; Adibekian, A. Clinical Antiviral Drug Arbidol Inhibits Infection by SARS-CoV-2 and Variants through Direct Binding to the Spike Protein. ACS Chem. Biol. 2021, 16, 2845–2851. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [Green Version]
- Kadam, R.U.; Wilson, I.A. A small-molecule fragment that emulates binding of receptor and broadly neutralizing antibodies to influenza A hemagglutinin. Proc. Natl. Acad. Sci. USA 2018, 115, 4240–4245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borisevich, S.S.; Khamitov, E.M.; Gureev, M.A.; Yarovaya, O.I.; Rudometova, N.B.; Zybkina, A.V.; Mordvinova, E.D.; Shcherbakov, D.N.; Maksyutov, R.A.; Salakhutdinov, N.F. Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses 2022, 14, 119. https://doi.org/10.3390/v14010119
Borisevich SS, Khamitov EM, Gureev MA, Yarovaya OI, Rudometova NB, Zybkina AV, Mordvinova ED, Shcherbakov DN, Maksyutov RA, Salakhutdinov NF. Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses. 2022; 14(1):119. https://doi.org/10.3390/v14010119
Chicago/Turabian StyleBorisevich, Sophia S., Edward M. Khamitov, Maxim A. Gureev, Olga I. Yarovaya, Nadezhda B. Rudometova, Anastasiya V. Zybkina, Ekaterina D. Mordvinova, Dmitriy N. Shcherbakov, Rinat A. Maksyutov, and Nariman F. Salakhutdinov. 2022. "Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights" Viruses 14, no. 1: 119. https://doi.org/10.3390/v14010119
APA StyleBorisevich, S. S., Khamitov, E. M., Gureev, M. A., Yarovaya, O. I., Rudometova, N. B., Zybkina, A. V., Mordvinova, E. D., Shcherbakov, D. N., Maksyutov, R. A., & Salakhutdinov, N. F. (2022). Simulation of Molecular Dynamics of SARS-CoV-2 S-Protein in the Presence of Multiple Arbidol Molecules: Interactions and Binding Mode Insights. Viruses, 14(1), 119. https://doi.org/10.3390/v14010119