
A Pre-Vaccination Baseline of SARS-CoV-2 Genetic Surveillance and Diversity in the United States

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sources for COVID-19 Cases and Deaths in the U.S. in 2020
2.2. Sources and Numbers of SARS-CoV-2 Sequences Used in Surveillance and Diversity Analyses
2.3. Analysis of SARS-CoV-2 Genetic Surveillance in the U.S. in 2020
2.4. Mutation Detection and Measurements of Genetic Diversity and Divergence
2.5. Analyses of Variants of Concern (VOCs)
2.6. Phylogenetic Analysis
2.7. Viral Genetic Surveillance Resources
3. Results and Discussion
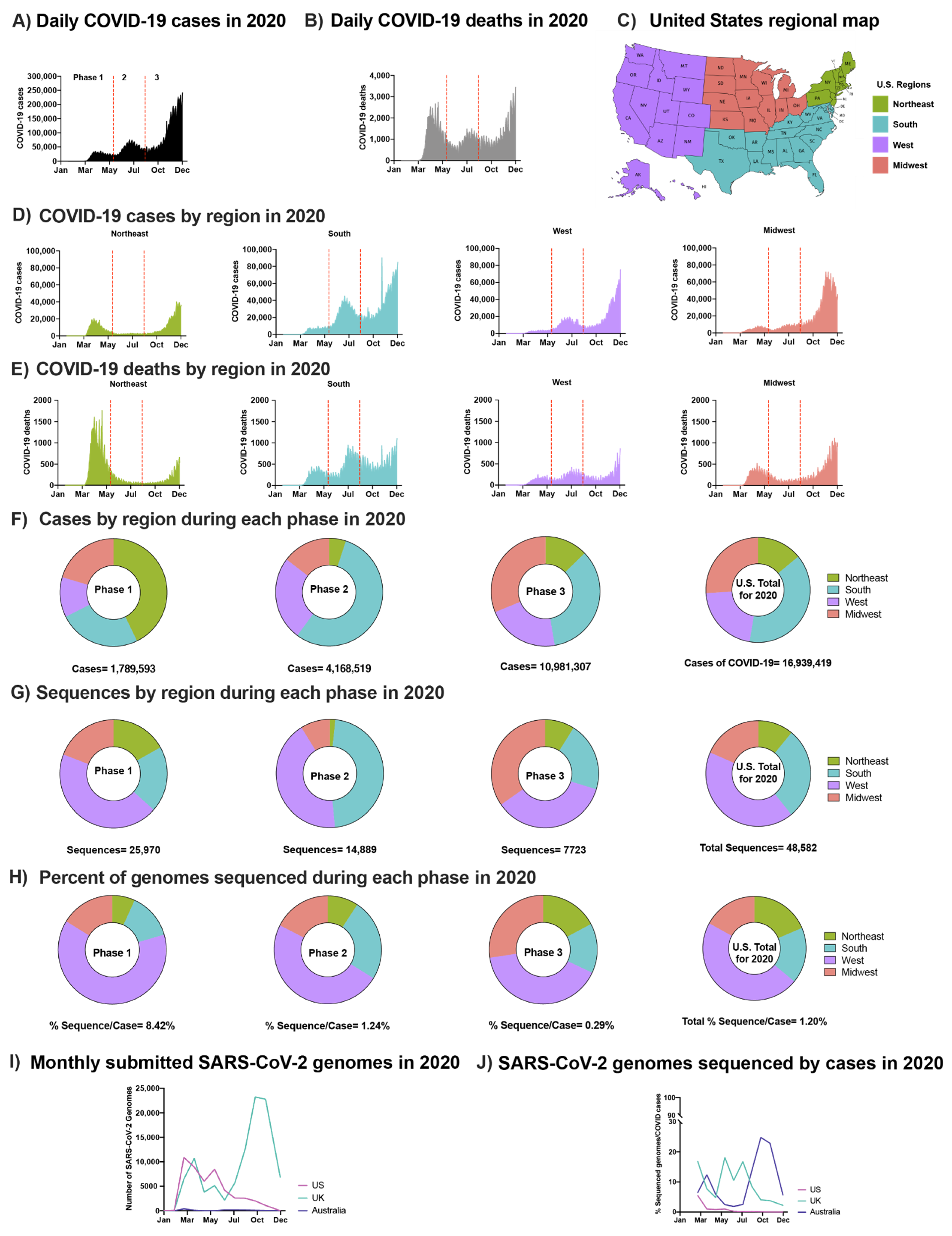
3.1. Spread of SARS-CoV-2 in the U.S. Pre-Vaccination Did Not Coorelate with Geographic or Levels of Sequence Surveillance
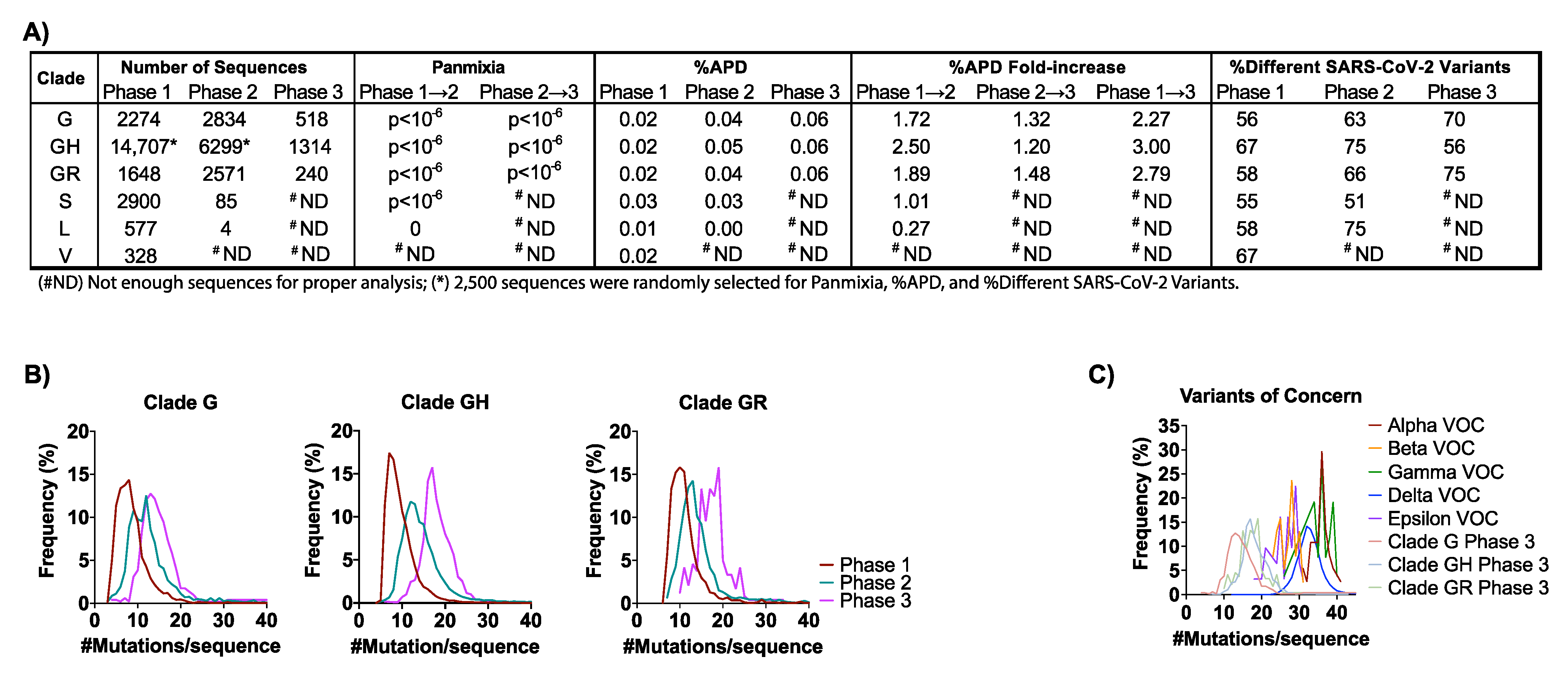
3.2. SARS-CoV-2 Genetic Diversity and Divergence Increased in the U.S. in 2020
3.3. Phylogenetic Analysis Reveals the Evolutionary Relationships of G Clades in the U.S. in 2020 to the VOCs
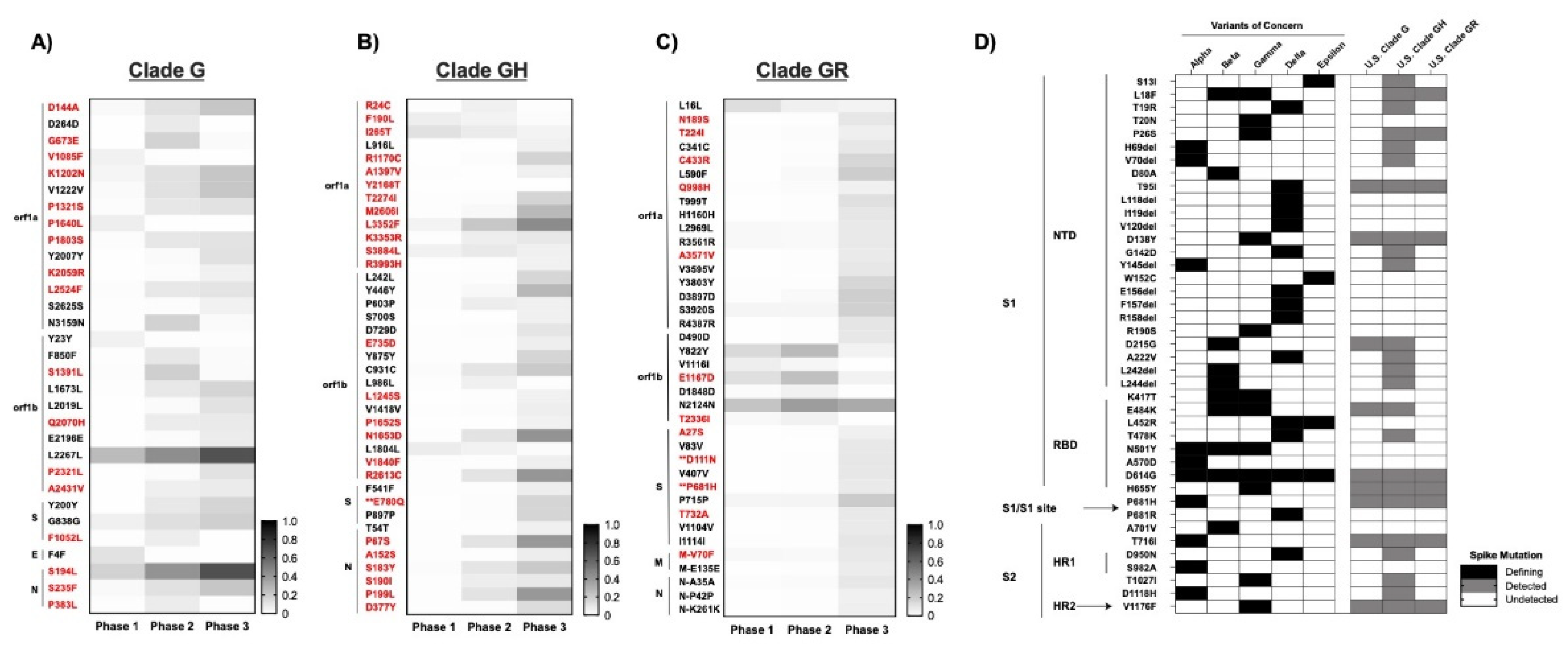
3.4. SARS-CoV-2 Mutations Present in ≥5% of the Population Detected in the U.S. in 2020
3.5. At Least 54 New Amino Acid Changes Emerged in 2020 and Persisted in ≥5% of the U.S. Population in at Least one Phase of the Spread
3.6. Mutations in Spike and Implications for COVID-19 Vaccines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First Case of 2019 Novel Coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. COVID-19 Response. COVID-19 Case Surveillance Public Data Access, Summary, and Limitations. Available online: https://data.cdc.gov/Case-Surveillance/United-States-COVID-19-Cases-and-Deaths-by-State-o/9mfq-cb36 (accessed on 11 June 2020).
- Wang, P.; Casner, R.G.; Nair, M.S.; Wang, M.; Yu, J.; Cerutti, G.; Liu, L.; Kwong, P.D.; Huang, Y.; Shapiro, L.; et al. Increased resistance of SARS-CoV-2 variant P.1 to antibody neutralization. Cell Host Microbe 2021, 29, 747–751.e4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Davis, B.D.; Chen, S.S.; Sincuir Martinez, J.M.; Plummer, J.T.; Vail, E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA 2021, 325, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wang, L.; Zhou, H.-Y.; Ji, C.-Y.; Xia, S.Z.; Cao, Y.; Meng, J.; Ding, X.; Gold, S.; Jiang, T.; et al. One year of SARS-CoV-2 evolution. Cell Host Microbe 2021, 29, 503–507. [Google Scholar] [CrossRef]
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The variant gambit: COVID-19’s next move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef]
- The COVID Tracking Project. Available online: https://covidtracking.com/about-data/data-summary (accessed on 17 December 2020).
- Available online: https://www.census.gov/geographies/reference-maps/2010/geo/2010-census-regions-and-divisions-of-the-united-states.html (accessed on 17 December 2020).
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Euro Surveill. Bull. Eur. Sur Les Mal. Transm. Eur. Commun. Dis. Bull. 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The biological and clinical significance of emerging SARS-CoV-2 variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- R Team. RStudio: Integrated Development for R; R CoreTeam: Vienna, Austria, 2020. [Google Scholar]
- Coronavirus (COVID-19) in the UK. Available online: https://coronavirus.data.gov.uk/ (accessed on 17 December 2020).
- Coronavirus (COVID-19) Case Numbers and Statistics. Available online: https://www.health.gov.au/news/health-alerts/novel-coronavirus-2019-ncov-health-alert/coronavirus-covid-19-case-numbers-and-statistics (accessed on 17 December 2020).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Tao, Q.; Weaver, S.; Sanderford, M.; Caraballo-Ortiz, M.A.; Sharma, S.; Pond, S.L.K.; Miura, S. An Evolutionary Portrait of the Progenitor SARS-CoV-2 and Its Dominant Offshoots in COVID-19 Pandemic. Mol. Biol. Evol. 2021, 38, 3046–3059. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.R.; Kearney, M.; Palmer, S.; Shao, W.; Maldarelli, F.; Coakley, E.P.; Chappey, C.; Wanke, C.; Coffin, J.M. Comparison of standard PCR/cloning to single genome sequencing for analysis of HIV-1 populations. J. Virol Methods 2010, 168, 114–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2019, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edler, D.; Klein, J.; Antonelli, A.; Silvestro, D. raxmlGUI 2.0: A graphical interface and toolkit for phylogenetic analyses using RAxML. Methods Ecol. Evol. 2021, 12, 373–377. [Google Scholar] [CrossRef]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B.; et al. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Julia, L.; Mullen, G.T.; Latif, A.A.; Alkuzweny, M.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. Outbreak.info. Available online: https://outbreak.info/ (accessed on 17 December 2020).
- Worobey, M.; Pekar, J.; Larsen, B.B.; Nelson, M.I.; Hill, V.; Joy, J.B.; Rambaut, A.; Suchard, M.A.; Wertheim, J.O.; Lemey, P. The emergence of SARS-CoV-2 in Europe and North America. Science 2020, 370, 564–570. [Google Scholar] [CrossRef]
- Chang, S.; Pierson, E.; Koh, P.W.; Gerardin, J.; Redbird, B.; Grusky, D.; Leskovec, J. Mobility network models of COVID-19 explain inequities and inform reopening. Nature 2021, 589, 82–87. [Google Scholar] [CrossRef]
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.J.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khan, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020, 369, 297–301. [Google Scholar] [CrossRef]
- Ladner, J.T.; Larsen, B.B.; Bowers, J.R.; Hepp, C.M.; Bolyen, E.; Folkerts, M.; Sheridan, K.; Pfeiffer, A.; Yaglom, H.; Lemmer, D.; et al. An Early Pandemic Analysis of SARS-CoV-2 Population Structure and Dynamics in Arizona. mBio 2020, 11, e02107-20. [Google Scholar] [CrossRef]
- Bedford, T.; Greninger, A.L.; Roychoudhury, P.; Starita, L.M.; Famulare, M.; Huang, M.L.; Nalla, A.; Pepper, G.; Reinhardt, A.; Xie, H.; et al. Cryptic transmission of SARS-CoV-2 in Washington state. Science 2020, 370, 571–575. [Google Scholar] [CrossRef]
- Lemieux, J.E.; Siddle, K.J.; Shaw, B.M.; Loreth, C.; Schaffner, S.F.; Gladden-Young, A.; Adams, G.; Fink, T.; Tomkins-Tinch, C.H.; Krasilnikova, L.A.; et al. Phylogenetic analysis of SARS-CoV-2 in Boston highlights the impact of superspreading events. Science 2021, 371, eabe3261. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Gao, K.; Hozumi, Y.; Yin, C.; Wei, G.W. Analysis of SARS-CoV-2 mutations in the United States suggests presence of four substrains and novel variants. Commun. Biol. 2021, 4, 228. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef] [PubMed]
- Dearlove, B.; Lewitus, E.; Bai, H.; Li, Y.; Reeves, D.B.; Joyce, M.G.; Scott, P.T.; Amare, M.F.; Vasan, S.; Michael, N.L.; et al. A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proc. Natl. Acad. Sci. USA 2020, 117, 23652–23662. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Alameh, M.G.; de Silva, T.; Collini, P.; Hornsby, H.; Brown, R.; LaBranche, C.C.; Edwards, R.J.; Sutherland, L.; Santra, S.; et al. D614G Spike Mutation Increases SARS CoV-2 Susceptibility to Neutralization. Cell Host Microbe 2021, 29, 23–31.e4. [Google Scholar] [CrossRef] [PubMed]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Achaz, G.; Palmer, S.; Kearney, M.; Maldarelli, F.; Mellors, J.W.; Coffin, J.M.; Wakeley, J. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol. Biol. Evol. 2004, 21, 1902–1912. [Google Scholar] [CrossRef] [PubMed]
- European Centre for Disease Prevention and Control. Guidance for Representative and Targeted Genomic SARS-Cov-2 Monitoring; European Centre for Disease Prevention and Control: Solna Municipality, Sweden, 2021. [Google Scholar]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Konings, F.; Perkins, M.D.; Kuhn, J.H.; Pallen, M.J.; Alm, E.J.; Archer, B.N.; Barakat, A.; Bedford, T.; Bhiman, J.N.; Caly, L.; et al. SARS-CoV-2 Variants of Interest and Concern naming scheme conducive for global discourse. Nat. Microbiol. 2021, 6, 821–823. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, V.A.; Matson, M.J.; Seifert, S.N.; Pryce, R.; Williamson, B.N.; Anzick, S.L.; Barbian, K.; Judson, S.D.; Fischer, E.R.; Martens, C.; et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell 2020, 183, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Alter, G.; Yu, J.; Liu, J.; Chandrashekar, A.; Borducchi, E.N.; Tostanoski, L.H.; McMahan, K.; Jacob-Dolan, C.; Martinez, D.R.; Chang, A.; et al. Immunogenicity of Ad26.COV2.S vaccine against SARS-CoV-2 variants in humans. Nature 2021, 596, 268–272. [Google Scholar] [CrossRef]
- Corbett, K.S.; Nason, M.C.; Flach, B.; Gagne, M.; O’Connell, S.; Johnston, T.S.; Shah, S.N.; Edara, V.V.; Floyd, K.; Lai, L.; et al. Immune correlates of protection by mRNA-1273 vaccine against SARS-CoV-2 in nonhuman primates. Science 2021, 373, eabj0299. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Lopez Bernal, J.; Andrews, N.; Gower, C.; Gallagher, E.; Simmons, R.; Thelwall, S.; Stowe, J.; Tessier, E.; Groves, N.; Dabrera, G.; et al. Effectiveness of Covid-19 Vaccines against the B.1.617.2 (Delta) Variant. N. Engl. J. Med. 2021, 385, 585–594. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, D.; Lee, C.D.; Wu, N.C.; Jackson, A.M.; Zhu, X.; Liu, H.; Peng, L.; van Gils, M.J.; Sanders, R.W.; et al. Structural and functional ramifications of antigenic drift in recent SARS-CoV-2 variants. Science 2021, 373, 818–823. [Google Scholar] [CrossRef]
- Garcia-Beltran, W.F.; Lam, E.C.; St. Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef] [PubMed]
- Edara, V.V.; Norwood, C.; Floyd, K.; Lai, L.; Davis-Gardner, M.E.; Hudson, W.H.; Mantus, G.; Nyhoff, L.E.; Adelman, M.W.; Fineman, R.; et al. Infection- and vaccine-induced antibody binding and neutralization of the B.1.351 SARS-CoV-2 variant. Cell Host Microbe 2021, 29, 516–521.e3. [Google Scholar] [CrossRef]
- Kuzmina, A.; Khalaila, Y.; Voloshin, O.; Keren-Naus, A.; Boehm-Cohen, L.; Raviv, Y.; Shemer-Avni, Y.; Rosenberg, E.; Taube, R. SARS-CoV-2 spike variants exhibit differential infectivity and neutralization resistance to convalescent or post-vaccination sera. Cell Host Microbe 2021, 29, 522–528.e2. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Tang, H.; McDanal, C.; Wagh, K.; Fischer, W.; Theiler, J.; Yoon, H.; Li, D.; Haynes, B.F.; Sanders, K.O.; et al. SARS-CoV-2 variant B.1.1.7 is susceptible to neutralizing antibodies elicited by ancestral spike vaccines. Cell Host Microbe 2021, 29, 529–539.e3. [Google Scholar] [CrossRef]
- Pegu, A.; O’Connell, S.; Schmidt, S.D.; O’Dell, S.; Talana, C.A.; Lai, L.; Albert, J.; Anderson, E.; Bennett, H.; Corbett, K.S.; et al. Durability of mRNA-1273 vaccine-induced antibodies against SARS-CoV-2 variants. Science 2021, 373, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Mbaeyi, S. The Advisory Committee on Immunization Practices’ Interim Recommendations for Additional Primary and Booster Doses of COVID-19 Vaccines—United States, 2021. MMWR Morb. Mortal Wkly. Rep. 2021, 70, 1545–1552. [Google Scholar] [CrossRef]
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 Covid-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189. [Google Scholar] [CrossRef]
- Haas, E.J.; Angulo, F.J.; McLaughlin, J.M.; Anis, E.; Singer, S.R.; Khan, F.; Brooks, N.; Smaja, M.; Mircus, G.; Pan, K.; et al. Impact and effectiveness of mRNA BNT162b2 vaccine against SARS-CoV-2 infections and COVID-19 cases, hospitalisations, and deaths following a nationwide vaccination campaign in Israel: An observational study using national surveillance data. Lancet 2021, 397, 1819–1829. [Google Scholar] [CrossRef]
- Sadoff, J.; Gray, G.; Vandebosch, A.; Cárdenas, V.; Shukarev, G.; Grinsztejn, B.; Goepfert, P.A.; Truyers, C.; Fennema, H.; Spiessens, B.; et al. Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19. N. Engl. J. Med. 2021, 384, 2187–2201. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C. SARS-CoV-2 Delta VOC in Scotland: Demographics, risk of hospital admission, and vaccine effectiveness. Lancet 2021, 397, 2461–2462. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236.e13. [Google Scholar] [CrossRef]
- Thompson, M.G.; Burgess, J.L.; Naleway, A.L.; Tyner, H.; Yoon, S.K.; Meece, J.; Olsho, L.E.W.; Caban-Martinez, A.J.; Fowlkes, A.L.; Lutrick, K.; et al. Prevention and Attenuation of Covid-19 with the BNT162b2 and mRNA-1273 Vaccines. N. Engl. J. Med. 2021, 385, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Rausch, J.W.; Capoferri, A.A.; Katusiime, M.G.; Patro, S.C.; Kearney, M.F. Low genetic diversity may be an Achilles heel of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 24614–24616. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capoferri, A.A.; Shao, W.; Spindler, J.; Coffin, J.M.; Rausch, J.W.; Kearney, M.F. A Pre-Vaccination Baseline of SARS-CoV-2 Genetic Surveillance and Diversity in the United States. Viruses 2022, 14, 104. https://doi.org/10.3390/v14010104
Capoferri AA, Shao W, Spindler J, Coffin JM, Rausch JW, Kearney MF. A Pre-Vaccination Baseline of SARS-CoV-2 Genetic Surveillance and Diversity in the United States. Viruses. 2022; 14(1):104. https://doi.org/10.3390/v14010104
Chicago/Turabian StyleCapoferri, Adam A., Wei Shao, Jon Spindler, John M. Coffin, Jason W. Rausch, and Mary F. Kearney. 2022. "A Pre-Vaccination Baseline of SARS-CoV-2 Genetic Surveillance and Diversity in the United States" Viruses 14, no. 1: 104. https://doi.org/10.3390/v14010104
APA StyleCapoferri, A. A., Shao, W., Spindler, J., Coffin, J. M., Rausch, J. W., & Kearney, M. F. (2022). A Pre-Vaccination Baseline of SARS-CoV-2 Genetic Surveillance and Diversity in the United States. Viruses, 14(1), 104. https://doi.org/10.3390/v14010104