Abstract
The error rate displayed during template copying to produce viral RNA progeny is a biologically relevant parameter of the replication complexes of viruses. It has consequences for virus–host interactions, and it represents the first step in the diversification of viruses in nature. Measurements during infections and with purified viral polymerases indicate that mutation rates for RNA viruses are in the range of 10−3 to 10−6 copying errors per nucleotide incorporated into the nascent RNA product. Although viruses are thought to exploit high error rates for adaptation to changing environments, some of them possess misincorporation correcting activities. One of them is a proofreading-repair 3′ to 5′ exonuclease present in coronaviruses that may decrease the error rate during replication. Here we review experimental evidence and models of information maintenance that explain why elevated mutation rates have been preserved during the evolution of RNA (and some DNA) viruses. The models also offer an interpretation of why error correction mechanisms have evolved to maintain the stability of genetic information carried out by large viral RNA genomes such as the coronaviruses.
Keywords:
RNA virus; virus diversification; quasispecies; error catastrophe; exonuclease; SARS-CoV-2 1. Introduction: Mistakes as Hallmark of the Evolution of Life and of Present Day Viruses
Studies of prebiotic nucleotide synthesis suggest that the origin of the high mutation rates exhibited by many present day viruses may be attributed to a limited template-copied fidelity that likely operated during the early stages of life development. There are several models of life emergence, and all of them include at some stage replication of molecules that carried inheritable information. This statement is supported both by theoretical and experimental results that the reader can find in References [1,2,3,4,5,6]. According to such results, the evolution of primitive life forms was possible thanks to mistakes that unavoidably and blindly occurred during copying of the template molecules that carried the information to be transmitted. Production of error copies is inherent to subcellular and cellular life forms [1,2,3,4,5,6]. Several lines of evidence suggest that viruses have played a prominent role in the organization of cellular genomes, mainly as a result of their capacity to penetrate into cells and to transport, integrate, and exchange genetic material with that of cellular organizations under construction [5]. A number of mechanisms of integration of viral genomes (or part of them) into cellular genomes have been characterized in present day viruses. They include retroviral DNA integration, insertion of temperate viral DNA of lysogenic bacteria such as phage lambda into E. coli DNA, and bacterial DNA rescue by transducing viruses. These well-characterized integration events should be added to the extensive evidence of the presence of DNA copies of many pathogenic RNA viral genomes in their animal or insect hosts, in most cases by mechanisms that are not well understood [5].
Together with other mobile elements, viruses are thought to have promoted cellular variation and differentiation, often through co-evolutionary processes (reviewed in [5,7,8,9]; among other accounts). According to these studies, the perception of viruses as blind, selfish, and invasive replicators should be regarded only a facet of their natural history, and probably not the one responsible for their maintenance and abundance in the biosphere. To effectively participate in the construction of a cellular world, genome variation of the primitive replicons must have played a prominent role to cope with an increasing cellular differentiation and to overcome barriers to penetration into cells, as again evidenced by independent studies [5,8,9].
Many of the viruses that have been isolated and studied exhibit an “error-prone replication”, as they multiply in cells and organisms, in some cases with associated disease manifestations. From the studies summarized above [2,3,4,5,6,7,8,9,10], it has been proposed that this feature is probably a beneficial remnant of virus origins, combined with its usefulness for sustained adaptability [10,11,12]. Production of error copies underlies quasispecies dynamics (continuous production of variant genomes subjected to competition, selection, and random drift, and which may act as a unit of selection), which is crucial for virus adaptation to changing environments and for the collective behavior of viral populations [10,11,12,13].
Awareness of the medical consequences of mutations for viruses arose during the last century when viral genome analyses entered the scene as a tool to understand and confront viral diseases. Many studies focused on diseases that acquired epidemic and pandemic proportions, such as poliomyelitis, influenza, and AIDS. The ongoing COVID-19 pandemic represents the most recent scourge, with again prominence of genome variations of the causative virus.
During the massive vaccination campaigns to control poliovirus using the Sabin live-attenuated vaccine in the middle of last century, it was observed that approximately one out of a million vaccine recipients (or their contacts) developed paralytic disease. In the afflicted individuals, poliovirus had mutated to lose its attenuation phenotype. Vaccine-associated mutant and recombinant polioviruses emerged in different world locations. Poliovirus mutants and recombinants became a major problem for the global eradication of poliomyelitis which, to this date, is still incomplete due both to the adaptive capacity of poliovirus and to socio-political impediments [13,14,15].
For influenza, mutations that promote antigenic drift of influenza virus (IV) (the gradual accumulation of amino acid substitutions in the surface antigens hemagglutinin and neuraminidase) and genome segment reassortments that result in antigenic shift (exchange of genome segments that encode the surface antigens particularly between human and animal IVs) are behind periodic human pandemics. One of the consequences of antigenic drift and shift is the need to update the composition of the inactivated anti-influenza vaccines, which generally display a limited efficacy [16].
In the case of AIDS, the capacity of immunodeficiency virus type 1 (HIV-1) to escape components of the human immune response through mutation, the debilitated response due to the HIV-1 tropism for cells involved in the immune response, and the proviral DNA integration as part of the virus life cycle are reasons why to date there are no effective anti-AIDS vaccines [17,18]. Antiretroviral therapy has been successful in reducing AIDS and HIV-1 infection-related mortality, but antiretroviral resistance through genetic variation of the virus and selection of escape mutants remains an issue.
For COVID-19, we are confronting the seemingly paradoxical situation of a coronavirus that is supposed to limit its mutation rate via a proofreading-repair activity (Section 5) but that is continuously producing new variants that circulate in the human population and that represent a threat to vaccine efficacy [19,20,21].
The observations with four pandemic viruses, here summarized briefly, render unquestionable an essential role of genetic variation as effector of virus–host interactions and of difficulties for viral disease control. There are numerous additional examples of the relevance of mutation, recombination, and genome segment reassortment for viruses to overcome selective constraints. The latter are ubiquitous during all stages of a virus life cycle. Remarkably, they also include medical interventions, such as administration of antiviral agents, vaccines, or immunotherapy with polyclonal or monoclonal antibodies [5]. Genetic variation of viruses provides the key molecular mechanism for responding to selective constraints. Several ways of trying to counteract such virus capacities (without guaranteed success) have been proposed: use of multi-epitopic vaccines (ideally live-attenuated, with limited reversion potential), combination therapies, and new broad-spectrum antiviral designs such as lethal mutagenesis in synergistic combinations, among others [5,11,12]. The scope of implications of error-prone replication of RNA (and many DNA) viruses justifies focusing on the origin of mutations and mechanisms to limit their frequency. Here we review some studies on viral mutation rates and frequencies, as well as the evidence that, as for any replicative system, there is a limit to the number of mutations that can be accepted to preserve the encoded genetic information. Therefore, we also review why and how mutation rates may be modulated by proofreading-repair and post-replicative repair (error-correcting) activities.
2. Mutation Rates and Frequencies
The term “mutation rate” is often used by geneticists as synonymous of “rate of evolution”. This indistinct use is misleading and hides relevant differences between these two parameters that describe evolutionary episodes that are only indirectly connected. “Mutation rate” should be used to mean the proportion of erroneous nucleotides introduced in a process of template copying to yield complementary strands and progeny viral genomes. It applies to events occurring during RNA or DNA synthesis in replication complexes. The newly arising mutations constitute the very first stage of virus diversification on which subsequent selective forces and random events act. “Rate of evolution” should be the term used to refer to the speed at which genomic sequences of viruses accumulate mutations in nature or in some experimental evolution setting as a function of time, leading to genome diversification. Within a replication complex, a specific mutation may arise at high rate (for example, under the influence of a particular sequence context in the template RNA) but be found at low frequency during subsequent rounds of intra-cellular template copying as a consequence of low replicative capacity (fitness). This gives rise to a third parameter termed “mutation frequency”. This parameter may be given for the average calculated for a set of mutations determined in a genome or genomic region, or for an individual mutation at a defined genomic site, in which case it is often termed a “mutant frequency”. The mutation frequency is dependent on (but not identical with) the mutation rate (Figure 1). Mutation frequencies dictate the repertoire of variant genomes that populate an infected host and that contribute the infectious particles that can be transmitted to other hosts.
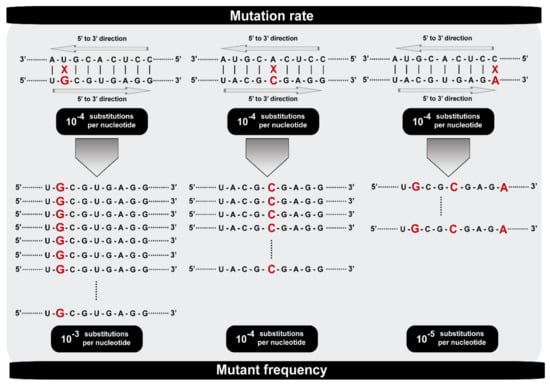
Figure 1.
Scheme that illustrates the difference between mutation rate and mutant frequency. Residue U (on the left), A (in the middle), and C (on the right) can be misread to incorporate a G (on the left), C (in the middle), or A (on the right) in the complementary strand at a rate of 10−4 substitutions per nucleotide in all cases. The replicative capacity of the newly G, C, and A templates determines the different mutant frequencies with 10−3, 10−4, and 10−5 substitutions per nucleotide, respectively.
A difference between a mutation rate and a mutation frequency is an example of conflict between a molecular instruction and a biological requirement, often encountered in the generation of mutant and recombinant genomes (reviewed in [5]). A pertinent example studied in our laboratory is the elongation of an internal oligoadenylate tract located between the two functional AUG triplets of the foot-and-mouth disease virus (FMDV) genome. The elongation was observed very frequently during plaque-to-plaque transfers of the virus in cell culture (an experimental design used to mimic the effect of repeated bottleneck events in viral populations), but it has never been observed in FMDV isolates from infected animals [22]. Fitness values of FMDV clones harboring the elongated oligoadenylate and quantification of the rate of reversion to its original length suggested that the site was a hot spot for homopolymeric tract elongation, likely through polymerase slippage (a molecular instruction). This genome modification was detectable probably because of the limited negative selection operating during plaque-to-plaque transfers, in contrast with the sieving effect (elimination of genomes harboring some types of mutations) of selection during standard virus evolution. In this manner, a high rate of oligonucleotide elongation translated into undetectable mutant frequency outside the bottleneck regimen ([22]; reviews in [5,23]). This example shows that calculation of a true mutation rate either for a specific mutation or as the average for a set of mutations or nucleotide positions is more difficult than the calculation of a mutation frequency.
Nuances of mutation rate calculations apply both to viruses and cells, including bacteria. Problems comprise (i) the use of different units (mutations per nucleotide versus mutations per genome), (ii) not considering the mode of viral genome replication in terms of template utilization (multiple copies produced from the same template versus each progeny molecule becoming a new template; probably an intermediate situation occurs in most viruses), and (iii) bias caused by selection intervening between the biochemical event that defines the mutation rate and the actual mutant quantification to give the mutation frequency [24,25,26,27,28,29,30,31,32].
Concerning (i), it is worth mentioning that the use of mutations per nucleotide as a unit of mutation rate does not take into consideration that accumulation of mutations in the same genome has a limit, thus generally decreasing the frequency of those genomes harboring multiple mutations. Regarding (ii), the problem of replication mode can become significant for comparative purposes if a virus displays marked asymmetry in template usage by positive or minus (complementary) strand and if the replicative polymerase complex has a different protein composition (that affects fidelity properties) when synthesizing plus or minus strands. In addition to amino acid substitutions in the protein subunit that harbors the catalytic polymerization domain, substitutions at other proteins (from the replication complex or associated with it) can also affect template copying fidelity (examples are the coronavirus nsp10 [33], alphavirus helicase/protease nsP2 [34], FMDV non-structural protein 2C [35], and some proteins of yellow fever virus [36]).
A selection-associated bias takes place when the presence of the mutation whose rate is to be quantified affects the multiplication of the virus or bacterium under study. The first mutation rate for an RNA virus that was calculated was for the direct reversion of an A to G mutation introduced at the 3′ extracistronic region of genomic Qβ RNA, which was part of the methodology that gave birth to reverse genetics [37,38]. The mutation rate was estimated in 10−4 mutational events per genome doubling. Both mutation occurrence and competition between the mutant and wild-type phage were considered in the calculation, thus eliminating selection bias. Its limitation was that it referred to only one mutation type at a specific genomic site.
Selection bias affects quantification of mutation rates based on genotypic and phenotypic markers, including the classic fluctuation test. Statistical methods have been developed for correction of selection bias. One of them is the Ma-Sandri-Sarkar maximum likelihood estimator, available at the Fluctuation Analysis Calculator (FALCOR) [39,40,41]. When no correction for selection bias is applied, the discrepancy between a mutation frequency determined experimentally and the underlying mutation rate will depend on the fitness effect of the mutation. The difference will increase with the number of viral or cellular multiplication rounds elapsed between the initial mutational event and the time of the mutant frequency measurement.
Some phenotypic transitions used as mutation frequency markers (i.e., resistance to a neutralizing monoclonal antibody or to an inhibitor of viral replication) may depend on more than one mutation, resulting in underestimation of the mutation frequency if assumed to be due to a single point mutation. This, together with fitness effects, probably explains the broad range of values calculated for the frequency of monoclonal antibody-resistant mutants in viral populations (as broad as 10−3 to 10−7 escape mutants per infectious unit with values that are independent of the serological diversity of the viruses in nature; reviewed in [5]).
Studies with several viruses and their polymerases suggest that the following structural and environmental factors can influence the actual mutation rate at a genomic nucleotide site:
- The structure of the protein that contains the catalytic domain for nucleotide polymerization because it can directly or indirectly affect the interaction of the catalytic residues (and their neighbors) with template and primer residues, as well as with the incoming nucleotide;
- Other subunits or proteins that functionally interact with the protein that contains the polymerization catalytic site;
- Micro-environment in which template copying takes place (ionic composition, temperature, presence of metabolites);
- The sequence context in the template;
- Presence of functional proofreading-repair activities either as part of the polymerase or in proteins that interact with the polymerase;
- Availability and functionality of post-replicative repair pathways;
- Host-coded editing enzymes that may introduce viral genome mutations, unrelated to attributes of the replication complex.
The above points summarize a (probably minimum) number of influences on mutation rates (overviews in [5,24,42]). Main support for the effect of these influences has come from characterization of fidelity mutants of several RNA viruses [43,44,45,46,47,48,49,50,51,52,53,54,55,56], studies with purified viral RNA-dependent RNA and DNA polymerases in vitro [57,58,59,60], and the recognition of the influence of minor tautomeric forms in mutagenesis [61,62].
Considering the several influences and uncertainties that preside mutation rate determinations, it is reassuring that there is a wide consensus in that the average mutation rates for RNA viruses fall in the range of 10−3 to 10−6 mutations incorporated per nucleotide copied. The range is one or more orders of magnitude higher than that estimated for replication of cellular DNA under normal metabolic conditions. High mutation rates stand as a general feature of RNA viruses, other RNA genetic elements, and many DNA viruses. They push viruses towards exploring portions of sequence space where mutations and combinations of mutations are tolerated, waiting to be selected when the environment so demands.
3. Limits to Mutation Rates: The Need of Repair
The difference in copying fidelity between the replication complexes of viruses and cells (the latter considered globally for replicative DNA polymerases) suggests that mutation rates have been modulated historically by biological requirements. The fact that mutation rates are affected by amino acid substitutions in viral polymerases and also in other viral proteins (Section 2)—together with the evidence of intra-population mutation rate heterogeneity [63]—implies that mutation rates are evolvable [64]. Interpretations of why their values have settled within the observed limits include: (i): they are a compromise to ensure long-term stability of the core genetic information (understood as the one that permits virus classification) and to cope with changing environments, in particular following bottleneck events that reduce genome diversity; (ii) they are the result of a trade-off between speed of RNA synthesis—assumed to confer a selective advantage to viruses—and copying fidelity; and (iii) they are a requirement of viral dynamics during infection of individual hosts—invasion of different intra-host compartments—and transmissibility [5,11,12,23,65,66].
These models are interconnected and not mutually exclusive. Bottleneck events of different intensity (given by the number of founder genomes) are particularly noteworthy because of their abundance in the course of virus life cycles. They occur within infected hosts when viruses transit from one compartment to another and during host-to-host transmission. Bottlenecks permit genomes to explore new regions of sequence space, facilitated by newly arising genome heterogeneity (see as examples [65,66]).
From the available evidence, we consider it unlikely that a specific trade-off—such as speed of RNA synthesis, energy cost of repair (see Section 5), or other—could impose high mutation rates if the latter were not in compliance with long-term core information stability and adaptability requirements. Mutants displaying higher or lower fidelity than their parental wild-type viruses (Section 2) are often attenuated, show defects in replication, and are not found as dominant genomes in mutant spectra evolving in nature. There are biochemical and evolutionary arguments that favor the view that primitive replicons displayed high error rates [67,68]. It is tempting to propose that error-prone replication is not a biological novelty of the cellular-viral world. Rather, it appears to be an inheritance of an ancient RNA (or RNA-like) world. The true novelty seems to be the development of error-correction mechanisms.
Mutation rates in the range of those observed with RNA viruses would not allow survival of complex genomes (complex in terms of amount of genetic information that they carry). John Drake calculated mutation rates for some DNA viruses and cellular microbes [69,70,71]. He obtained evidence that DNA viruses and organisms mutate at an approximately constant rate of 0.003 mutations per genome (note the units) and multiplication round [71], an observation known as Drake’s rule. It suggests a limitation of the mutation load tolerated by complex genomes, expected from fitness losses of genomes with multiple mutations, with the exception of constellations of either tolerated combinations of mutations or of positive epistasis (fitness gain by the presence of two or more mutations in the same genome as compared with their effect when present individually). In line with genome size-dependent mutation intolerance, a reduction in the bacterial genome size resulted in an increase in mutation frequency [72].
Limitation of mutation acceptability by complex genomes is recapitulated in the error threshold relationship of quasispecies theory. This relationship sets a limit to the maximum permissible error rate, depending on the complexity of the information to be maintained. More genetic information—embodied in large genomes—implies vulnerability to mutations [73,74,75,76]. Terminal (irreversible) deterioration of the genetic information due to mutations is known as entry into error catastrophe. Molecularly it can be visualized as a transition (sometimes referred to as “melting”) from a quasispecies distribution into random sequences (without biological meaning). To avoid error catastrophe, replicative systems have developed proofreading repair (occurring at the replication forks of polymerase complexes) and post-replicative repair activities (operating on finalized error copies once they have been synthesized). Types of DNA lesions that can be corrected by these mechanisms include base substitutions, single and double strand breaks, DNA adducts, DNA-protein crosslinks, and other types of double helix perturbations [77,78]. There are several lines of evidence that suggest that the achieved preservation of genome integrity may protect from accelerated aging and cellular transformation (cancer development). The importance of error correction is also suggested by the more than 100 proteins encoded by the human genome that are devoted to repair functions [78]. Post-replicative repair activities are essential for life stability. They operate efficiently only on double stranded DNA but not on double stranded RNA or DNA-RNA hybrids. Therefore, the cellular post-replicative repair functions that have been characterized to date are ineffective for RNA virus replication products whose error input limitation has to rely on proofreading repair at the step of genome synthesis [77,78,79].
4. Repair Mechanisms in Viruses
Viruses are no exception to the need of repair functions, as evidenced by the presence of 3′ to 5′ exonuclease (Exo N) activities in complex bacterial and animal DNA viruses and some RNA viruses. Several decades of study have established the role of such activities in DNA copying fidelity, in determining mutator phenotypes when they have been inactivated, and at least in some cases, in promoting DNA recombination ([80,81,82,83,84]; see also other articles of the same Virus Research issue [84], among many studies). The extent of functional interdependence between polymerase and Exo N activities varies depending on the virus. In vaccinia virus, the two activities appear to be coupled [84], while in herpes simplex virus 1, mutations in the exonuclease do not necessarily impair the polymerase activity [85]. It appears to be a subtle relationship, as also suggested by studies with coronaviruses (Section 5).
Repair functions have also been described for RNA viruses. In this case, a distinction can be made between repair activities that compensate specific lesions in primer or terminal sequences relevant to replication functions and those, such as Exo N, that mediate a bona fide reduction in mutation rate (Figure 2). When elongation of IV RNA was attempted with a primer to which 3′ guanosine residues had been added, elongation did not proceed until the excess of GMP residues had been removed [86]. NTP-dependent excision of the 3′ nucleotide at the growing nucleic acid product has been identified for hepatitis C virus and HIV-1 [87,88,89]. A 3′-end repair mechanism has been described for satellite RNAs of some plant viruses [90,91]. Coronaviruses exhibit an Exo N activity that increases template copying fidelity and confers decreased sensitivity to some antiviral agents [92]. Its implication in SARS-CoV-2 biology and diversification has become a focus of interest with the COVID-19 pandemic.
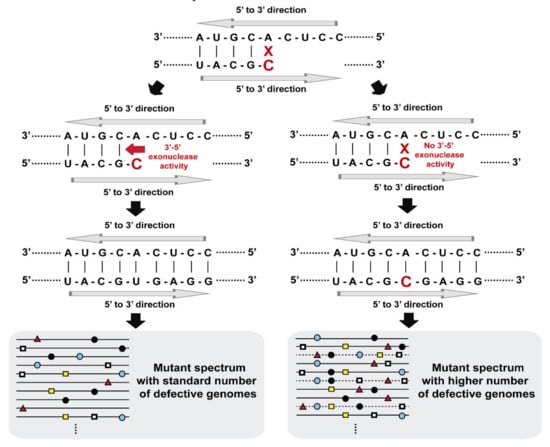
Figure 2.
Scheme that illustrates mutant spectrum complexities in the absence and presence of a 3′-5′ exonuclease activity during viral replication. Residue A (on the top) can be misread to incorporate a C in the complementary strand. In the presence of a proofreading activity, the mutant spectrum (represented by lines with mutations as colored symbols) contains a standard number of defective genomes (depicted on the left). In the absence of a proofreading activity, there is an accumulation of mutations per genome that can eliminate viral replication (indicated by discontinuous lines of mutant spectrum on the right).
6. Summary, Conclusions, and Further Comments
The range of mutation rates and frequencies exhibited by RNA viruses, with or without a functional proofreading-repair activity in their replication complexes, is compatible with the rapid development of mutant spectra that are an asset for adaptability. So great is the tendency towards diversification that, at least according to studies with HCV, long-term, monotonous replication in a cell culture environment does not deter the system from continuous exploration of sequence space via expansion of the mutant spectrum [115,116,117,118]. An unknown in viral population dynamics is the time required for a set of infected cells to produce a sufficiently diverse population of infectious particles so as to confer adaptive value to the mutant swarm. Particularly relevant is the relationship between that time and the time it takes for environmental fluctuations to impact the viral population. Although viruses are indeed efficient in adapting, the time span between stimulus and response may mark differences in the scope of adaptability.
In the present review, based on theoretical and experimental studies on the stability of genetic information, we have suggested that a proofreading-repair function of the type described for coronaviruses may have a positive impact on production of a sufficient number of viable genomes to sustain the infection in host cells and organisms. However, it appears less likely that the presence of a repair activity has a significant influence on virus diversification in an epidemiological context.
Either with repair activity or not, some important problems for disease control that arise from viral population dynamics are likely to apply to many viral pathogens. One is a sustained population disequilibrium even when a virus replicates extensively in an invariant biological environment (reviewed in [118]). The second problem is that limited attention has been paid to mutant spectrum dynamics in comparison with temporal variations of consensus sequences. This would be a minor omission if the analysis of consensus sequences had epidemiological studies as its main focus and a survey of circulating antigenic types to update the composition of antiviral vaccines. The problem arises when the information in data banks is used to prepare universal vaccines or antiviral agents. Efforts along these lines are ongoing for highly variable RNA viruses, including attempts to design pan-coronavirus vaccines. If our results with HCV apply to other variable pathogens, the degree of residue conservation in mutant spectra is far less strict than conservation as inferred from sequence alignments in data banks [119,120]. Since the target of antiviral agents are mutant spectra rather than consensus sequences (which embody limited biological information [12]), prospects of success of universal ligands or immunological stimulants such as vaccines are contingent upon minority variants remaining at low frequency due to their limited fitness. When fitness-enhancing pathways are available, universal ligands are unlikely to impede selection of escape mutants. For this reason, we have proposed expansions of sequence information in data banks by including mutant spectra sequences in addition to consensus sequences [120]. The proposal appears feasible, given the available computer power and big data methodology.
In summary, the potential of many viral pathogens to vary genetically and antigenically remains an important challenge for disease control, but means are now available to try to limit the selection of treatment-resistant viral mutants.
Author Contributions
Conceptualization, E.D. and C.P.; writing—original draft preparation, E.D. and C.P.; writing—review and editing, C.G.-C. and R.L.-V.; funding acquisition, E.D. and C.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Ministerio de Economía y Competitividad (MINECO), grant number SAF2014-52400-R, SAF2017-87846-R and BFU2017-91384-EXP from Ministerio de Ciencia, Innovación y Universidades (MCIU), PI18/00210 from Instituto de Salud Carlos III, S2013/ABI-2906 (PLATESA from Comunidad de Madrid/FEDER), and S2018/BAA-4370 (PLATESA2 from Comunidad de Madrid/FEDER). C.P. is supported by the Miguel Servet program of the Instituto de Salud Carlos III (CPII19/00001), cofinanced by the European Regional Development Fund (ERDF). CIBERehd (Centro de Investigación en Red de Enfermedades Hepáticas y Digestivas) is funded by Instituto de Salud Carlos III. Institutional grants from the Fundación Ramón Areces and Banco Santander to the CBMSO are also acknowledged. The team at CBMSO belongs to the Global Virus Network (GVN). C.G.-C. is supported by predoctoral contract PRE2018-083422 from MCIU. R. L.-V. is supported by predoctoral contract PEJD-2019-PRE/BMD-16414 from Comunidad de Madrid.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eigen, M.; Schuster, P. Stages of emerging life—five principles of early organization. J. Mol. Evol. 1982, 19, 47–61. [Google Scholar] [CrossRef]
- Eigen, M. Steps towards Life. Oxford University Press: Oxford, UK, 1992. [Google Scholar]
- Kauffman, S.A. The Origins of Order. In Self-Organization and Selection in Evolution; Oxford University Press: New York, NY, USA; Oxford, UK, 1993. [Google Scholar]
- De Duve, C. Life Evolving. Molecules, Mind and Meaning; Oxford University Press: Oxford, UK, 2002. [Google Scholar]
- Domingo, E. Virus as Populations, 2nd ed.; Academic Press, Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Szostak, J.W. The Narrow Road to the Deep Past: In Search of the Chemistry of the Origin of Life. Angew. Chem. Int. Ed. Engl. 2017, 56, 11037–11043. [Google Scholar] [CrossRef] [PubMed]
- Jalasvuori, M.; Bamford, J.K. Structural co-evolution of viruses and cells in the primordial world. Orig. Life Evol. Biosph. 2008, 38, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P. The Widespread Evolutionary Significance of Viruses. In Origin and Evolution of Viruses, 2nd ed.; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Elsevier: Oxford, UK, 2008; Volume 393–416, pp. 477–516. [Google Scholar]
- Villarreal, L.P.; Witzany, G. Rethinking quasispecies theory: From fittest type to cooperative consortia. World J. Biol. Chem. 2013, 4, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Perales, C. Viral quasispecies. PLoS Genet. 2019, 15, e1008271. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; García-Crespo, C.; Perales, C. Historical perspective on the discovery of the quasispecies concept. Annu. Rev. Virol. 2021, in press. [Google Scholar]
- Domingo, E.; Schuster, P. Quasispecies: From Theory to Experimental systems. Current Topics in Microbiology and Immunology; Springer: Berlin, Germany, 2016; Volume 392. [Google Scholar]
- Chumakov, K.; Kew, O. The poliovirus eradication iniciative. In The Picornaviruses; Ehrenfeld, E., Domingo, E., Roos, R.P., Eds.; ASM Press: Washington, DC, USA, 2010; pp. 449–459. [Google Scholar]
- Roberts, L. Polio eradication campaign loses ground. Science 2019, 365, 106–107. [Google Scholar]
- Abraham, P. Polio: The Odyssey of Eradication; C. Hurst & Co. Ltd.: London, UK, 2018. [Google Scholar]
- Estrada, L.D.; Schultz-Cherry, S. Development of a Universal Influenza Vaccine. J. Immunol. 2019, 202, 392–398. [Google Scholar] [CrossRef]
- Richman, D.D.; Wrin, T.; Little, S.J.; Petropoulos, C.J. Rapid evolution of the neutralizing antibody response to HIV type 1 infection. Proc. Natl. Acad. Sci. USA 2003, 100, 4144–4149. [Google Scholar] [CrossRef] [PubMed]
- Lopez Angel, C.J.; Tomaras, G.D. Bringing the path toward an HIV-1 vaccine into focus. PLoS Pathog. 2020, 16, e1008663. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Badua, C.; Baldo, K.A.T.; Medina, P.M.B. Genomic and proteomic mutation landscapes of SARS-CoV-2. J. Med. Virol. 2021, 93, 1702–1721. [Google Scholar] [CrossRef] [PubMed]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Escarmís, C.; Dávila, M.; Charpentier, N.; Bracho, A.; Moya, A.; Domingo, E. Genetic lesions associated with Muller’s ratchet in an RNA virus. J. Mol. Biol. 1996, 264, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Agol, V.I.; Gmyl, A.P. Emergency Services of Viral RNAs: Repair and Remodeling. Microbiol. Mol. Biol. Rev. 2018, 82, e00067-17. [Google Scholar] [CrossRef]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef]
- Mansky, L.M.; Temin, H.M. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 1995, 69, 5087–5094. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Peck, K.M.; Lauring, A.S. Complexities of Viral Mutation Rates. J. Virol. 2018, 92, e01031-17. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, T. Irregularities in genetic variation and mutation rates with environmental stresses. Environ. Microbiol. 2019, 21, 3979–3988. [Google Scholar] [CrossRef]
- Rosche, W.A.; Foster, P.L. Determining mutation rates in bacterial populations. Methods 2000, 20, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Yeo, J.Y.; Koh, D.W.; Yap, P.; Goh, G.R.; Gan, S.K. Spontaneous Mutations in HIV-1 Gag, Protease, RT p66 in the First Replication Cycle and How They Appear: Insights from an In Vitro Assay on Mutation Rates and Types. Int. J. Mol. Sci. 2020, 22, 370. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Case, J.B.; Blanc, H.; Isakov, O.; Shomron, N.; Vignuzzi, M.; Denison, M.R. Mutations in coronavirus nonstructural protein 10 decrease virus replication fidelity. J. Virol. 2015, 89, 6418–6426. [Google Scholar] [CrossRef] [PubMed]
- Stapleford, K.A.; Rozen-Gagnon, K.; Das, P.K.; Saul, S.; Poirier, E.Z.; Blanc, H.; Vidalain, P.O.; Merits, A.; Vignuzzi, M. Viral polymerase-helicase complexes regulate replication fidelity to overcome intracellular nucleotide depletion. J. Virol. 2015, 89, 11233–11244. [Google Scholar] [CrossRef] [PubMed]
- Agudo, R.; de la Higuera, I.; Arias, A.; Grande-Perez, A.; Domingo, E. Involvement of a joker mutation in a polymerase-independent lethal mutagenesis escape mechanism. Virology 2016, 494, 257–266. [Google Scholar] [CrossRef]
- Collins, N.D.; Beck, A.S.; Widen, S.G.; Wood, T.G.; Higgs, S.; Barrett, A.D.T. Structural and Nonstructural Genes Contribute to the Genetic Diversity of RNA Viruses. mBio 2018, 9, e01871-18. [Google Scholar] [CrossRef]
- Batschelet, E.; Domingo, E.; Weissmann, C. The proportion of revertant and mutant phage in a growing population, as a function of mutation and growth rate. Gene 1976, 1, 27–32. [Google Scholar] [CrossRef]
- Weissmann, C.; Taniguchi, T.; Domingo, E.; Sabo, D.; Flavell, R.A. Site-directed mutagenesis as a tool in genetics. In Genetic Manipulation as It Affects the Cancer Problem; Schultz, J., Brada, Z., Eds.; Academic Press: New York, NY, USA, 1977; pp. 11–36. [Google Scholar]
- Jones, M.E.; Thomas, S.M.; Rogers, A. Luria-Delbruck fluctuation experiments: Design and analysis. Genetics 1994, 136, 1209–1216. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.M.; Ma, C.X.; Liang, P.; Singh, K.K. Fluctuation analysis CalculatOR: A web tool for the determination of mutation rate using Luria-Delbruck fluctuation analysis. Bioinformatics 2009, 25, 1564–1565. [Google Scholar] [CrossRef]
- Nyinoh, I.W. Spontaneous mutations conferring antibiotic resistance to antitubercular drugs at a range of concentrations in Mycobacterium smegmatis. Drug Dev. Res. 2019, 80, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef]
- Wainberg, M.A.; Drosopoulos, W.C.; Salomon, H.; Hsu, M.; Borkow, G.; Parniak, M.; Gu, Z.; Song, Q.; Manne, J.; Islam, S.; et al. Enhanced fidelity of 3TC-selected mutant HIV-1 reverse transcriptase. Science 1996, 271, 1282–1285. [Google Scholar] [CrossRef] [PubMed]
- Menéndez-Arias, L. Molecular basis of fidelity of DNA synthesis and nucleotide specificity of retroviral reverse transcriptases. Prog. Nucl. Acid Res. Mol. Biol. 2002, 71, 91–147. [Google Scholar]
- Mansky, L.M.; Le Rouzic, E.; Benichou, S.; Gajary, L.C. Influence of reverse transcriptase variants, drugs, and Vpr on human immunodeficiency virus type 1 mutant frequencies. J. Virol. 2003, 77, 2071–2080. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [PubMed]
- Rezende, L.F.; Prasad, V.R. Nucleoside-analog resistance mutations in HIV-1 reverse transcriptase and their influence on polymerase fidelity and viral mutation rates. Int. J. Biochem. Cell Biol. 2004, 36, 1716–1734. [Google Scholar] [CrossRef] [PubMed]
- Vignuzzi, M.; Stone, J.K.; Arnold, J.J.; Cameron, C.E.; Andino, R. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 2006, 439, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Vignuzzi, M. Host alternation of chikungunya virus increases fitness while restricting population diversity and adaptability to novel selective pressures. J. Virol. 2011, 85, 1025–1035. [Google Scholar] [CrossRef]
- Gnadig, N.F.; Beaucourt, S.; Campagnola, G.; Borderia, A.V.; Sanz-Ramos, M.; Gong, P.; Blanc, H.; Peersen, O.B.; Vignuzzi, M. Coxsackievirus B3 mutator strains are attenuated in vivo. Proc. Natl. Acad. Sci. USA 2012, 109, E2294–E2303. [Google Scholar] [CrossRef]
- Meng, T.; Kwang, J. Attenuation of human enterovirus 71 high-replication-fidelity variants in AG129 mice. J. Virol. 2014, 88, 5803–5815. [Google Scholar] [CrossRef]
- Rozen-Gagnon, K.; Stapleford, K.A.; Mongelli, V.; Blanc, H.; Failloux, A.B.; Saleh, M.C.; Vignuzzi, M. Alphavirus mutator variants present host-specific defects and attenuation in mammalian and insect models. PLoS Pathog. 2014, 10, e1003877. [Google Scholar] [CrossRef]
- Borderia, A.V.; Rozen-Gagnon, K.; Vignuzzi, M. Fidelity Variants and RNA Quasispecies. Curr. Top Microbiol. Immunol. 2016, 392, 303–322. [Google Scholar] [PubMed]
- Lloyd, S.B.; Lichtfuss, M.; Amarasena, T.H.; Alcantara, S.; De Rose, R.; Tachedjian, G.; Alinejad-Rokny, H.; Venturi, V.; Davenport, M.P.; Winnall, W.R.; et al. High fidelity simian immunodeficiency virus reverse transcriptase mutants have impaired replication in vitro and in vivo. Virology 2016, 492, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kautz, T.F.; Guerbois, M.; Khanipov, K.; Patterson, E.I.; Langsjoen, R.M.; Yun, R.; Warmbrod, K.L.; Fofanov, Y.; Weaver, S.C.; Forrester, N.L. Low-fidelity Venezuelan equine encephalitis virus polymerase mutants to improve live-attenuated vaccine safety and efficacy. Virus Evol. 2018, 4, vey004. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.I.; Khanipov, K.; Swetnam, D.M.; Walsdorf, S.; Kautz, T.F.; Thangamani, S.; Fofanov, Y.; Forrester, N.L. Measuring Alphavirus Fidelity Using Non-Infectious Virus Particles. Viruses 2020, 12, 546. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.D.; Stokes, M.A.; Flanegan, J.B. Direct measurement of the poliovirus RNA polymerase error frequency in vitro. J. Virol. 1988, 62, 558–562. [Google Scholar] [CrossRef]
- Menendez-Arias, L. Mutation rates and intrinsic fidelity of retroviral reverse transcriptases. Viruses 2009, 1, 1137–1165. [Google Scholar] [CrossRef] [PubMed]
- Cameron, C.E.; Moustafa, I.M.; Arnold, J.J. Fidelity of Nucleotide Incorporation by the RNA-Dependent RNA Polymerase from Poliovirus. Enzymes 2016, 39, 293–323. [Google Scholar]
- Yang, X.; Liu, X.; Musser, D.M.; Moustafa, I.M.; Arnold, J.J.; Cameron, C.E.; Boehr, D.D. Triphosphate Reorientation of the Incoming Nucleotide as a Fidelity Checkpoint in Viral RNA-dependent RNA Polymerases. J. Biol. Chem. 2017, 292, 3810–3826. [Google Scholar] [CrossRef]
- Harris, V.H.; Smith, C.L.; Jonathan Cummins, W.; Hamilton, A.L.; Adams, H.; Dickman, M.; Hornby, D.P.; Williams, D.M. The effect of tautomeric constant on the specificity of nucleotide incorporation during DNA replication: Support for the rare tautomer hypothesis of substitution mutagenesis. J. Mol. Biol. 2003, 326, 1389–1401. [Google Scholar] [CrossRef]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; American Society for Microbiology: Washington, DC, USA, 2006. [Google Scholar]
- Suarez, P.; Valcarcel, J.; Ortin, J. Heterogeneity of the mutation rates of influenza A viruses: Isolation of mutator mutants. J. Virol. 1992, 66, 2491–2494. [Google Scholar] [CrossRef]
- Earl, D.J.; Deem, M.W. Evolvability is a selectable trait. Proc. Natl. Acad. Sci. USA 2004, 101, 11531–11536. [Google Scholar] [CrossRef] [PubMed]
- Lequime, S.; Fontaine, A.; Ar Gouilh, M.; Moltini-Conclois, I.; Lambrechts, L. Genetic Drift, Purifying Selection and Vector Genotype Shape Dengue Virus Intra-host Genetic Diversity in Mosquitoes. PLoS Genet. 2016, 12, e1006111. [Google Scholar] [CrossRef]
- Kadoya, S.S.; Urayama, S.I.; Nunoura, T.; Hirai, M.; Takaki, Y.; Kitajima, M.; Nakagomi, T.; Nakagomi, O.; Okabe, S.; Nishimura, O.; et al. Bottleneck Size-Dependent Changes in the Genetic Diversity and Specific Growth Rate of a Rotavirus A Strain. J. Virol. 2020, 94, e02083-19. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Orgel, L.E. A nonenzymatic RNA polymerase model. Science 1983, 219, 859–862. [Google Scholar] [CrossRef]
- Kun, A.; Szilagyi, A.; Konnyu, B.; Boza, G.; Zachar, I.; Szathmary, E. The dynamics of the RNA world: Insights and challenges. Ann. N. Y. Acad. Sci. 2015, 1341, 75–95. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. Comparative rates of spontaneous mutation. Nature 1969, 221, 1132. [Google Scholar] [CrossRef]
- Drake, J.W. The Molecular Basis of Mutation; Holden-Day: San Francisco, CA, USA, 1970. [Google Scholar]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, I.; Kurokawa, M.; Liu, L.; Ying, B.W. Coordinated Changes in Mutation and Growth Rates Induced by Genome Reduction. mBio 2017, 8, e00676-17. [Google Scholar] [CrossRef]
- Eigen, M.; Schuster, P. The Hypercycle. A Principle of Natural Self-Organization; Springer: Berlin, Germany, 1979. [Google Scholar]
- Swetina, J.; Schuster, P. Self-replication with errors. A model for polynucleotide replication. Biophys. Chem. 1982, 16, 329–345. [Google Scholar] [CrossRef]
- Schuster, P. Quasispecies on fitness landscapes. Curr. Top. Microbiol. Immunol. 2016, 392, 61–120. [Google Scholar]
- Spivak, G. Nucleotide excision repair in humans. DNA Repair 2015, 36, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, J.; Bowerman, S.; Luger, K. Quantitating repair protein accumulation at DNA lesions: Past, present, and future. DNA Repair 2019, 81, 102650. [Google Scholar] [CrossRef]
- Liu, D.; Keijzers, G.; Rasmussen, L.J. DNA mismatch repair and its many roles in eukaryotic cells. Mutat. Res. 2017, 773, 174–187. [Google Scholar] [CrossRef] [PubMed]
- Reha-Krantz, L.J.; Marquez, L.A.; Elisseeva, E.; Baker, R.P.; Bloom, L.B.; Dunford, H.B.; Goodman, M.F. The proofreading pathway of bacteriophage T4 DNA polymerase. J. Biol. Chem. 1998, 273, 22969–22976. [Google Scholar] [CrossRef]
- Reha-Krantz, L.J. Regulation of DNA polymerase exonucleolytic proofreading activity: Studies of bacteriophage T4 “antimutator” DNA polymerases. Genetics 1998, 148, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.F.; Fygenson, K.D. DNA polymerase fidelity: From genetics toward a biochemical understanding. Genetics 1998, 148, 1475–1482. [Google Scholar] [CrossRef]
- Knopf, C.W. Evolution of viral DNA-dependent DNA polymerases. Virus Genes 1998, 16, 47–58. [Google Scholar] [CrossRef]
- Gammon, D.B.; Evans, D.H. The 3′-to-5′ exonuclease activity of vaccinia virus DNA polymerase is essential and plays a role in promoting virus genetic recombination. J. Virol. 2009, 83, 4236–4250. [Google Scholar] [CrossRef] [PubMed]
- Czarnecki, M.W.; Traktman, P. The vaccinia virus DNA polymerase and its processivity factor. Virus Res. 2017, 234, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J.L.; Mukherjee, P.; Coen, D.M. Herpes Simplex Virus 1 DNA Polymerase RNase H Activity Acts in a 3′-to-5′ Direction and Is Dependent on the 3′-to-5′ Exonuclease Active Site. J. Virol. 2018, 92, e01813-17. [Google Scholar] [CrossRef]
- Ishihama, A.; Mizumoto, K.; Kawakami, K.; Kato, A.; Honda, A. Proofreading function associated with the RNA-dependent RNA polymerase from influenza virus. J. Biol. Chem. 1986, 261, 10417–10421. [Google Scholar] [CrossRef]
- Jin, Z.; Leveque, V.; Ma, H.; Johnson, K.A.; Klumpp, K. NTP-mediated nucleotide excision activity of hepatitis C virus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2013, 110, E348–E357. [Google Scholar] [CrossRef]
- Meyer, P.R.; Matsuura, S.E.; So, A.G.; Scott, W.A. Unblocking of chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. USA 1998, 95, 13471–13476. [Google Scholar] [CrossRef]
- Kharytonchyk, S.; King, S.R.; Ndongmo, C.B.; Stilger, K.L.; An, W.; Telesnitsky, A. Resolution of Specific Nucleotide Mismatches by Wild-Type and AZT-Resistant Reverse Transcriptases during HIV-1 Replication. J. Mol. Biol. 2016, 428, 2275–2288. [Google Scholar] [CrossRef]
- Nagy, P.D.; Carpenter, C.D.; Simon, A.E. A novel 3′-end repair mechanism in an RNA virus. Proc. Natl. Acad. Sci. USA 1997, 94, 1113–1118. [Google Scholar] [CrossRef]
- Kwon, S.J.; Chaturvedi, S.; Rao, A.L. Repair of the 3′ proximal and internal deletions of a satellite RNA associated with Cucumber mosaic virus is directed toward restoring structural integrity. Virology 2014, 450–451, 222–232. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Steinhauer, D.A.; Domingo, E.; Holland, J.J. Lack of evidence for proofreading mechanisms associated with an RNA virus polymerase. Gene 1992, 122, 281–288. [Google Scholar] [CrossRef]
- Minskaia, E.; Hertzig, T.; Gorbalenya, A.E.; Campanacci, V.; Cambillau, C.; Canard, B.; Ziebuhr, J. Discovery of an RNA virus 3’->5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5108–5113. [Google Scholar] [CrossRef]
- Ogando, N.S.; Zevenhoven-Dobbe, J.C.; van der Meer, Y.; Bredenbeek, P.J.; Posthuma, C.C.; Snijder, E.J. The Enzymatic Activity of the nsp14 Exoribonuclease Is Critical for Replication of MERS-CoV and SARS-CoV-2. J. Virol. 2020, 94, e01246-20. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Chen, H.; Chen, Z.; Yang, F.; Ye, F.; Zheng, Y.; Yang, J.; Lin, X.; Sun, H.; Wang, L.; et al. Crystal structure of SARS-CoV-2 nsp10 bound to nsp14-ExoN domain reveals an exoribonuclease with both structural and functional integrity. Nucleic Acids Res. 2021, 49, 5382–5392. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Pourfarjam, Y.; Kim, I.K. Reconstitution and functional characterization of SARS-CoV-2 proofreading complex. Protein Expr. Purif. 2021, 185, 105894. [Google Scholar] [CrossRef]
- Scholle, M.D.; Liu, C.; Deval, J.; Gurard-Levin, Z.A. Label-Free Screening of SARS-CoV-2 NSP14 Exonuclease Activity Using SAMDI Mass Spectrometry. SLAS Discov. 2021, 26, 766–774. [Google Scholar]
- Eckerle, L.D.; Becker, M.M.; Halpin, R.A.; Li, K.; Venter, E.; Lu, X.; Scherbakova, S.; Graham, R.L.; Baric, R.S.; Stockwell, T.B.; et al. Infidelity of SARS-CoV Nsp14-exonuclease mutant virus replication is revealed by complete genome sequencing. PLoS Pathog. 2010, 6, e1000896. [Google Scholar] [CrossRef]
- Eckerle, L.D.; Lu, X.; Sperry, S.M.; Choi, L.; Denison, M.R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 2007, 81, 12135–12144. [Google Scholar] [CrossRef]
- Graham, R.L.; Becker, M.M.; Eckerle, L.D.; Bolles, M.; Denison, M.R.; Baric, R.S. A live, impaired-fidelity coronavirus vaccine protects in an aged, immunocompromised mouse model of lethal disease. Nat. Med. 2012, 18, 1820–1826. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.C.; Blanc, H.; Vignuzzi, M.; Denison, M.R. Coronaviruses lacking exoribonuclease activity are susceptible to lethal mutagenesis: Evidence for proofreading and potential therapeutics. PLoS Pathog. 2013, 9, e1003565. [Google Scholar] [CrossRef]
- Smith, E.C.; Denison, M.R. Coronaviruses as DNA wannabes: A new model for the regulation of RNA virus replication fidelity. PLoS Pathog. 2013, 9, e1003760. [Google Scholar] [CrossRef]
- Graepel, K.W.; Lu, X.; Case, J.B.; Sexton, N.R.; Smith, E.C.; Denison, M.R. Proofreading-Deficient Coronaviruses Adapt for Increased Fitness over Long-Term Passage without Reversion of Exoribonuclease-Inactivating Mutations. mBio 2017, 8, e01503-17. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Mallory, J.D.; Mallory, X.F.; Kolomeisky, A.B.; Igoshin, O.A. Theoretical Analysis Reveals the Cost and Benefit of Proofreading in Coronavirus Genome Replication. J. Phys. Chem. Lett. 2021, 12, 2691–2698. [Google Scholar] [CrossRef] [PubMed]
- Ellefson, J.W.; Gollihar, J.; Shroff, R.; Shivram, H.; Iyer, V.R.; Ellington, A.D. Synthetic evolutionary origin of a proofreading reverse transcriptase. Science 2016, 352, 1590–1593. [Google Scholar] [CrossRef] [PubMed]
- Trypsteen, W.; Van Cleemput, J.; Snippenberg, W.V.; Gerlo, S.; Vandekerckhove, L. On the whereabouts of SARS-CoV-2 in the human body: A systematic review. PLoS Pathog. 2020, 16, e1009037. [Google Scholar] [CrossRef]
- Sender, R.; Bar-On, Y.M.; Gleizer, S.; Bernshtein, B.; Flamholz, A.; Phillips, R.; Milo, R. The total number and mass of SARS-CoV-2 virions. Proc. Natl. Acad. Sci. USA 2021, 118, e2024815118. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.A.; Read, A.F. Monitor for COVID-19 vaccine resistance evolution during clinical trials. PLoS Biol. 2020, 18, e3001000. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef]
- Hacisuleyman, E.; Hale, C.; Saito, Y.; Blachere, N.E.; Bergh, M.; Conlon, E.G.; Schaefer-Babajew, D.J.; DaSilva, J.; Muecksch, F.; Gaebler, C.; et al. Vaccine Breakthrough Infections with SARS-CoV-2 Variants. N. Engl. J. Med. 2021, 384, 2212–2218. [Google Scholar] [CrossRef] [PubMed]
- Rolland, M.; Gilbert, P.B. Sieve analysis to understand how SARS-CoV-2 diversity can impact vaccine protection. PLoS Pathog. 2021, 17, e1009406. [Google Scholar] [CrossRef]
- Domingo, E. RNA virus evolution and the control of viral disease. Prog. Drug Res. 1989, 33, 93–133. [Google Scholar]
- Moreno, E.; Gallego, I.; Gregori, J.; Lucia-Sanz, A.; Soria, M.E.; Castro, V.; Beach, N.M.; Manrubia, S.; Quer, J.; Esteban, J.I.; et al. Internal Disequilibria and Phenotypic Diversification during Replication of Hepatitis C Virus in a Noncoevolving Cellular Environment. J. Virol. 2017, 91, e02505-16. [Google Scholar] [CrossRef] [PubMed]
- Gallego, I.; Soria, M.E.; Garcia-Crespo, C.; Chen, Q.; Martinez-Barragan, P.; Khalfaoui, S.; Martinez-Gonzalez, B.; Sanchez-Martin, I.; Palacios-Blanco, I.; de Avila, A.I.; et al. Broad and Dynamic Diversification of Infectious Hepatitis C Virus in a Cell Culture Environment. J. Virol. 2020, 94, e01856-19. [Google Scholar] [CrossRef]
- Domingo, E.; Soria, M.E.; Gallego, I.; de Avila, A.I.; Garcia-Crespo, C.; Martinez-Gonzalez, B.; Gomez, J.; Briones, C.; Gregori, J.; Quer, J.; et al. A new implication of quasispecies dynamics: Broad virus diversification in absence of external perturbations. Infect. Genet. Evol. 2020, 82, 104278. [Google Scholar] [CrossRef] [PubMed]
- García-Crespo, C.; Gallego, I.; Soria, M.E.; De Ávila, A.I.; Martínez-González, B.; Vázquez-Sirvent, L.; Lobo-Vega, R.; Moreno, E.; Gómez, J.; Briones, C.; et al. Population disequilibrium as promoter of adaptive explorations in hepatitis C virus. Viruses 2021, 13, 616. [Google Scholar] [CrossRef] [PubMed]
- Delgado, S.; Perales, C.; García-Crespo, C.; Soria, M.E.; Gallego, I.; De Ávila, A.I.; Martínez-González, B.; Vázquez-Sirvent, L.; López-Galíndez, C.; Morán, F.; et al. A Two-Level, Dynamic Fitness Landscape of Hepatitis C Virus Revealed by Self-Organized Haplotype Maps. 2021. Available online: https://www.biorxiv.org/content/10.1101/2021.04.22.441053v1 (accessed on 24 April 2021).
- Garcia-Crespo, C.; Soria, M.E.; Gallego, I.; Avila, A.I.; Martinez-Gonzalez, B.; Vazquez-Sirvent, L.; Gomez, J.; Briones, C.; Gregori, J.; Quer, J.; et al. Dissimilar Conservation Pattern in Hepatitis C Virus Mutant Spectra, Consensus Sequences, and Data Banks. J. Clin. Med. 2020, 9, 3450. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).