
Peptide Platform as a Powerful Tool in the Fight against COVID-19



 ,
,  and
and 
Abstract
:1. Introduction
- In this paper, we have demonstrated the applicability of a peptide-based strategy, focused both to a preventive as well as a therapeutic purpose.
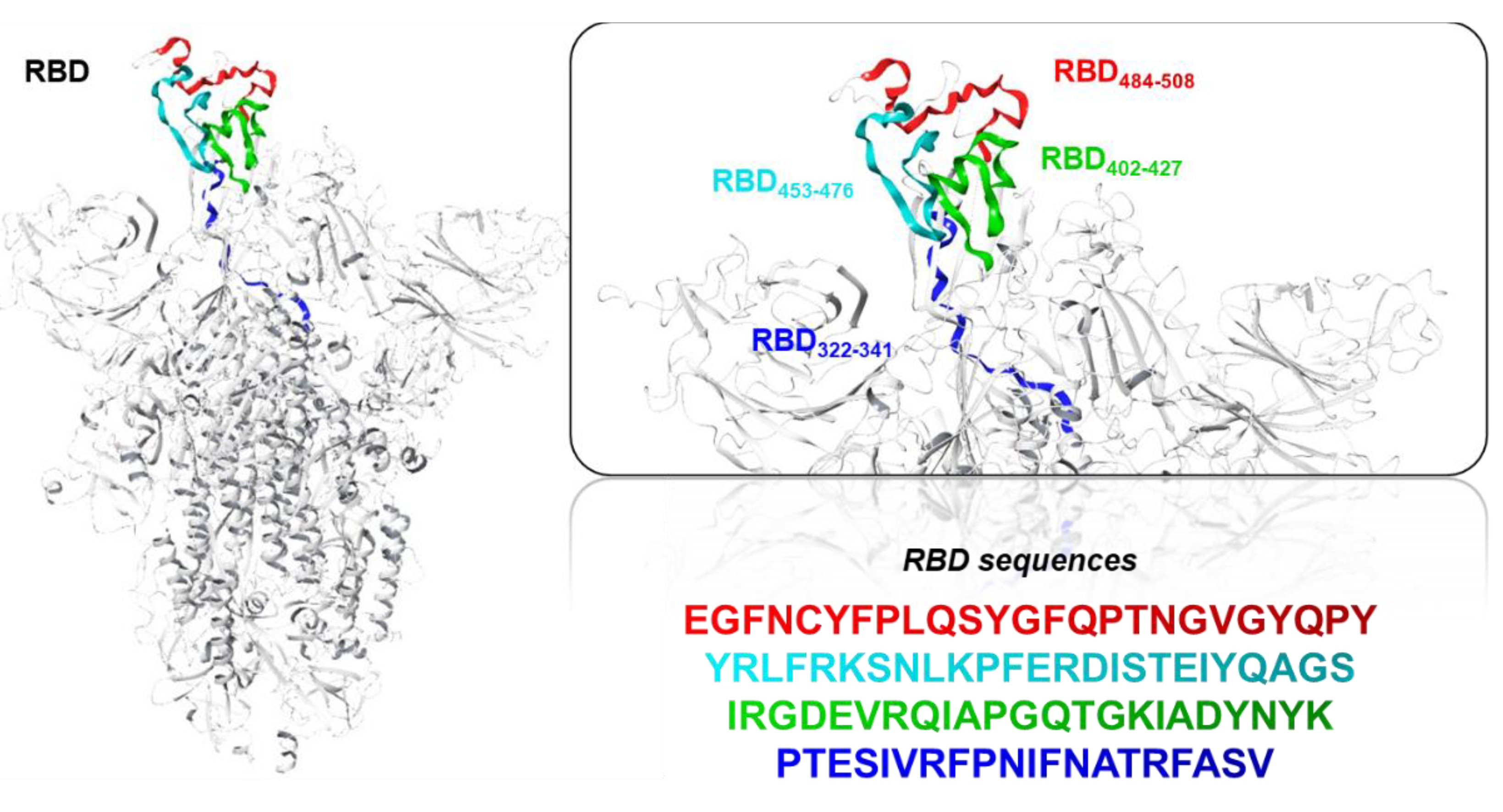
- Specifically, we have selected and synthetized peptide sequences located within the RBD conserved region [25]. Four 25-residue sequences (comprising amino-acids 322–508), were predicted as highly potential antiviral peptides (T-cell epitopes) to impede the pathogenic process of SARS-CoV-2 through immunoinformatics methodologies. Then, we administered the peptides in vivo in mice in order to stimulate the production of IgG antibodies specific against the regions of interest, and sera have been tested to evaluate their neutralizing activity.
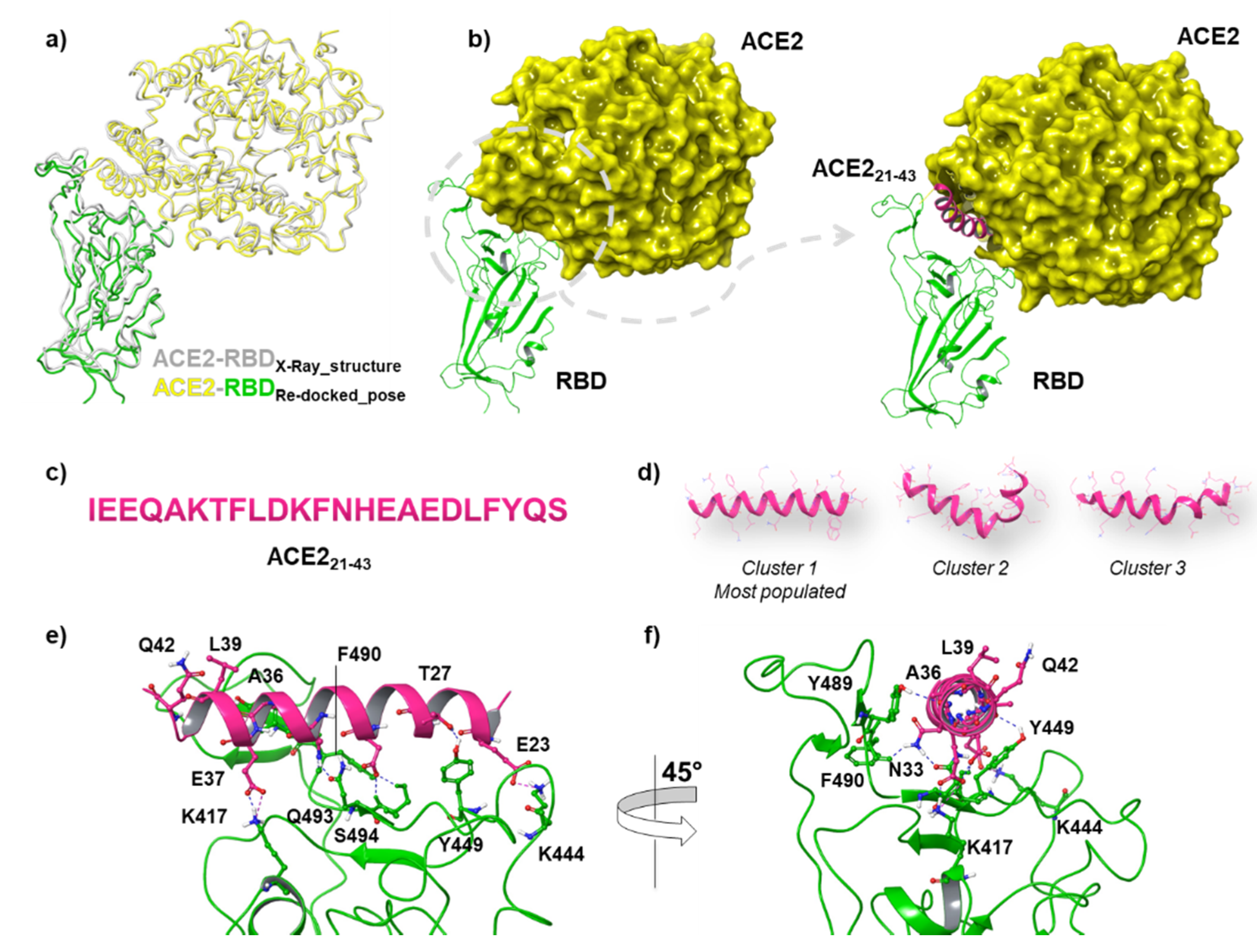
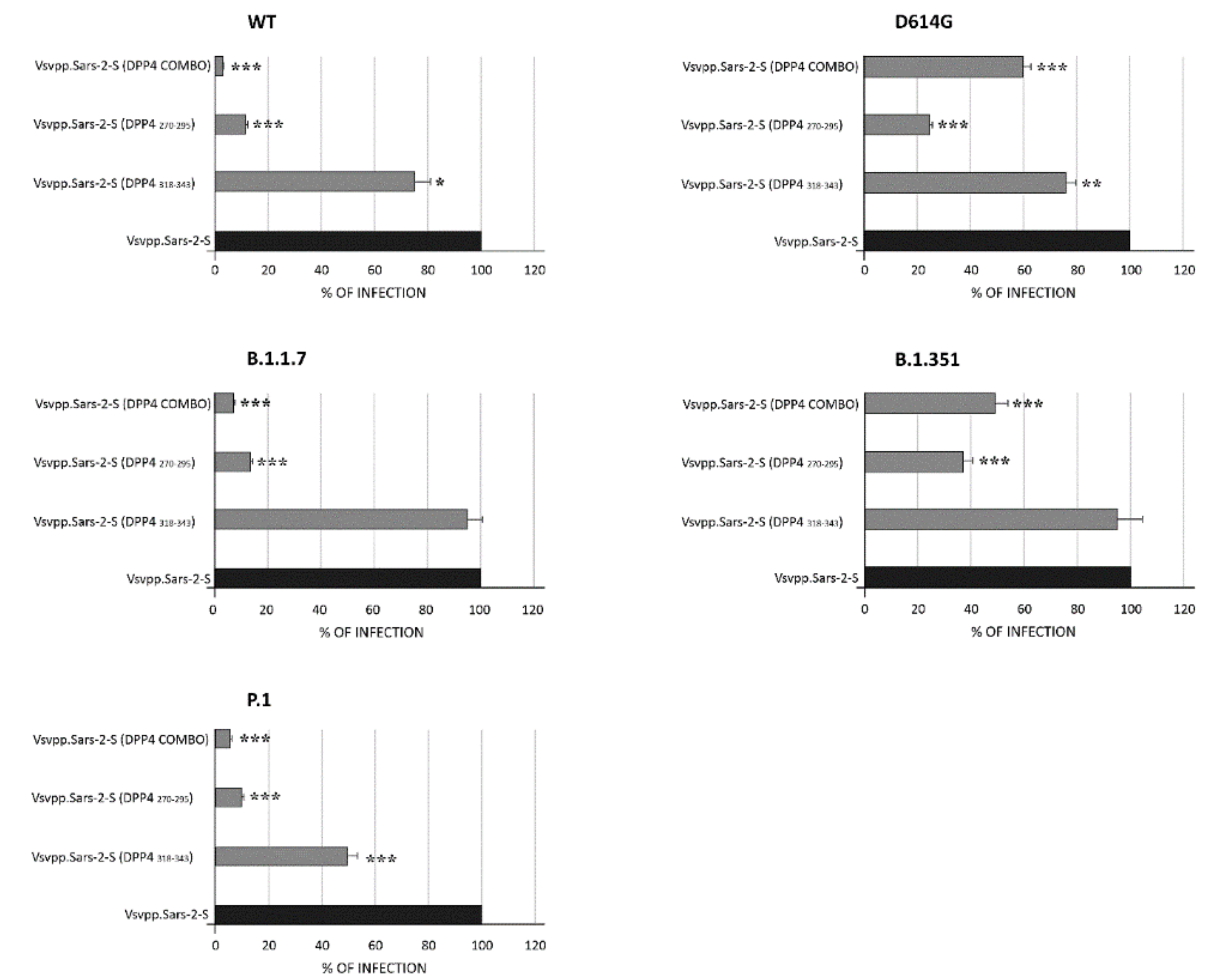
- In parallel, based on the established involvement of the DPP4 receptor, a computational approach helped to rationalize the ability of two peptides to bind the S protein by carrying out a protein–protein docking analysis in order to investigate the binding affinity for RBD. Afterwards, the peptides bearing DPP4 sequences have been produced and used in vitro to assess their capacity to block the VSV* ∆G-Fluc pseudovirus infection.
2. Materials and Methods
2.1. Peptide Design and Production
2.2. Protein Preparation
2.3. Preparation of Peptides and Molecular Dynamic Simulations (MDs)
2.4. Protein–Protein Binding Affinity Prediction
2.5. T-Cell Epitope Identification
2.6. Cells
2.7. Preparation of Pseudotyped Particles
2.8. Serum Anti-Peptide Production
2.9. Determination of Anti-SARS-CoV-2 S-Protein RBD IgG Antibody Circulating Levels
2.10. In Vitro Neutralization Assay of DPP4 Peptide
2.11. Quantification and Statistical Analysis
3. Results
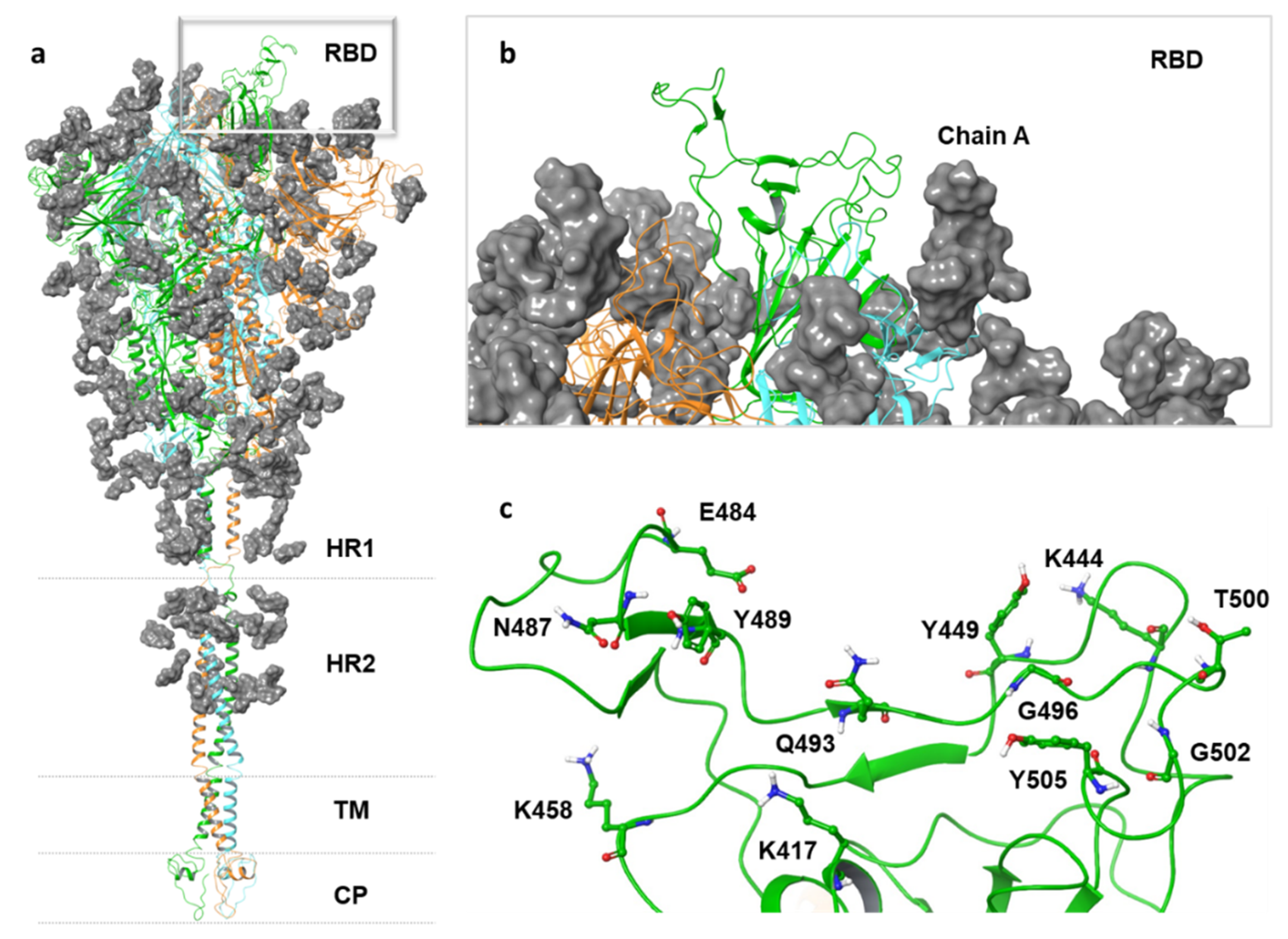
3.1. Structural Comparison among DPP4 Peptides Docked into RBD Binding Pocket
3.2. DPP4 Peptides Block SARS-CoV-2 Entry into Cells
3.3. T Cell Epitope Identification
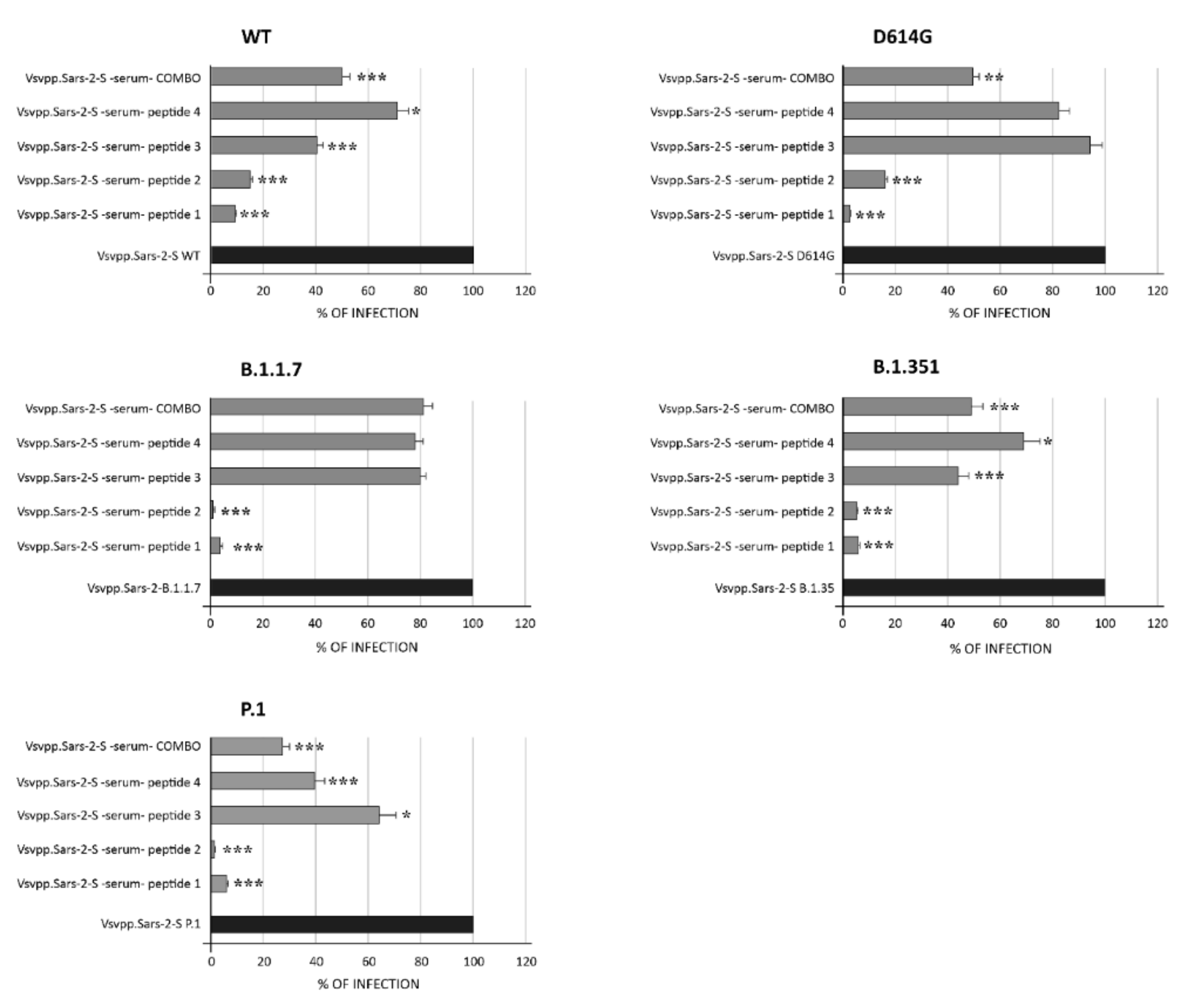
3.4. Murine Humoral Response and Neutralizing Activity of IgG Elicited by RBD Peptide
4. Discussion
- Thus, our results indicate the potential use of the peptide platform for the production of small peptides able to inhibit early virus entry into host cells.
- On the other side, this platform allows producing specific antibodies (polyclonal and monoclonal) against the S protein of SARS-CoV-2 [68,69]. In particular, using peptides focused on the conserved RBD region, the antibody produced will be effective for preventing current and future variants that ineluctably will arise during pandemics.
- Future in vivo experiments will be performed to immunize transgenic animals (human ACE2 protein expression in the lung) or hamster models [70] before the administration of isovirus carrying the known different variants. Animal experiments make it possible to identify the dose, timing, and method of administration of the vaccine and also to evaluate any unwanted pathological effects.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell. Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [Green Version]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. Plos Pathogen 2018, 14, e1007236. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K.Y. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Werner Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hornich, B.F.; Hahn, A.S.; Kruger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393. [Google Scholar] [CrossRef]
- Kidd, M.; Richter, A.; Best, A.; Cumley, N.; Mirza, J.; Percival, B.; Mayhew, M.; Megram, O.; Ashford, F.; Thomas White, T.; et al. S-Variant SARS-CoV-2 Lineage B1.1.7 is Associated with significantly higher viral load in Samples Tested by TaqPath Polymerase Chain Reaction. J. Infect. Dis. 2021, 223, 1666–1670. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.; Shum, M.H.; Leung, G.M.; Lam, T.T.; Wu, J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill. 2021, 26, 2002106. [Google Scholar] [CrossRef]
- Mwenda, M.; Saasa, N.; Sinyange, N.; Busby, G.; Chipimo, P.J.; Hendry, J.; Kapona, O.; Yingst, S.; Hines, J.Z.; Minchella, P.; et al. Detection of B.1.351 SARS-CoV-2 Variant Strain-Zambia, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 280–282. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Francisco, R., Jr.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; de C Guimarães, A.P.; Antunes, A.K.E.; et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef] [PubMed]
- Shanga, J.; Wana, Y.; Luoa, C.; Yea, G.; Genga, Q.; Auerbacha, A.; Lia, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Tannock, G.A.; Kim, H.; Xue, L. Why are vaccines against many human viral diseases still unavailable; an historic perspective? J. Med. Virol. 2020, 92, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Schütz, D.; Ruiz-Blanco, Y.B.; Münch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Müller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Delivery Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Heydari, H.; Golmohammadi, R.; Mirnejad, R.; Tebyanian, H.; Fasihi-Ramandi, M.; Moghaddam, M.M. Antiviral peptides against Coronaviridae family: A review. Peptides 2021, 139, 170526. [Google Scholar] [CrossRef]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 1700–2707. [Google Scholar] [CrossRef]
- Han, Y.; Petr Král, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Li, J.; Du, L.; Yan, X.; Hu, G.; Zhou, Y.; Jiang, S. Identification and characterization of novel neutralizing epitopes in the receptor-binding domain of SARS-CoV spike protein: Revealing the critical antigenic determinants in inactivated SARS-CoV vaccine. Vaccine 2006, 24, 5498–5508. [Google Scholar] [CrossRef] [PubMed]
- Tripet, B.; Howard, M.W.; Jobling, M.; Holmes, R.K.; Holmes, K.V.; Hodges, R.S. Structural characterization of the SARS-coronavirus spike S fusion protein core. J. Biol. Chem. 2004, 279, 20836–20849. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [Green Version]
- Holmes, K.V. SARS coronavirus: A new challenge for prevention and therapy. J. Clin. Invest. 2003, 111, 1605–1609. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.; Hasan, A.; Babadaei, M.M.N.; Bloukh, S.H.; Chowdhury, M.E.H.; Sharifi, M.; Haghighat, S.; Falahati, M. Targeting SARS-CoV2 Spike Protein Receptor Binding Domain by Therapeutic Antibodies. Biomed. Pharmacother. 2020, 130, 110559. [Google Scholar] [CrossRef]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Ye Cui, Y.; Xi Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-em structure of the 2019-ncov spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.; Park, S.J.; Choi, Y.K.; Park, T.; Tanveer, M.; Cao, Y.; Kern, N.R.; Lee, J.; Yeom, M.S.; Croll, T.I.; et al. Developing a fully glycosylated full-length sars-cov-2 spike protein model in a viral membrane. J. Phys. Chem. 2020, 124, 7128–7137. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM Structures of SARS-CoV-2 Spike without and with ACE2 Reveal a pH-Dependent Switch to Mediate Endosomal Positioning of Receptor-Binding Domains. Cell Host Microbe 2020, 28, 867–879. [Google Scholar] [CrossRef] [PubMed]
- RCSB Protein Data Bank. Available online: https://www.rcsb.org/structure/2G63 (accessed on 22 August 2021).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Pall, S.; Smith, J.C.; Hess, B.; Lindah, E. GROMACS: High performance molecular simulations through multi-levelparallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Sapay, N.; Tieleman, D.P. Combination of the CHARMM27 force field with united-atom lipid force fields. J. Comput. Chem. 2011, 32, 1400–1410. [Google Scholar] [CrossRef] [PubMed]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; Van Gunsteren, W.F.; Mark, A.E. Peptide folding: When simulation meets experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Vangone, A.; Bonvin, A.M.J.J. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4, e074554. [Google Scholar] [CrossRef]
- Xue, L.; Rodrigues, J.; Kastritis, P.; Bonvin, A.M.J.J.; Vangone, A. PRODIGY: A web-server for predicting the binding affinity in protein-protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Schrödinger Release: Maestro, version 11.5.011, Schrödinger, LLC, NY 2018–1. Available online: https://www.schrodinger.com/products/maestro (accessed on 3 May 2021).
- Sidney, J.; Assarsson, E.; Moore, C.; Ngo, S.; Pinilla, C.; Sette, A.; Peters, B. Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res. 2008, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Lundegaard, C.; Lamberth, K.; Buus, S.; Lund, O.; Nielsen, M. Large Scale validation of methods for cytotoxic T-Lymphocyte epitope prediction. BMC Bioinf. 2007, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Backert, L.; Kohlbacher, O. Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med. 2015, 7, 119. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhang, J.; Li, S.; Sun, J.; Teng, Y.; Wu, M.; Li, J.; Li, Y.; Hu, N.; Wang, H.; et al. Epitope-Based Vaccine Target Screening against Highly Pathogenic MERS-CoV: An In Silico Approach Applied to Emerging Infectious Diseases. PLoS ONE 2015, 10, e0144475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 2020, 6, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Tan, X.; Loas, A.; Pentelute, B.L. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. ACS Cent. Sci. 2020, 6, 1722–1734, Preprint. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Lesk, A.M.; Janin, J.; Chothia, C. The accessible surface area and stability of oligomeric proteins. Nature 1987, 328, 834–836. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Protein-protein interactions: A review of protein dimer structures. Prog. Biophys. Mol. Biol. 1995, 63, 31–65. [Google Scholar] [CrossRef]
- Nilofer, C.; Sukhwal, A.; Mohanapriya, A.; Kangueane, P. Protein-protein interfaces are vdW dominant with selective H-bonds and (or) electrostatics towards broad functional specificity. Bioinformation 2017, 13, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastritis, P.L.; Bonvin, A.M.J.J. Are scoring functions in protein-protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J. Proteome Res. 2010, 9, 2216–2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.C.L.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Stanfield, R.L.; Wilson, I.A. Protein-peptide interactions. Curr Opin Struct Biol. 1995, 5, 103–113. [Google Scholar] [CrossRef]
- Vanhee, M.; Delputte, P.; Delrue, I.; Geldhof., M.; Nauwynck, H. Development of an experimental inactivated PRRSV vaccine that induces virus-neutralizing antibodies. Vet. Res 2009, 40, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A protein-peptide docking tool based on interaction similarity and energy optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [Green Version]
- Neduva, V.; Russell, R.B. Peptides mediating interaction networks: New leads at last. Curr Opin Biotechnol. 2006, 17, 465–471. [Google Scholar] [CrossRef]
- De Vries, S.J.; Rey, J.; Schindler, C.E.M.; Zacharias, M.; Tuffery, P. The pepATTRACT web server for blind, large-scale peptide–protein docking. Nucl. Acids Res. [CrossRef] [PubMed]
- Angeletti, D.; Yeweil, J.W. Understanding and manipulating viral immunity: Antibody immunodominance enters center stage. Trends in immunology 2018, 39, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Dale, G.A.; Shartouny, J.R.; Jacob, J. Quantifying the shifting landscape of B cell immunodominance. Nat. Immunol. 2017, 18, 367–368. [Google Scholar] [CrossRef]
- Angeletti, D.; Gibbs, J.S.; Angel, M.; Kosik, I.; Hickman, H.D.; Frank, G.M.; Das, S.R.; Wheatley, A.K.; Prabhakaran, M.; Leggat, D.J.; et al. Defining B cell immunodominance to viruses. Nat. Immunol. 2017, 18, 456–463. [Google Scholar] [CrossRef]
- Cirelli, K.M.; Carnathan, D.G.; Nogal, B.; Martin, J.T.; Rodriguez, O.L.; Upadhyay, A.A.; Enemuo, C.A.; Gebru, E.H.; Choe, Y.; Viviano, F.; et al. Slow Delivery Immunization Enhances HIV Neutralizing Antibody and Germinal Center Responses via Modulation of Immunodominance. Cell 2019, 177, 1153–1171.e28. [Google Scholar] [CrossRef]
- Qiu, T.; Mao, T.; Wang, Y.; Zhou, M.; Qiu, J.; Wang, J.; Xu, J.; Cao, Z. Identification of potential cross-protective epitope between a new type of coronavirus (2019-nCoV) and severe acute respiratory syndrome virus. J. Genet. Genom. 2020, 47, 115–117. [Google Scholar] [CrossRef] [PubMed]
- Xin-Meng Wang, X.M.; Wu, X.W.; Zhao, X.Y.; Wang, C.W.; Jiang-Ning Zhou, J.N. Exposure-time-dependent subcellular staging of gold nanoparticles deposition and vesicle destruction in mice livers. Nanom. Nanotechnol. Biol. Med. 2021, 34, 102393. [Google Scholar] [CrossRef]
- Farfán-Castro, S.; García-Sotoa, M.J.; Comas-García, M.; Arévalo-Villalobos, J.I.; Palestinoa, G.; ù González-Ortega, O.; Rosales-Mendoza, S. Synthesis and immunogenicity assessment of a gold nanoparticle conjugate for the delivery of a peptide from SARS-CoV-2. Nanom. Nanotechnol. Biol. Med. 2021, 31, 102372. [Google Scholar] [CrossRef]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.W.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.A.; Zaki, A.; Fouchier, R.A.M.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Chen, P.; Wang, J.; Feng, J.; Zhou, H.; Li, X.; Zhong, W.; Hao, P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020, 63, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Vankadari, N.; Wilce, J.A. Emerging WuHan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef] [PubMed]
- Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Prokscha, A.; Naim, H.Y.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Polymorphisms in dipeptidyl peptidase 4 reduce host cell entry of Middle East respiratory syndrome coronavirus. Emerg. Microbes Infect. 2020, 9, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Huang, J.; Jayathilaka, L.P.; Jenny Lee, J.; Lee, B.; Gupta, S. Antibody Production with Synthetic Peptides. Methods Mol. Biol. 2016, 1474, 25–47. [Google Scholar] [CrossRef]
- Miersch, S.; Ustav, M.; Li, Z.; Case, J.; Ganaie, S.; Matusali, G.; Colavita, F.; Lapa, D.; Capobianchi, M.R. Synthetic antibodies neutralize SARS-CoV-2 infection of mammalian cells. BioRxiv 2020. Preprint. [Google Scholar] [CrossRef]
- Longlong Si, L.; Bai, H.; Rodas, M.; Cao, W.; Oh, C.Y.; Jiang, A.; Moller, R. A human-airway-on-a-chip for the rapid identification of candidate antiviral therapeutics and prophylactics. Nat. Biomed. Eng. 2021. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | Sequence | |
|---|---|---|
| ACE2 | 21–45 | IEEQAKTFLDKFNHEAEDLFYQS |
| DPP4 | 270–295 | VVNTDSLSSVTNATSIQITAPASMLI |
| 318–343 | RIQNYSVMDICDYDESSGRWNCLVAR | |
| SPIKE | ||
| 1 | 484–508 | EGFNCYFPLQSYGFQPTNGVGYQPY |
| 2 | 453–476 | YRLFRKSNLKPFERDISTEIYQAGS |
| 3 | 402–427 | IRGDEVRQIAPGQTGKIADYNYK |
| 4 | 322–341 | PTESIVRFPNIFNATRFASV |
| HADDOCK Score * | Cluster Size | RMSD ** | VdW Energy | Electrostatic Energy | Desolvation Energy | BSA *** | Z-Score **** | |
|---|---|---|---|---|---|---|---|---|
| DPP4270-295 | −98.1 +/− 2.9 | 53 | 0.2 +/− 0.2 | −59.6 +/− 2.8 | −108.5 +/− 15.9 | −17.8 +/− 2.8 | 1441.4 +/− 68.1 | −2.5 |
| DPP4318-343 | −90.5 +/− 4.2 | 10 | 2.2 +/− 0.1 | −48.4 +/− 8.5 | −127.4 +/− 16.5 | −18.1 +/− 2.6 | 1270.0 +/− 23.4 | −1.3 |
| DPP4270–295 | DPP4318–343 | |||
|---|---|---|---|---|
| Temperature (°C) | Binding affinity ΔG (kcal mol−1) | Dissociation constant Kd (M) | Binding affinity ΔG (kcal mol−1) | Dissociation constant Kd (M) |
| 37° | −11.8 | 4.5 × 10−9 | −9.0 | 4.8 × 10−7 |
| HADDOCK Score * | Cluster Size | RMSD ** | VdW Energy | Electrostatic Energy | Desolvation Energy | BSA *** | Z-Score **** | |
|---|---|---|---|---|---|---|---|---|
| B.1.1.7 DPP4270–295 | −89.5 +/− 1.9 | 24 | 0.4 +/− 0.2 | −55.7 +/− 3.0 | −94.8 +/− 12.1 | −15.1 +/− 2.2 | 1424.0 +/− 107.7 | −2.4 |
| B.1.351 DPP4270–295 | −84.3 +/− 5.6 | 26 | 2.2 +/− 0.0 | −50.7 +/− 7.7 | −85.2 +/− 18.5 | −18.9 +/− 2.5 | 1338.5 +/− 69.0 | −2.2 |
| P.1 DPP4270–295 | −95.3 +/− 2.4 | 52 | 0.4 +/− 0.2 | −62.0 +/− 2.1 | −115.8 +/− 26.7 | −10.3 +/− 2.3 | 1485.9 +/− 65.5 | −2.6 |
| Allele | Spike Sequences | Start | End | Length | Peptide | Predicted IC50 | Rank |
|---|---|---|---|---|---|---|---|
| H-2-Dd | 1 | 6 | 14 | 9 | YFPLQSYGF | 0.000033 | 0.8 |
| H-2-Dd | 1 | 1 | 9 | 9 | EGFNCYFPL | 0.000035 | 0.8 |
| H-2-Kd | 2 | 1 | 9 | 9 | YRLFRKSNL | 0.000042 | 1.6 |
| H-2-Kd | 1 | 7 | 15 | 9 | FPLQSYGFQ | 0.000050 | 1.9 |
| H-2-Kd | 4 | 11 | 19 | 9 | IFNATRFAS | 0.000065 | 2.3 |
| H-2-Dd | 4 | 7 | 15 | 9 | RFPNIFNAT | 0.000104 | 2.6 |
| H-2-Kd | 2 | 12 | 20 | 9 | FERDISTEI | 0.000121 | 3.8 |
| H-2-Dd | 4 | 12 | 20 | 9 | FNATRFASV | 0.000129 | 3.2 |
| H-2-Kd | 1 | 5 | 13 | 9 | CYFPLQSYG | 0.000134 | 4.2 |
| H-2-Kd | 1 | 11 | 19 | 9 | SYGFQPTNG | 0.000158 | 4.9 |
| H-2-Kd | 1 | 12 | 20 | 9 | YGFQPTNGV | 0.000197 | 6 |
| H-2-Dd | 1 | 12 | 20 | 9 | YGFQPTNGV | 0.000206 | 5 |
| H-2-Dd | 4 | 11 | 19 | 9 | IFNATRFAS | 0.000300 | 7.2 |
| H-2-Dd | 2 | 1 | 9 | 9 | YRLFRKSNL | 0.000309 | 7.4 |
| H-2-Dd | 4 | 2 | 10 | 9 | TESIVRFPN | 0.000342 | 8 |
| H-2-Dd | 2 | 4 | 12 | 9 | FRKSNLKPF | 0.000382 | 8.7 |
| H-2-Dd | 1 | 14 | 22 | 9 | FQPTNGVGY | 0.000425 | 9.7 |
| H-2-Dd | 3 | 14 | 22 | 9 | TGKIADYNY | 0.000511 | 13 |
| H-2-Dd | 4 | 9 | 17 | 9 | PNIFNATRF | 0.000542 | 14 |
| H-2-Dd | 3 | 9 | 17 | 9 | IAPGQTGKI | 0.000555 | 14 |
| H-2-Kd | 2 | 7 | 15 | 9 | SNLKPFERD | 0.000578 | 16 |
| H-2-Dd | 4 | 3 | 11 | 9 | ESIVRFPNI | 0.000608 | 15 |
| H-2-Kd | 3 | 15 | 23 | 9 | GKIADYNYK | 0.000613 | 16 |
| H-2-Dd | 3 | 11 | 19 | 9 | PGQTGKIAD | 0.000691 | 17 |
| H-2-Dd | 4 | 6 | 14 | 9 | VRFPNIFNA | 0.000703 | 17 |
| H-2-Dd | 3 | 3 | 11 | 9 | GDEVRQIAP | 0.000725 | 17 |
| H-2-Dd | 2 | 9 | 17 | 9 | LKPFERDIS | 0.000740 | 18 |
| H-2-Dd | 1 | 8 | 16 | 9 | PLQSYGFQP | 0.000745 | 18 |
| H-2-Dd | 2 | 2 | 10 | 9 | RLFRKSNLK | 0.000849 | 20 |
| H-2-Dd | 1 | 9 | 17 | 9 | LQSYGFQPT | 0.000992 | 22 |
| H-2-Dd | 2 | 10 | 18 | 9 | KPFERDIST | 0.001060 | 23 |
| H-2-Kd | 2 | 8 | 16 | 9 | NLKPFERDI | 0.001126 | 25 |
| H-2-Kd | 4 | 2 | 10 | 9 | TESIVRFPN | 0.001152 | 25 |
| H-2-Dd | 2 | 16 | 24 | 9 | ISTEIYQAG | 0.001178 | 25 |
| H-2-Dd | 2 | 6 | 14 | 9 | KSNLKPFER | 0.001218 | 25 |
| H-2-Kd | 4 | 7 | 15 | 9 | RFPNIFNAT | 0.001245 | 26 |
| H-2-Kd | 3 | 8 | 16 | 9 | QIAPGQTGK | 0.001282 | 27 |
| H-2-Dd | 3 | 6 | 14 | 9 | VRQIAPGQT | 0.001547 | 30 |
| H-2-Kd | 3 | 7 | 15 | 9 | RQIAPGQTG | 0.001553 | 30 |
| H-2-Dd | 1 | 4 | 12 | 9 | NCYFPLQSY | 0.001557 | 30 |
| H-2-Dd | 3 | 2 | 10 | 9 | RGDEVRQIA | 0.001560 | 30 |
| H-2-Dd | 4 | 10 | 18 | 9 | NIFNATRFA | 0.001674 | 31 |
| H-2-Dd | 1 | 7 | 15 | 9 | FPLQSYGFQ | 0.001885 | 34 |
| H-2-Dd | 1 | 2 | 10 | 9 | GFNCYFPLQ | 0.002278 | 38 |
| H-2-Dd | 4 | 5 | 13 | 9 | IVRFPNIFN | 0.002596 | 41 |
| H-2-Kd | 4 | 4 | 12 | 9 | SIVRFPNIF | 0.002697 | 41 |
| H-2-Kd | 3 | 1 | 9 | 9 | IRGDEVRQI | 0.002699 | 41 |
| H-2-Kd | 2 | 3 | 11 | 9 | LFRKSNLKP | 0.002716 | 41 |
| H-2-Kd | 4 | 12 | 20 | 9 | FNATRFASV | 0.002880 | 43 |
| H-2-Kd | 2 | 4 | 12 | 9 | FRKSNLKPF | 0.002885 | 43 |
| H-2-Kd | 3 | 2 | 10 | 9 | RGDEVRQIA | 0.002892 | 43 |
| H-2-Dd | 2 | 13 | 21 | 9 | ERDISTEIY | 0.002938 | 44 |
| H-2-Kd | 1 | 9 | 17 | 9 | LQSYGFQPT | 0.003065 | 44 |
| H-2-Dd | 1 | 17 | 25 | 9 | TNGVGYQPY | 0.003155 | 46 |
| H-2-Dd | 2 | 12 | 20 | 9 | FERDISTEI | 0.003191 | 47 |
| H-2-Kd | 1 | 6 | 14 | 9 | YFPLQSYGF | 0.003251 | 46 |
| H-2-Dd | 4 | 4 | 12 | 9 | SIVRFPNIF | 0.003263 | 47 |
| H-2-Kd | 4 | 9 | 17 | 9 | PNIFNATRF | 0.003361 | 46 |
| H-2-Dd | 2 | 17 | 25 | 9 | STEIYQAGS | 0.003431 | 49 |
| H-2-Kd | 3 | 9 | 17 | 9 | IAPGQTGKI | 0.003609 | 48 |
| H-2-Kd | 2 | 16 | 24 | 9 | ISTEIYQAG | 0.003758 | 49 |
| H-2-Kd | 1 | 1 | 9 | 9 | EGFNCYFPL | 0.004132 | 51 |
| H-2-Dd | 4 | 1 | 9 | 9 | PTESIVRFP | 0.004211 | 55 |
| H-2-Kd | 2 | 11 | 19 | 9 | PFERDISTE | 0.004236 | 52 |
| H-2-Dd | 4 | 8 | 16 | 9 | FPNIFNATR | 0.004272 | 55 |
| H-2-Dd | 3 | 1 | 9 | 9 | IRGDEVRQI | 0.004426 | 56 |
| H-2-Kd | 1 | 13 | 21 | 9 | GFQPTNGVG | 0.004526 | 53 |
| H-2-Kd | 4 | 3 | 11 | 9 | ESIVRFPNI | 0.004574 | 53 |
| H-2-Dd | 1 | 3 | 11 | 9 | FNCYFPLQS | 0.004609 | 57 |
| H-2-Kd | 1 | 17 | 25 | 9 | TNGVGYQPY | 0.004972 | 55 |
| H-2-Kd | 4 | 10 | 18 | 9 | NIFNATRFA | 0.004999 | 55 |
| H-2-Dd | 2 | 5 | 13 | 9 | RKSNLKPFE | 0.005308 | 62 |
| H-2-Kd | 2 | 17 | 25 | 9 | STEIYQAGS | 0.006395 | 61 |
| H-2-Kd | 4 | 6 | 14 | 9 | VRFPNIFNA | 0.006504 | 62 |
| H-2-Kd | 1 | 14 | 22 | 9 | FQPTNGVGY | 0.006524 | 62 |
| H-2-Dd | 3 | 15 | 23 | 9 | GKIADYNYK | 0.006682 | 69 |
| H-2-Kd | 4 | 8 | 16 | 9 | FPNIFNATR | 0.006707 | 62 |
| H-2-Kd | 3 | 11 | 19 | 9 | PGQTGKIAD | 0.006785 | 63 |
| H-2-Kd | 2 | 2 | 10 | 9 | RLFRKSNLK | 0.006786 | 63 |
| H-2-Dd | 3 | 7 | 15 | 9 | RQIAPGQTG | 0.007514 | 72 |
| H-2-Dd | 3 | 4 | 12 | 9 | DEVRQIAPG | 0.007684 | 73 |
| H-2-Kd | 1 | 15 | 23 | 9 | QPTNGVGYQ | 0.007849 | 66 |
| H-2-Dd | 3 | 5 | 13 | 9 | EVRQIAPGQ | 0.007858 | 73 |
| H-2-Kd | 2 | 13 | 21 | 9 | ERDISTEIY | 0.007966 | 66 |
| H-2-Kd | 3 | 6 | 14 | 9 | VRQIAPGQT | 0.008057 | 66 |
| H-2-Dd | 3 | 13 | 21 | 9 | QTGKIADYN | 0.008880 | 77 |
| H-2-Dd | 2 | 15 | 23 | 9 | DISTEIYQA | 0.008993 | 77 |
| H-2-Dd | 1 | 15 | 23 | 9 | QPTNGVGYQ | 0.009217 | 78 |
| H-2-Kd | 2 | 14 | 22 | 9 | RDISTEIYQ | 0.009264 | 70 |
| H-2-Kd | 3 | 10 | 18 | 9 | APGQTGKIA | 0.009298 | 70 |
| H-2-Kd | 2 | 5 | 13 | 9 | RKSNLKPFE | 0.009471 | 70 |
| H-2-Dd | 1 | 11 | 19 | 9 | SYGFQPTNG | 0.009661 | 79 |
| H-2-Dd | 2 | 7 | 15 | 9 | SNLKPFERD | 0.010219 | 81 |
| H-2-Kd | 2 | 9 | 17 | 9 | LKPFERDIS | 0.010252 | 72 |
| H-2-Dd | 2 | 14 | 22 | 9 | RDISTEIYQ | 0.011202 | 83 |
| H-2-Dd | 2 | 11 | 19 | 9 | PFERDISTE | 0.011614 | 84 |
| H-2-Kd | 3 | 3 | 11 | 9 | GDEVRQIAP | 0.012688 | 77 |
| H-2-Kd | 1 | 4 | 12 | 9 | NCYFPLQSY | 0.012694 | 77 |
| H-2-Kd | 3 | 4 | 12 | 9 | DEVRQIAPG | 0.012768 | 77 |
| H-2-Kd | 1 | 3 | 11 | 9 | FNCYFPLQS | 0.013268 | 78 |
| H-2-Dd | 3 | 8 | 16 | 9 | QIAPGQTGK | 0.013684 | 88 |
| H-2-Dd | 1 | 10 | 18 | 9 | QSYGFQPTN | 0.013791 | 88 |
| H-2-Kd | 3 | 14 | 22 | 9 | TGKIADYNY | 0.016227 | 82 |
| H-2-Kd | 3 | 12 | 20 | 9 | GQTGKIADY | 0.016425 | 83 |
| H-2-Dd | 1 | 16 | 24 | 9 | PTNGVGYQP | 0.016657 | 91 |
| H-2-Kd | 4 | 1 | 9 | 9 | PTESIVRFP | 0.017077 | 84 |
| H-2-Dd | 3 | 12 | 20 | 9 | GQTGKIADY | 0.018247 | 93 |
| H-2-Kd | 3 | 5 | 13 | 9 | EVRQIAPGQ | 0.019432 | 86 |
| H-2-Dd | 3 | 10 | 18 | 9 | APGQTGKIA | 0.019829 | 94 |
| H-2-Kd | 1 | 16 | 24 | 9 | PTNGVGYQP | 0.020156 | 87 |
| H-2-Dd | 1 | 5 | 13 | 9 | CYFPLQSYG | 0.020267 | 94 |
| H-2-Dd | 2 | 3 | 11 | 9 | LFRKSNLKP | 0.022977 | 96 |
| H-2-Kd | 4 | 5 | 13 | 9 | IVRFPNIFN | 0.023020 | 89 |
| H-2-Kd | 3 | 13 | 21 | 9 | QTGKIADYN | 0.023357 | 90 |
| H-2-Kd | 2 | 6 | 14 | 9 | KSNLKPFER | 0.026144 | 92 |
| H-2-Kd | 1 | 2 | 10 | 9 | GFNCYFPLQ | 0.026266 | 92 |
| H-2-Kd | 2 | 10 | 18 | 9 | KPFERDIST | 0.027065 | 92 |
| H-2-Kd | 2 | 15 | 23 | 9 | DISTEIYQA | 0.027473 | 92 |
| H-2-Kd | 1 | 8 | 16 | 9 | PLQSYGFQP | 0.027964 | 93 |
| H-2-Dd | 2 | 8 | 16 | 9 | NLKPFERDI | 0.028100 | 98 |
| H-2-Dd | 1 | 13 | 21 | 9 | GFQPTNGVG | 0.028708 | 98 |
| H-2-Kd | 1 | 10 | 18 | 9 | QSYGFQPTN | 0.040577 | 97 |
| Supertype | Peptide | Binding Affinity | Rescale Binding Affinity | Proteosomal C-Terminal Cleavage | Transport Affinity | Prediction Score | MHC-I Binding |
|---|---|---|---|---|---|---|---|
| A1 | FQPTNGVGY | 0.1117 | 0.4741 | 0.7859 | 2.8600 | 0.7583 | < -E |
| A1 | ERDISTEIY | 0.2097 | 0.8903 | 0.9734 | 2.6460 | 1.1686 | < -E |
| A26 | FQPTNGVGY | 0.2611 | 0.7007 | 0.9409 | 2.8600 | 0.9848 | < -E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murdocca, M.; Citro, G.; Romeo, I.; Lupia, A.; Miersch, S.; Amadio, B.; Bonomo, A.; Rossi, A.; Sidhu, S.S.; Pandolfi, P.P.; et al. Peptide Platform as a Powerful Tool in the Fight against COVID-19. Viruses 2021, 13, 1667. https://doi.org/10.3390/v13081667
Murdocca M, Citro G, Romeo I, Lupia A, Miersch S, Amadio B, Bonomo A, Rossi A, Sidhu SS, Pandolfi PP, et al. Peptide Platform as a Powerful Tool in the Fight against COVID-19. Viruses. 2021; 13(8):1667. https://doi.org/10.3390/v13081667
Chicago/Turabian StyleMurdocca, Michela, Gennaro Citro, Isabella Romeo, Antonio Lupia, Shane Miersch, Bruno Amadio, Alessia Bonomo, Antonio Rossi, Sachdev S. Sidhu, Pier Paolo Pandolfi, and et al. 2021. "Peptide Platform as a Powerful Tool in the Fight against COVID-19" Viruses 13, no. 8: 1667. https://doi.org/10.3390/v13081667
APA StyleMurdocca, M., Citro, G., Romeo, I., Lupia, A., Miersch, S., Amadio, B., Bonomo, A., Rossi, A., Sidhu, S. S., Pandolfi, P. P., Alcaro, S., Sangiuolo, F. C., & Novelli, G. (2021). Peptide Platform as a Powerful Tool in the Fight against COVID-19. Viruses, 13(8), 1667. https://doi.org/10.3390/v13081667