
NIgPred: Class-Specific Antibody Prediction for Linear B-Cell Epitopes Based on Heterogeneous Features and Machine-Learning Approaches


Abstract
:1. Introduction
2. Materials and Methods
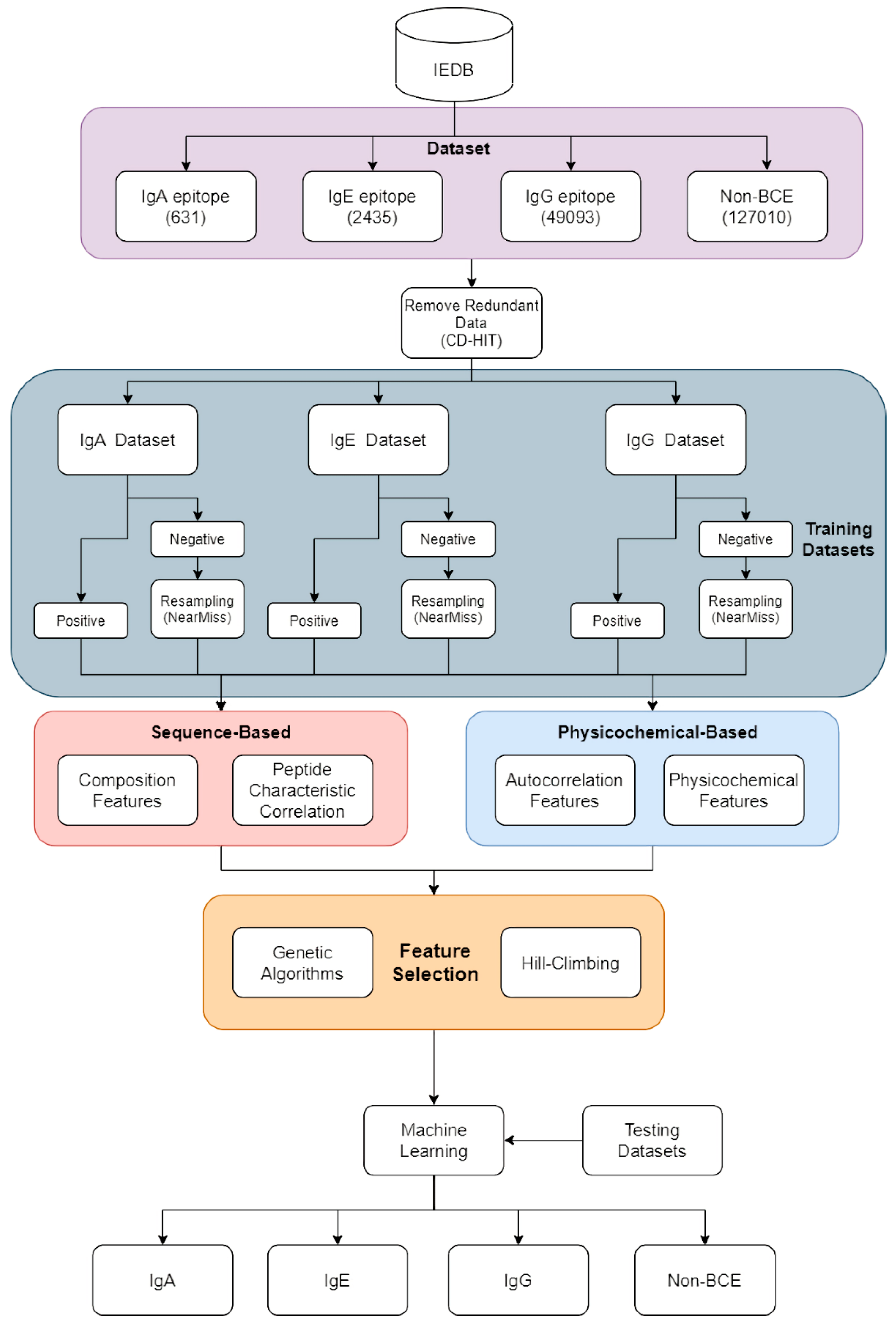
2.1. Overview of Our Study
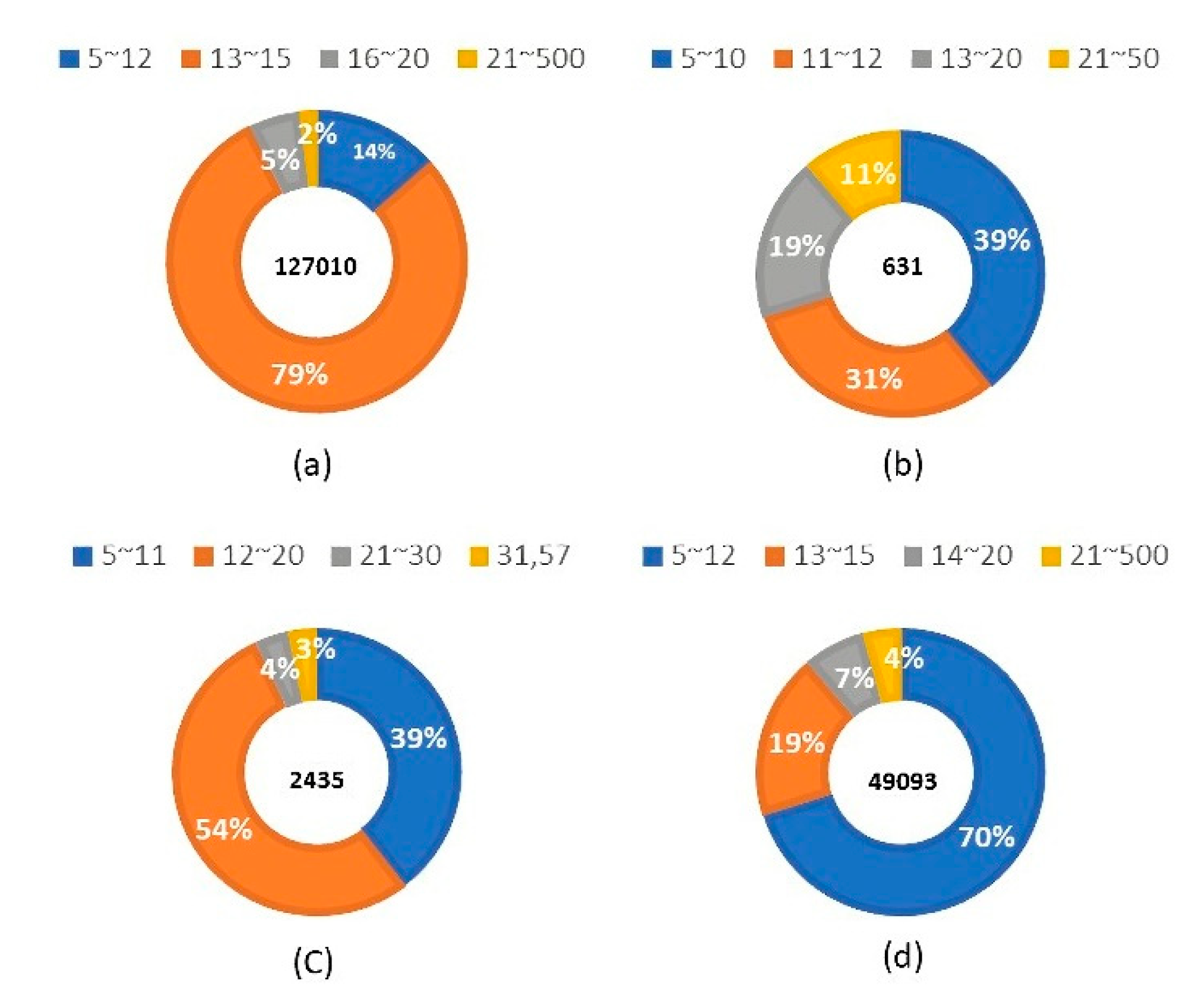
2.2. Data Set and Pre-Processing
2.3. Feature Construction
2.4. Machine Learning and Feature Selection Algorithms
2.5. Validation Measures
3. Results and Conclusions
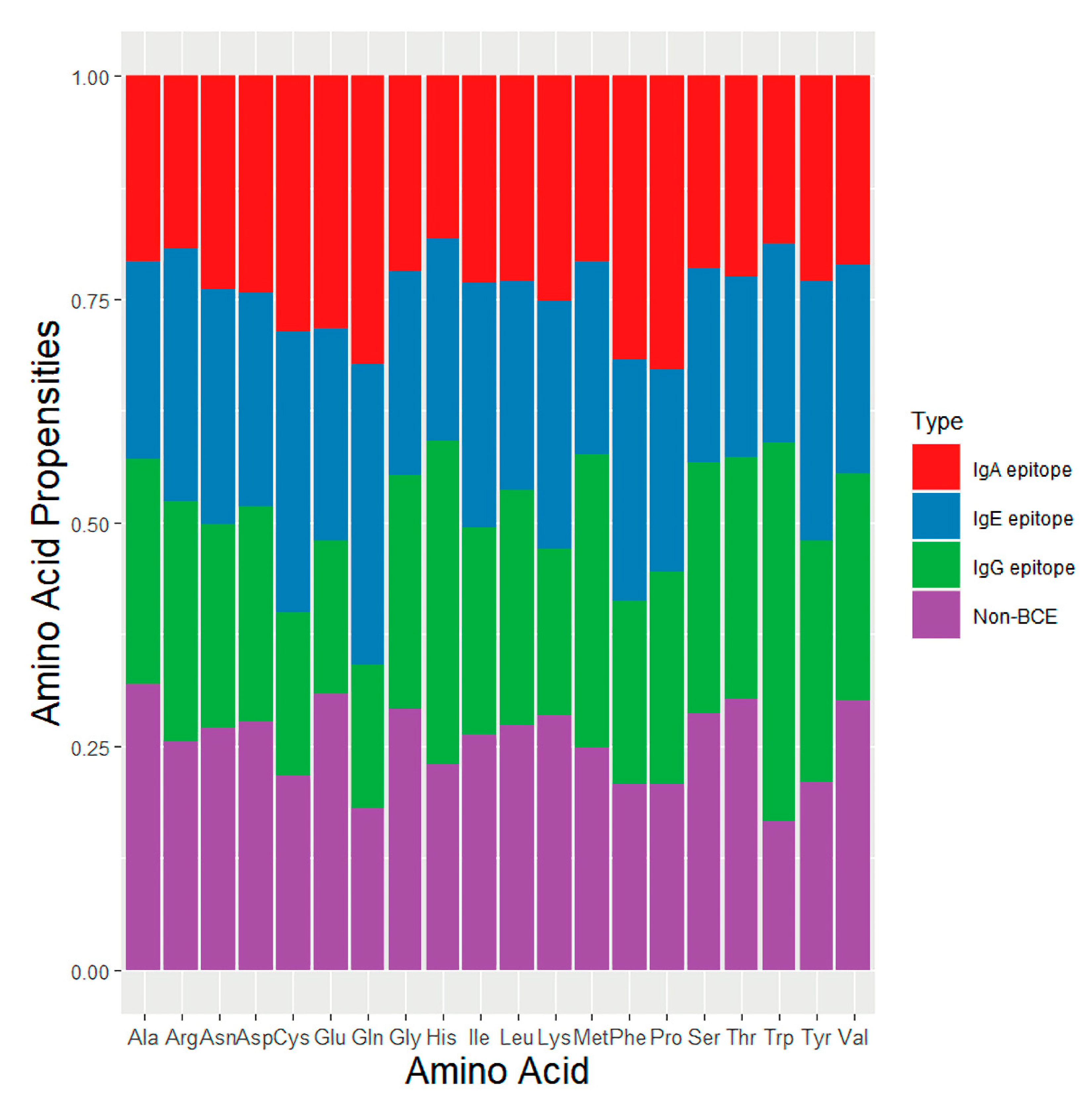
3.1. Amino Acid Composition
3.2. Performance Comparisons for Various Features
3.3. Performance Comparison Based on Epitopes of Different Lengths
3.4. Ability of the Model on an Independent Data Set
3.5. Performances of the Models Trained on Data from Prior Years with Regard to Predicting Specific Epitopes Collected in Later Years
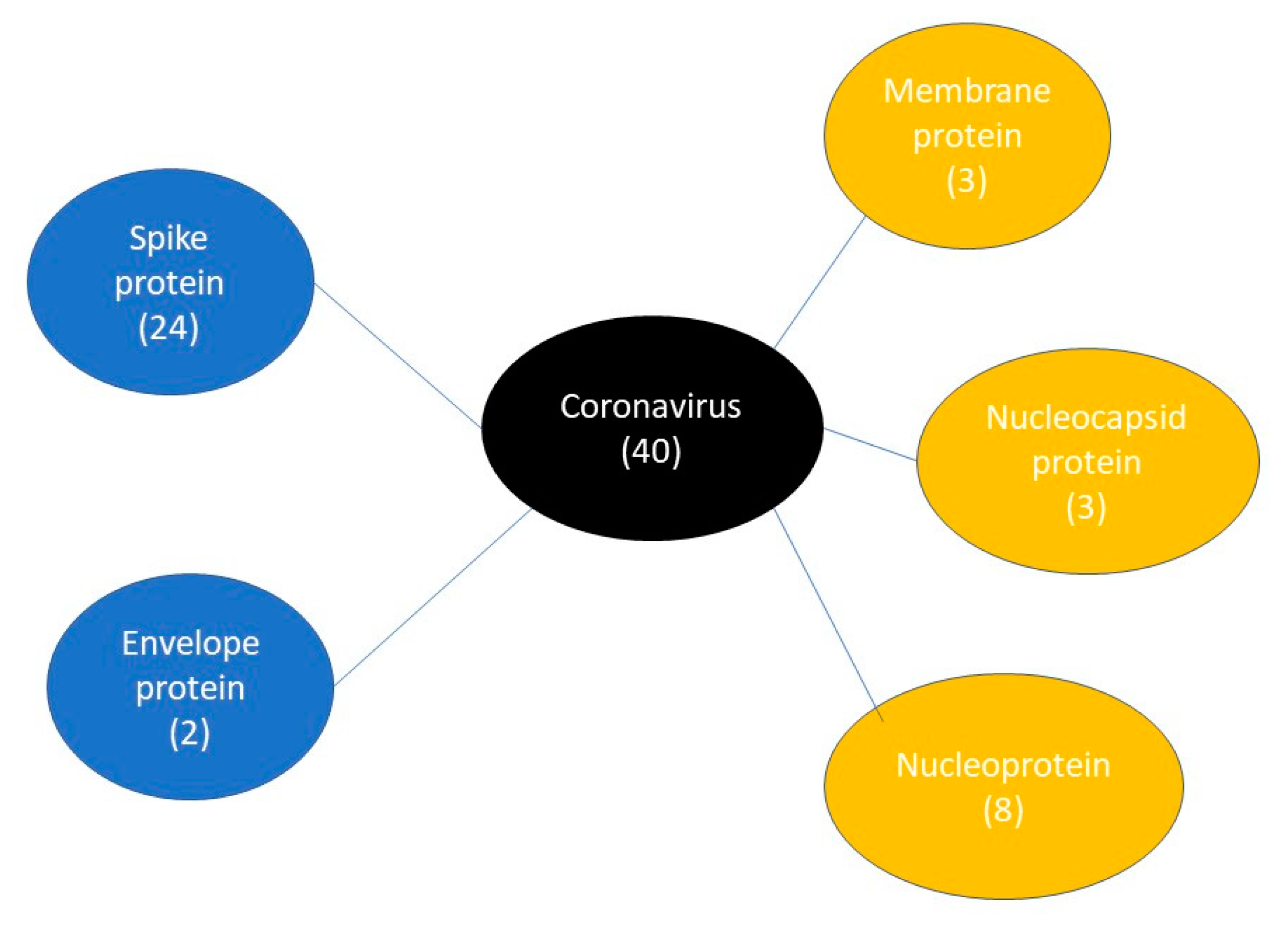
3.6. Case Studies
3.6.1. IgE Epitope Case Study
3.6.2. IgG Epitope Case Study
3.7. Developing the NIgPred Tool
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, D.R.; Cohen, G.H. Interactions of Protein Antigens with Antibodies. Proc. Natl. Acad. Sci. USA 1996, 93, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.S.; Gilman, M.S.A.; McLellan, J.S. Structure-Based Vaccine Antigen Design. Annu. Rev. Med. 2019, 70, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Yu, L.; Wu, Q.; Lin, X.; Liu, S.; Hu, S.; Rosa, C.; Eichinger, D.; Pino, I.; Zhu, H. Integration of IgA and IgG Autoantigens Improves Performance of Biomarker Panels for Early Diagnosis of Lung Cancer. Mol. Cell. Proteom. 2020, 19, 490–500. [Google Scholar] [CrossRef]
- Long, Q.-X.; Liu, B.-Z.; Deng, H.-J.; Wu, G.-C.; Deng, K.; Chen, Y.-K.; Liao, P.; Qiu, J.-F.; Lin, Y.; Cai, X.-F. Antibody Responses to SARS-CoV-2 in Patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Shan, M.; Carrillo, J.; Yeste, A.; Gutzeit, C.; Segura-Garzón, D.; Walland, A.C.; Pybus, M.; Grasset, E.K.; Yeiser, J.R.; Matthews, D.B. Secreted IgD Amplifies Humoral T Helper 2 Cell Responses by Binding Basophils via Galectin-9 and CD44. Immunity 2018, 49, 709–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waserman, S.; Bégin, P.; Watson, W. IgE-Mediated Food Allergy. Allergy Asthma Clin. Immunol. 2018, 14, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Waserman, S.; Watson, W. Food Allergy. Allergy Asthma Clin. Immunol. 2011, 7, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, V.; Gautham, N. BCIgEPRED-a Dual-Layer Approach for Predicting Linear IgE Epitopes. Mol. Biol. 2018, 52, 285–293. [Google Scholar] [CrossRef]
- Pellequer, J.L.; Westhof, E.; Van Regenmortel, M.H.V. [8] Predicting Location of Continuous Epitopes in Proteins from Their Primary Structures. Methods Enzymol. 1991, 203, 176–201. [Google Scholar]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving Sequence-Based B-Cell Epitope Prediction Using Conformational Epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Raghava, G.P.S. BcePred: Prediction of Continuous B-Cell Epitopes in Antigenic Sequences Using Physico-Chemical Properties. In International Conference on Artificial Immune Systems; Springer: Berlin/Heidelberg, Germany, 2004; pp. 197–204. [Google Scholar]
- EL-Manzalawy, Y.; Dobbs, D.; Honavar, V. Predicting Linear B-Cell Epitopes Using String Kernels. J. Mol. Recognit. Interdiscip. J. 2008, 21, 243–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Cao, Y.; Cha, L.; Zhang, X.; Ying, X.; Zhang, W.; Ge, K.; Li, W.; Zhong, L. Predicting Linear B-Cell Epitopes Using Amino Acid Anchoring Pair Composition. BioData Min. 2015, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manavalan, B.; Govindaraj, R.G.; Shin, T.H.; Kim, M.O.; Lee, G. IBCE-EL: A New Ensemble Learning Framework for Improved Linear B-Cell Epitope Prediction. Front. Immunol. 2018, 9, 1695. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Ansari, H.R.; Raghava, G.P. Improved Method for Linear B-Cell Epitope Prediction Using Antigen’s Primary Sequence. PLoS ONE 2013, 8, e62216. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Shi, K.; Li, W. Deep Learning Methods Improve Linear B-Cell Epitope Prediction. BioData Min. 2020, 13, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Raghava, G.P.S. AlgPred: Prediction of Allergenic Proteins and Mapping of IgE Epitopes. Nucleic Acids Res. 2006, 34, W202–W209. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Ansari, H.R.; Gautam, A.; Raghava, G.P. Identification of B-Cell Epitopes in an Antigen for Inducing Specific Class of Antibodies. Biol. Direct 2013, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA Data Mining Software: An Update. ACM SIGKDD Explor. Newsl. 2009, 11, 10–18. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Mani, I.; Zhang, I. KNN Approach to Unbalanced Data Distributions: A Case Study Involving Information Extraction. In Proceedings of the Workshop on Learning from Imbalanced Datasets ICML, Washinton DC, DC, USA, 18–24 July 2021; Volume 126. [Google Scholar]
- Lemaître, G.; Nogueira, F.; Aridas, C.K. Imbalanced-Learn: A Python Toolbox to Tackle the Curse of Imbalanced Datasets in Machine Learning. J. Mach. Learn. Res. 2017, 18, 559–563. [Google Scholar]
- Nakamura, R.; Teshima, R.; Takagi, K.; Sawada, J. Development of Allergen Database for Food Safety (ADFS): An Integrated Database to Search Allergens and Predict Allergenicity. Kokuritsu Iyakuhin Shokuhin Eisei Kenkyusho Hokoku 2005, 123, 32–36. [Google Scholar]
- Kadam, K.; Karbhal, R.; Jayaraman, V.K.; Sawant, S.; Kulkarni-Kale, U. AllerBase: A Comprehensive Allergen Knowledgebase. Database 2017, 2017, 1–12. [Google Scholar] [CrossRef] [PubMed]
- COVID-19—The Antibody Registry. Available online: https://antibodyregistry.org/covid19 (accessed on 4 July 2021).
- Osorio, D.; Rondón-Villarreal, P.; Torres, R. Peptides: A Package for Data Mining of Antimicrobial Peptides. Small 2015, 12, 44–444. [Google Scholar] [CrossRef]
- Xiao, N.; Cao, D.-S.; Zhu, M.-F.; Xu, Q.-S. Protr/ProtrWeb: R Package and Web Server for Generating Various Numerical Representation Schemes of Protein Sequences. Bioinformatics 2015, 31, 1857–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.-P.; Zhang, C.-T. Prediction of Membrane Protein Types Based on the Hydrophobic Index of Amino Acids. J. Protein Chem. 2000, 19, 269–275. [Google Scholar] [CrossRef]
- Horne, D.S. Prediction of Protein Helix Content from an Autocorrelation Analysis of Sequence Hydrophobicities. Biopolym. Orig. Res. Biomol. 1988, 27, 451–477. [Google Scholar] [CrossRef]
- Sokal, R.R.; Thomson, B.A. Population Structure Inferred by Local Spatial Autocorrelation: An Example from an Amerindian Tribal Population. Am. J. Phys. Anthropol. Off. Publ. Am. Assoc. Phys. Anthropol. 2006, 129, 121–131. [Google Scholar] [CrossRef]
- Man, K.-F.; Tang, K.-S.; Kwong, S. Genetic Algorithms: Concepts and Applications [in Engineering Design]. IEEE Trans. Ind. Electron. 1996, 43, 519–534. [Google Scholar] [CrossRef]
- Hall, M.A. Correlation-Based Feature Selection of Discrete and Numeric Class Machine Learning. Master’s Thesis, University of Waikato, Hamilton, New Zealand, 2000. [Google Scholar]
- Keerthi, S.S.; Shevade, S.K.; Bhattacharyya, C.; Murthy, K.R.K. Improvements to Platt’s SMO Algorithm for SVM Classifier Design. Neural Comput. 2001, 13, 637–649. [Google Scholar] [CrossRef]
- Kim, S.-J.; Magnani, A.; Boyd, S. Robust Fisher Discriminant Analysis. In Advances in Neural Information Processing Systems; MIT Press: Cambridge, MA, USA, 2006; pp. 659–666. [Google Scholar]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Lever, J.; Krzywinski, M.; Altman, N. Points of significance: Model selection and overfitting. Nat. Methods 2016, 13, 703–705. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature Type | Function |
|---|---|
| Composition properties | extractAAC |
| extractDC | |
| extractCTDD | |
| length | |
| Physico-chemical properties | aindex |
| boman | |
| mw | |
| crucianiProperties | |
| hydrophobicity | |
| aacomp | |
| blosumIndices | |
| PI | |
| mswhimScores | |
| kideraFactors | |
| fasgaiVectors | |
| vhseScales | |
| Zscales | |
| Autocorrelation | extractMoreauBroto |
| extractMoran | |
| extractGeary | |
| Peptide characteristic correlation | extractSOCN |
| extractQSO | |
| extractPAAC | |
| extractAPAAC | |
| extractCTriad |
| Feature Type | ||||
|---|---|---|---|---|
| Tool | Composition | Physico-Chemical | Autocorrelation | Peptide-Characteristic Correlation |
| IgPred | ✓ | ✓ | ||
| BCIgEPred | ✓ | |||
| NIgPred | ✓ | ✓ | ✓ | ✓ |
| Feature Type | Features (bits) | IgA Epitope | IgE Epitope | IgG Epitope | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Precision (%) | Recall (%) | ACC (%) | MCC (%) | Precision (%) | Recall (%) | ACC (%) | MCC (%) | Precision (%) | Recall (%) | ACC (%) | MCC (%) | ||
| Composition | 526 | 79.5 | 79.5 | 79.5 | 59.0 | 79.0 | 79.0 | 79.0 | 58.0 | 82.9 | 82.5 | 82.5 | 65.4 |
| Physico-chemical | 59 | 80.3 | 80.3 | 80.3 | 60.6 | 73.4 | 73.1 | 73.1 | 46.5 | 81.3 | 81.1 | 81.0 | 62.4 |
| Autocorrelation | 96 | 72.9 | 72.9 | 72.8 | 45.7 | 67.0 | 67.0 | 66.9 | 33.9 | 69.5 | 69.5 | 69.4 | 38.9 |
| Peptide characteristic correlation | 451 | 85.2 | 85.0 | 85.0 | 70.2 | 79.9 | 79.9 | 79.9 | 59.9 | 84.7 | 84.6 | 84.6 | 69.4 |
| All | 1132 | 85.4 | 85.3 | 85.3 | 70.7 | 83.9 | 83.9 | 83.9 | 67.9 | 98.2 | 98.2 | 98.1 | 91.1 |
| Model | Model | Precision (%) | Recall (%) | ACC (%) | MCC (%) |
|---|---|---|---|---|---|
| IgA | Training | 85.4 | 85.3 | 85.3 | 70.7 |
| Testing | 85.8 | 85.8 | 85.7 | 71.5 | |
| IgE | Training | 83.9 | 83.9 | 83.9 | 67.9 |
| Testing | 82.0 | 82.0 | 81.9 | 64.0 | |
| BCIgEPred | 68.6 | 31.6 | 56.9 | 18.7 | |
| IgG | Training | 98.2 | 98.2 | 98.1 | 91.1 |
| Testing | 87.2 | 87.2 | 87.2 | 74.5 |
| Model | Train (Year) | Precision (%) | Recall (%) | ACC (%) | MCC (%) | Test (Year) | Precision (%) | Recall (%) | ACC (%) | MCC (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| IgA | 1985–2017 | 89.5 | 89.5 | 89.4 | 79.0 | 2018–2020 | 76.9 | 75.1 | 75.1 | 47.4 |
| IgE | 1976–2015 | 81.1 | 80.8 | 80.8 | 61.5 | 2015–2020 | 89.7 | 89.2 | 89.2 | 76.8 |
| IgG | 1970–2017 | 89.4 | 89.0 | 88.9 | 76.8 | 2017–2020 | 88.1 | 84.3 | 84.2 | 52.2 |
| Database\Tool | IgPred | BCIgEPred | NIgPred |
|---|---|---|---|
| ADFS | 0.10 | 0.52 | 0.84 |
| Allerbase | 0.46 | 0.25 | 0.74 |
| Antibody ID a | PMID | Protein Sequence | Sub-Class Type | Actual | IgG Prediction |
|---|---|---|---|---|---|
| AB_2833115 | 27627203 | TDVSTAIHADQLTPAWRIYSTG | Spike protein | IgG | IgG |
| AB_2833108 | 2464703 | LLGCIGSTC | Spike protein | IgG | IgG |
| AB_2833113 | 16731915 | GVLTPSSKRFQPFQQFG | Spike protein | IgG | IgG |
| AB_2833123 | 1711257 | MKRSGYGQPIA | Spike protein | IgG | IgG |
| AB_2833135 | 16378996 | ISGINASVVNIQKEIDRLNEVAKNLNESLIDLQELGKYEQYI | Spike protein | IgG | IgG |
| AB_2833139 | 16378996 | FFSPQIITTDNTFVSGNCDVVIGIINNTVYDPLQPELDSF | Spike protein | IgG | IgG |
| AB_2833187 | 15184071 | PDPLKPTKR | Spike protein | IgG | IgG |
| AB_2833188 | 15184071 | KLRPFERDI | Spike protein | IgG | IgG |
| AB_2833192 | 20168090 | FSPDGKPCTPPALNCYW | Spike protein | IgG | IgG |
| AB_2833197 | 1281870 | TTGYRFTNFEPFTV | Spike protein | IgG | IgG |
| AB_2833217 | 16725238 | SVYAWERKKISNCVADY | Spike protein | IgG | IgG |
| AB_2833224 | 19951177 | SNVYADSFVVKGDDVRQIAP | Spike protein | IgG | IgG |
| AB_2833227 | 15194798 | FQQFGRDVSDFTDSVRDPKT | Spike protein | IgG | IgG |
| AB_2847999 | 30355437 | TKPLKYSYINKCSRLLSDDRTEVPQ | Spike protein | IgG | IgG |
| AB_2847979 | 16132115 | SPDVDLGDASGINAS | Spike protein | IgG | IgG |
| AB_2847997 | 19951177 | VYAWERKKISNCVADYSVLYNSTF | Spike protein | IgG | IgG |
| AB_2833160 | 16920216 | DMDDFSRQLQ | Nucleoprotein | IgG | IgG |
| AB_2833161 | 23123213 | DLIARAAKI | Nucleoprotein | IgG | IgG |
| AB_2833162 | 16920216 | KKKKTDEAQP | Nucleoprotein | IgG | IgG |
| AB_2833173 | 23123213 | FGPRTK | Nucleoprotein | IgG | IgG |
| AB_2833179 | 16920216 | GNSRNSTPGS | Nucleoprotein | IgG | IgG |
| AB_2833222 | 15528730 | KKQPTVTLLPAADMDDF | Nucleoprotein | IgG | IgG |
| AB_2833126 | 27973413 | YSTEART | Membrane protein | IgG | IgG |
| AB_2848022 | 19851732 | FATFVYAK | Membrane protein | IgG | IgG |
| AB_2833158 | 23025700 | WAFYVR | Membrane protein | IgG | IgG |
| AB_2848062 | 31518629 | DAPVFTPAP | Nucleocapsid protein | IgG | IgG |
| AB_2848076 | 7516595 | PDMAEEIAALVLAKLGKDA | Nucleocapsid protein | IgG | IgG |
| AB_2848083 | 7516595 | SHEAIPTRFAPGTVLPQGFYVEGSGR | Nucleocapsid protein | IgG | IgG |
| AB_2833199 | 7516595 | AGQPKQVTKQSAKEVRQKILNKPRQKRTP | Nucleoprotein | IgG | IgG |
| AB_2833150 | 30877355 | VAAVKDALKSLGI | Nucleoprotein | IgG | IgG |
| AB_2847971 | 1693663 | SSFSSYGEI | Spike protein | IgG | IgG |
| AB_2847978 | 26292945 | HDFVADMYQLAQ | Spike protein | IgG | IgG |
| AB_2847978 | 26292945 | LMQINPTYYQIM | Spike protein | IgG | IgG |
| AB_2847978 | 26292945 | MQYVYTPTYYML | Spike protein | IgG | IgG |
| AB_2847978 | 26292945 | WSFNPSTYTIAG | Spike protein | IgG | IgG |
| AB_2848046 | 18400422 | YSNIGVCK | Spike protein | IgG | IgG |
| AB_2848064 | 18400422 | LQDGQVKI | Spike protein | IgG | IgG |
| AB_2848109 | 28647506 | PVLVYSNIGVCKS | Spike protein | IgG | IgG |
| AB_2848088 | 9874660 | MADPNRFRG | Envelope protein | IgG | IgG |
| AB_2848090 | 9874660 | DPEDSALL | Envelope protein | IgG | IgG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tung, C.-H.; Chang, Y.-S.; Chang, K.-P.; Chu, Y.-W. NIgPred: Class-Specific Antibody Prediction for Linear B-Cell Epitopes Based on Heterogeneous Features and Machine-Learning Approaches. Viruses 2021, 13, 1531. https://doi.org/10.3390/v13081531
Tung C-H, Chang Y-S, Chang K-P, Chu Y-W. NIgPred: Class-Specific Antibody Prediction for Linear B-Cell Epitopes Based on Heterogeneous Features and Machine-Learning Approaches. Viruses. 2021; 13(8):1531. https://doi.org/10.3390/v13081531
Chicago/Turabian StyleTung, Chi-Hua, Yi-Sheng Chang, Kai-Po Chang, and Yen-Wei Chu. 2021. "NIgPred: Class-Specific Antibody Prediction for Linear B-Cell Epitopes Based on Heterogeneous Features and Machine-Learning Approaches" Viruses 13, no. 8: 1531. https://doi.org/10.3390/v13081531
APA StyleTung, C.-H., Chang, Y.-S., Chang, K.-P., & Chu, Y.-W. (2021). NIgPred: Class-Specific Antibody Prediction for Linear B-Cell Epitopes Based on Heterogeneous Features and Machine-Learning Approaches. Viruses, 13(8), 1531. https://doi.org/10.3390/v13081531