
Phylogenetic Classification of Global Porcine Deltacoronavirus (PDCoV) Reference Strains and Molecular Characterization of PDCoV in Taiwan

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preliminary Examination by Real-Time PCR
2.2. Whole-Genome Sequencing
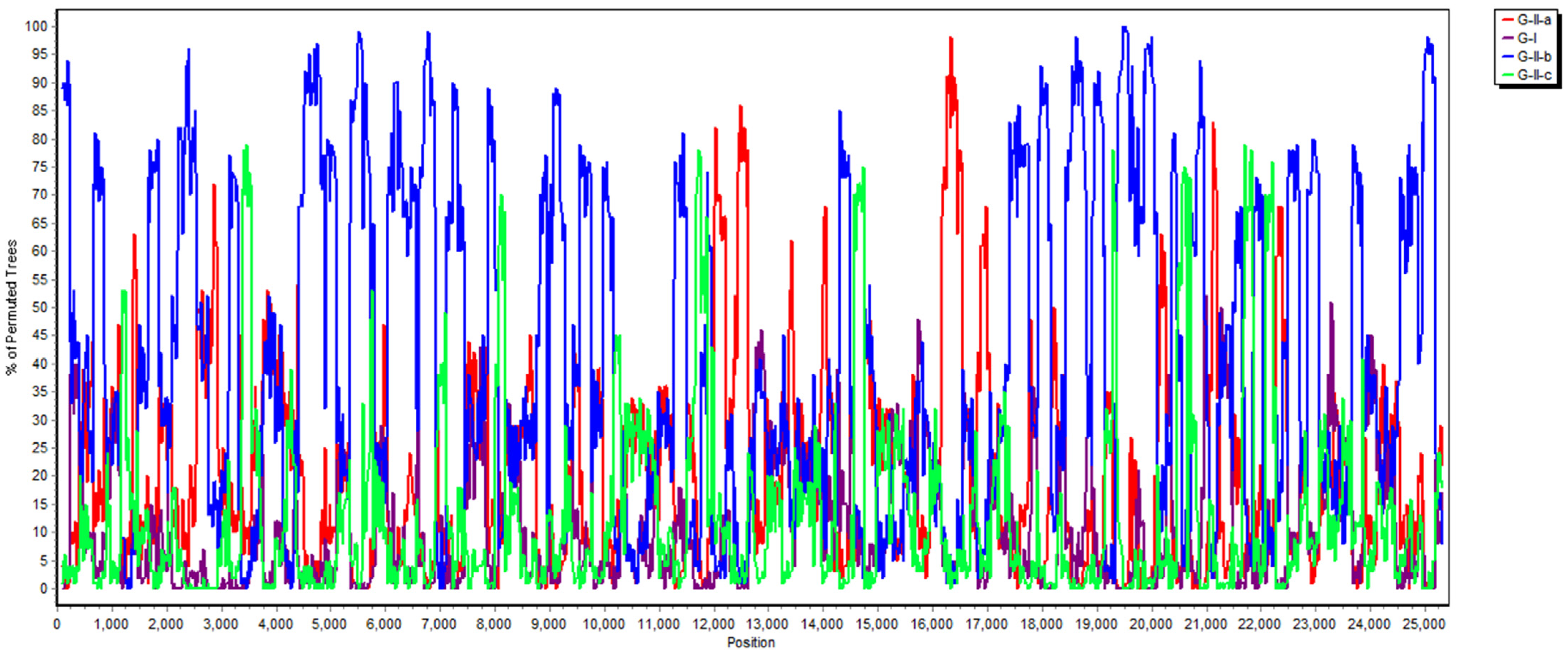
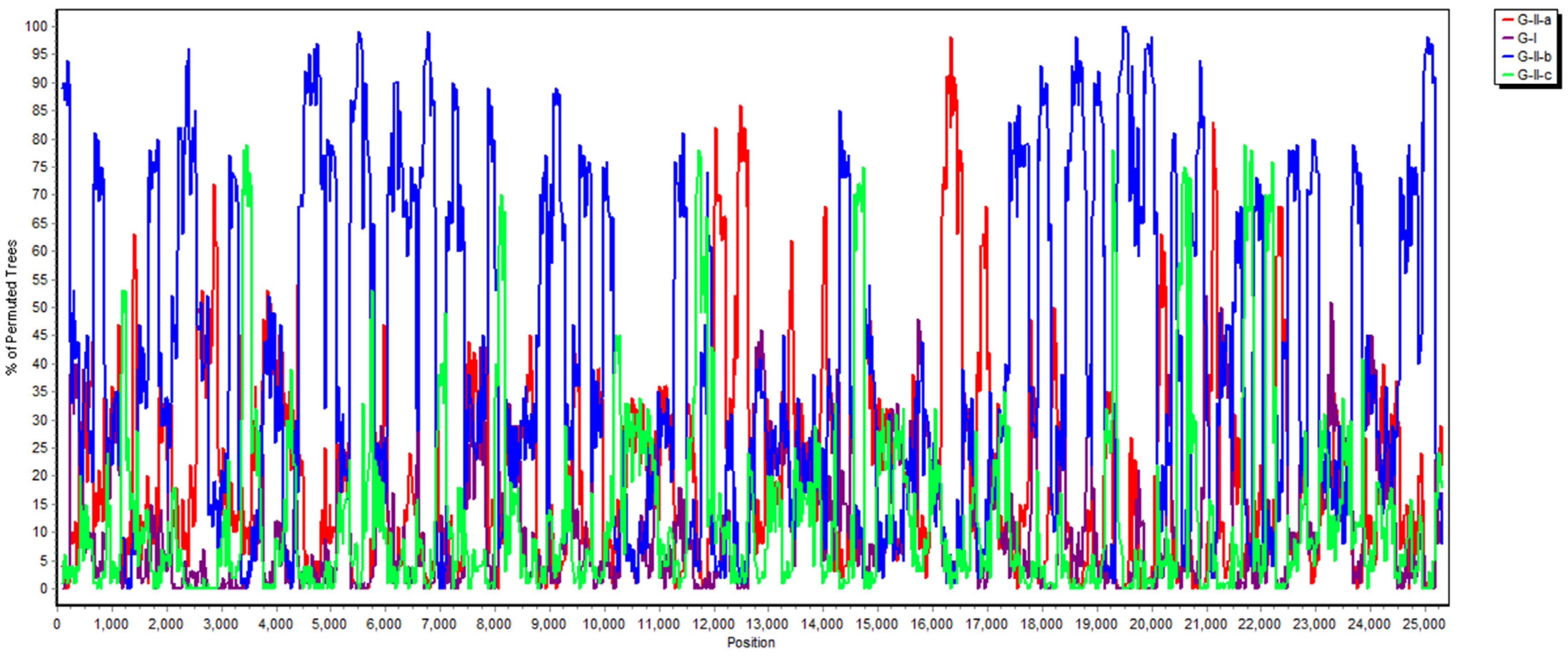
2.3. Phylogenetic and Recombination Analysis
3. Results
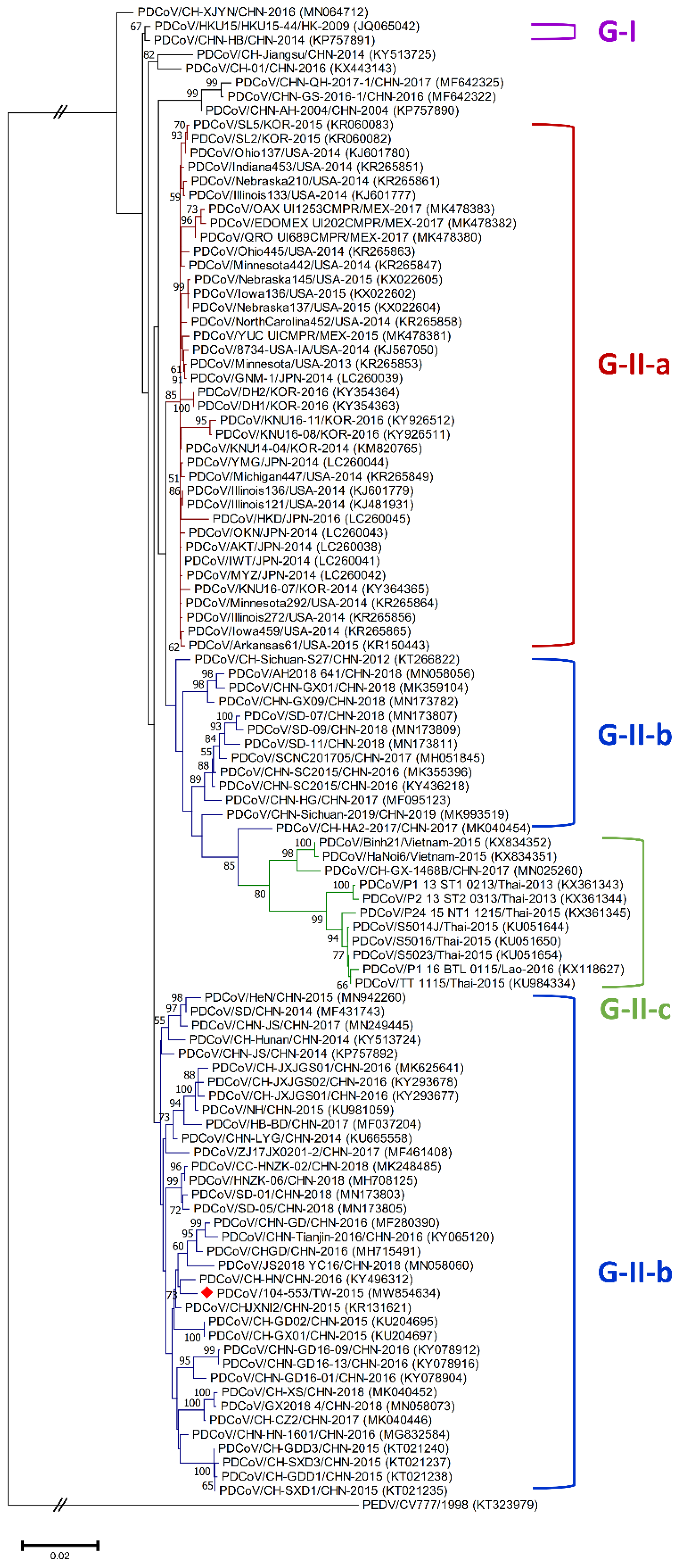
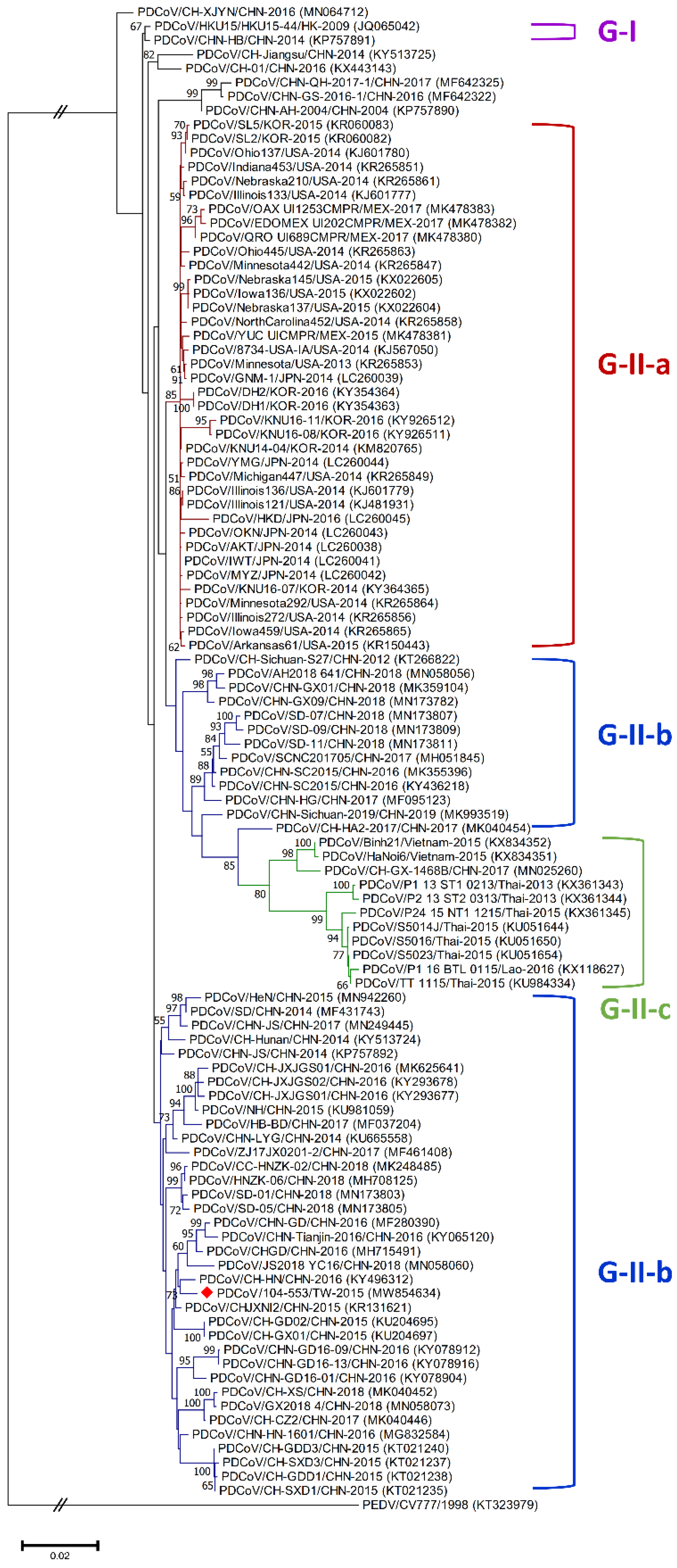
3.1. Classification Criteria of Global Historical PDCoV Strains
3.2. PDCoV Genomic Sequencing
3.3. Phylogenetic Analysis of the Whole Genome, S, M and N Genes
3.4. Amino Acid Analysis of the S Protein of the PDCoV Variant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar]
- Wang, L.; Byrum, B.; Zhang, Y. Detection and genetic characterization of deltacoronavirus in pigs, Ohio, USA, 2014. Emerg. Infect. Dis. 2014, 20, 1227–1230. [Google Scholar] [CrossRef]
- Marthaler, D.; Raymond, L.; Jiang, Y.; Collins, J.; Rossow, K.; Rovira, A. Rapid detection, complete genome sequencing, and phylogenetic analysis of porcine deltacoronavirus. Emerg. Infect. Dis. 2014, 20, 1347–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, N.; Fang, L.; Zeng, S.; Sun, Q.; Chen, H.; Xiao, S. Porcine deltacoronavirus in mainland China. Emerg. Infect. Dis. 2015, 21, 2254–2255. [Google Scholar] [CrossRef]
- Janetanakit, T.; Lumyai, M.; Bunpapong, N.; Boonyapisitsopa, S.; Chaiyawong, S.; Nonthabenjawan, N.; Kesdaengsakonwut, S.; Amonsin, A. Porcine deltacoronavirus, Thailand, 2015. Emerg. Infect. Dis. 2016, 22, 757–759. [Google Scholar] [CrossRef] [Green Version]
- Saeng-Chuto, K.; Lorsirigool, A.; Temeeyasen, G.; Vui, D.T.; Stott, C.J.; Madapong, A.; Tripipat, T.; Wegner, M.; Intrakamhaeng, M.; Chongcharoen, W.; et al. Different lineage of porcine deltacoronavirus in Thailand, Vietnam and Lao PDR in 2015. Transbound. Emerg. Dis. 2017, 64, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.; Lee, K.K.; Kim, S.H.; Lee, C. Prevalence, complete genome sequencing and phylogenetic analysis of porcine deltacoronavirus in South Korea, 2014–2016. Transbound. Emerg. Dis. 2017, 64, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shibahara, T.; Imai, N.; Yamamoto, T.; Ohashi, S. Genetic characterization and pathogenicity of Japanese porcine deltacoronavirus. Infect. Genet. Evol. 2018, 61, 176–182. [Google Scholar] [CrossRef]
- Hsu, T.H.; Liu, H.P.; Chin, C.Y.; Wang, C.; Zhu, W.Z.; Wu, B.L.; Chang, Y.C. Detection, sequence analysis, and antibody prevalence of porcine deltacoronavirus in Taiwan. Arch. Virol. 2018, 163, 3113–3117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Rivera, C.; Ramírez-Mendoza, H.; Mendoza-Elvira, S.; Segura-Velázquez, R.; Sánchez-Betancourt, J.I. First report and phylogenetic analysis of porcine deltacoronavirus in Mexico. Transbound. Emerg. Dis. 2019, 66, 1436–1441. [Google Scholar] [CrossRef]
- Dong, N.; Fang, L.; Yang, H.; Liu, H.; Du, T.; Fang, P.; Wang, D.; Chen, H.; Xiao, S. Isolation, genomic characterization, and pathogenicity of a Chinese porcine deltacoronavirus strain CHN-HN-2014. Vet. Microbiol. 2016, 196, 98–106. [Google Scholar] [CrossRef]
- Hu, H.; Jung, K.; Vlasova, A.N.; Chepngeno, J.; Lu, Z.; Wang, Q.; Saif, L.J. Isolation and characterization of porcine deltacoronavirus from pigs with diarrhea in the United States. J. Clin. Microbiol. 2015, 53, 1537–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsueh, F.C.; Lin, C.N.; Chiou, H.Y.; Chia, M.Y.; Chiou, M.T.; Haga, T.; Kao, C.F.; Chang, Y.C.; Chang, C.Y.; Jeng, C.R.; et al. Updated phylogenetic analysis of the spike gene and identification of a novel recombinant porcine epidemic diarrhoea virus strain in Taiwan. Transbound. Emerg. Dis. 2020, 67, 417–430. [Google Scholar] [CrossRef]
- Zhang, H.; Liang, Q.; Li, B.; Cui, X.; Wei, X.; Ding, Q.; Wang, Y.; Hu, H. Prevalence, phylogenetic and evolutionary analysis of porcine deltacoronavirus in Henan province, China. Prev. Vet. Med. 2019, 166, 8–15. [Google Scholar] [CrossRef]
- Sun, W.; Wang, L.; Huang, H.; Wang, W.; Cao, L.; Zhang, J.; Zheng, M.; Lu, H. Genetic characterization and phylogenetic analysis of porcine deltacoronavirus (PDCoV) in Shandong Province, China. Virus Res. 2020, 278, 197869. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, S.; Wang, X.; Luo, Z.; Shi, Y.; Wang, D.; Peng, G.; Chen, H.; Fang, L.; Xiao, S. Contribution of porcine aminopeptidase N to porcine deltacoronavirus infection. Emerg. Microbes Infect. 2018, 7, 65. [Google Scholar] [CrossRef]
- Wang, B.; Liu, Y.; Ji, C.M.; Yang, Y.L.; Liang, Q.Z.; Zhao, P.; Xu, L.D.; Lei, X.M.; Luo, W.T.; Qin, P.; et al. Porcine deltacoronavirus engages the transmissible gastroenteritis virus functional receptor porcine aminopeptidase N for infectious cellular entry. J. Virol. 2018, 92, e00318-18. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, K.; Feng, J.; Chen, G.; Li, D.; Zhou, L.; Bai, Y.; Wu, Q.; Ma, J. The detection and phylogenetic analysis of porcine deltacoronavirus from Guangdong Province in Southern China. Transbound. Emerg. Dis. 2018, 65, 166–173. [Google Scholar] [CrossRef]
- Fu, J.; Chen, R.; Hu, J.; Qu, H.; Zhao, Y.; Cao, S.; Wen, X.; Wen, Y.; Wu, R.; Zhao, Q.; et al. Identification of a novel linear B-Cell epitope on the nucleocapsid protein of porcine deltacoronavirus. Int. J. Mol. Sci. 2020, 21, 648. [Google Scholar] [CrossRef] [Green Version]
- Ajayi, T.; Dara, R.; Misener, M.; Pasma, T.; Moser, L.; Poljak, Z. Herd-level prevalence and incidence of porcine epidemic diarrhoea virus (PEDV) and porcine deltacoronavirus (PDCoV) in swine herds in Ontario, Canada. Transbound. Emerg. Dis. 2018, 65, 1197–1207. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Chiou, M.T.; Hsueh, F.C.; Lin, W.H.; Lin, C.F.; Yang, C.Y.; Pan, W.P. Development of a UPL probe based real-time PCR assay of porcine deltacoronavirus in Taiwan. Taiwan Vet. J. 2021, 46, 5. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Lee, C. Porcine epidemic diarrhea virus: An emerging and re-emerging epizootic swine virus. Virol. J. 2016, 12, 193. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Luo, S.; Gu, J.; Li, Z.; Li, K.; Yuan, W.; Ye, Y.; Li, H.; Ding, Z.; Song, D.; et al. Prevalence and phylogenetic analysis of porcine diarrhea associated viruses in southern China from 2012 to 2018. BMC Vet. Res. 2019, 15, 470. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Fan, B.; Chang, X.; Zhou, J.; Zhao, Y.; Shi, D.; Yu, Z.; He, K.; Li, B. Characterization and evaluation of the pathogenicity of a natural recombinant transmissible gastroenteritis virus in China. Virology. 2020, 545, 24–32. [Google Scholar] [CrossRef]
- Zhang, X.; Zhu, Y.; Zhu, X.; Shi, H.; Chen, J.; Shi, D.; Yuan, J.; Cao, L.; Liu, J.; Dong, H.; et al. Identification of a natural recombinant transmissible gastroenteritis virus between Purdue and Miller clusters in China. Emerg. Microbes Infect. 2017, 6, e74. [Google Scholar] [CrossRef] [Green Version]
- Nefedeva, M.; Titov, I.; Malogolovkin, A. Molecular characteristics of a novel recombinant of porcine epidemic diarrhea virus. Arch. Virol. 2019, 164, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qu, H.; Hu, J.; Fu, J.; Chen, R.; Li, C.; Cao, S.; Wen, Y.; Wu, R.; Zhao, Q.; et al. Characterization and pathogenicity of the porcine deltacoronavirus isolated in southwest China. Viruses. 2019, 11, 1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.T.; Ji, X.; He, W.; Dellicour, S.; Wang, S.; Li, G.; Zhang, L.; Gilbert, M.; Zhu, H.; Xing, G.; et al. Genomic epidemiology, evolution, and transmission dynamics of porcine deltacoronavirus. Mol. Biol. Evol. 2020, 37, 2641–2654. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Yin, Y.; Wang, W.; Cao, L.; Sun, W.; Shi, K.; Lu, H.; Jin, N. Emergence of Thailand-like strains of porcine deltacoronavirus in Guangxi Province, China. Vet. Med. Sci. 2020, 6, 854–859. [Google Scholar] [CrossRef]
- Lin, C.N.; Chang, R.Y.; Su, B.L.; Chueh, L.L. Full genome analysis of a novel type II feline coronavirus NTU156. Virus Genes 2013, 46, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Potocnakova, L.; Bhide, M.; Pulzova, L.B. An introduction to B-cell epitope mapping and in silico epitope prediction. J. Immunol. Res. 2016, 2016, 6760830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigerust, D.J.; Shepherd, V.L. Virus glycosylation: Role in virulence and immune interactions. Trends Microbiol. 2007, 15, 211–218. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDCoV/104-553/ TW-2015 | Genogroups | ||||
|---|---|---|---|---|---|
| G-I | G-II-a | G-II-b | G-II-c | Taiwan Strains | |
| Full-length genome | 98.54–98.74 | 98.50–98.93 | 98.79–99.42 | 97.22–97.80 | - |
| S gene | 97.14–98.70 | 97.46–98.43 | 97.46–99.32 | 95.53–96.51 | - |
| M gene | 99.16–99.44 | 98.88–99.16 | 98.87–99.44 | 98.44–99.30 | - |
| N gene | 98.30–98.91 | 98.50–99.01 | 98.50–99.70 | 97.08–98.71 | 98.40–98.61 |
| Mutations and/or Deletions of Amino Acids | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S1 Region | S2 Region | |||||||||||||||
| PDCoV Strains | 14 | 23 | 38 | 40 | 52 | 123 | 137 | 234 | 302 | 527 | 551 | 630 | 670 | 698 | 907 | 1016 |
| HKU15-44/HK-2009 | V | F | P | R | N | Y | A | N | L | A | A | A | L | A | P | V |
| CHN-HB/CHN-2014 | . | L | . | . | - | . | . | I | . | . | . | . | . | . | . | . |
| CH-01/CHN-2016 | . | L | L | S | - | . | . | S | . | . | . | . | . | . | S | . |
| CHN-QH-2017-1/CHN-2017 | . | L | . | . | . | . | . | S | . | . | . | . | V | S | S | . |
| CHN-AH-2004/CHN-2004 | . | L | . | . | . | . | . | S | . | . | . | . | V | S | S | . |
| Iowa136/USA-2015 | . | L | . | . | . | . | . | R | . | . | V | . | I | S | S | I |
| Nebraska210/USA-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| Ohio137/USA-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| Indiana453/USA-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| Minnesota292/USA-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| Michigan447/USA-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| HKD/JPN-2016 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| AKT/JPN-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| KNU14-04/KOR-2014 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| HKU14-11/KOR-2016 | . | . | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| YUC_UICMPR/MEX-2015 | . | L | . | . | . | . | . | S | . | . | V | . | I | S | S | I |
| CHN-HN-1601/CHN-2016 | . | L | L | S | - | . | V | S | . | . | . | . | I | . | S | I |
| CH-XS/CHN-2018 | . | L | L | S | - | . | V | S | . | . | . | L | I | S | S | . |
| CHJXNI2/CHN-2015 | . | L | L | S | - | . | V | S | . | . | . | . | . | . | S | I |
| CH-GX01/CHN2015 | . | L | L | S | - | . | V | S | . | . | . | . | . | . | S | . |
| CH-HN/CHN-2016 | . | L | L | S | - | . | V | S | . | . | . | . | . | . | S | I |
| HB-BD/CHN-2017 | . | L | L | S | - | H | . | S | . | . | . | . | . | . | S | I |
| AH2018_641/CHN-2018 | . | L | L | S | - | . | . | S | . | . | V | . | . | . | . | . |
| CHN-SC2015/CHN-2016 | . | L | L | S | - | . | . | S | . | . | V | . | I | . | S | . |
| CHN-HG/CHN-2017 | . | L | L | S | - | . | . | S | . | . | V | . | I | . | S | . |
| CH-HA2-2017/CHN-2017 | . | L | L | S | - | . | V | S | . | . | V | . | V | S | T | . |
| Binh21/Vietnam-2015 | A | L | . | . | . | . | . | S | . | . | V | . | V | S | . | . |
| P1_13_ST1_0213/Thai-2013 | A | L | . | . | . | H | . | S | . | . | V | . | V | S | . | . |
| S5016/Thai-2015 | A | L | . | . | . | . | . | S | . | . | . | . | V | S | . | . |
| P1_16_BTL_0115/Lao-2016 | . | L | . | . | . | . | . | S | . | . | . | . | V | S | . | . |
| 104-553/TW-2015 | A | L | L | S | - | H | V | S | V | G | . | V | . | . | S | I |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsueh, F.-C.; Hsu, F.-Y.; Chen, Y.-H.; Shih, H.-C.; Lin, W.-H.; Yang, C.-Y.; Lin, C.-F.; Chiou, M.-T.; Lin, C.-N. Phylogenetic Classification of Global Porcine Deltacoronavirus (PDCoV) Reference Strains and Molecular Characterization of PDCoV in Taiwan. Viruses 2021, 13, 1337. https://doi.org/10.3390/v13071337
Hsueh F-C, Hsu F-Y, Chen Y-H, Shih H-C, Lin W-H, Yang C-Y, Lin C-F, Chiou M-T, Lin C-N. Phylogenetic Classification of Global Porcine Deltacoronavirus (PDCoV) Reference Strains and Molecular Characterization of PDCoV in Taiwan. Viruses. 2021; 13(7):1337. https://doi.org/10.3390/v13071337
Chicago/Turabian StyleHsueh, Fu-Chun, Feng-Yang Hsu, Yu-Hsuan Chen, Hsing-Chun Shih, Wei-Hao Lin, Cheng-Yao Yang, Chuen-Fu Lin, Ming-Tang Chiou, and Chao-Nan Lin. 2021. "Phylogenetic Classification of Global Porcine Deltacoronavirus (PDCoV) Reference Strains and Molecular Characterization of PDCoV in Taiwan" Viruses 13, no. 7: 1337. https://doi.org/10.3390/v13071337
APA StyleHsueh, F.-C., Hsu, F.-Y., Chen, Y.-H., Shih, H.-C., Lin, W.-H., Yang, C.-Y., Lin, C.-F., Chiou, M.-T., & Lin, C.-N. (2021). Phylogenetic Classification of Global Porcine Deltacoronavirus (PDCoV) Reference Strains and Molecular Characterization of PDCoV in Taiwan. Viruses, 13(7), 1337. https://doi.org/10.3390/v13071337