
Low Genetic Diversity of Hepatitis B Virus Surface Gene amongst Australian Blood Donors

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasma Samples
2.2. DNA Extraction and HBV Viral Quantification
2.3. HBV Nested Polymerase Chain Reactions (PCRs)
2.4. HBV Sequencing and Preliminary Analysis
2.5. Genotyping by Phylogenetic Analysis
2.6. Comparative Analysis of Nucleotide Diversity of HBV Small Surface Gene
2.7. Variant Calling
2.8. Identification of HBV Small Surface AA Variations
3. Results
3.1. Demographic and Clinical Characteristics of Study Samples
3.2. HBV Viral Load and PCR Amplicon Quantification
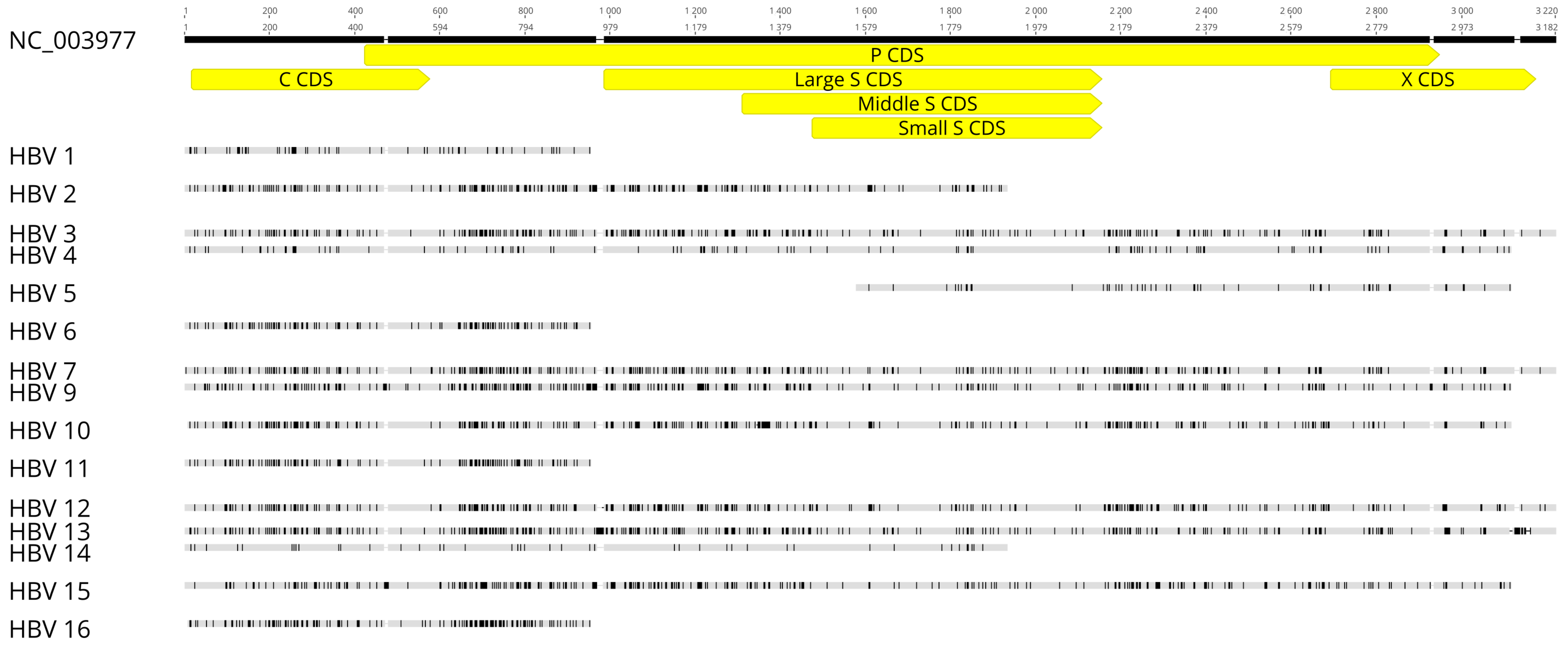
3.3. HBV Sequencing and Preliminary Analysis
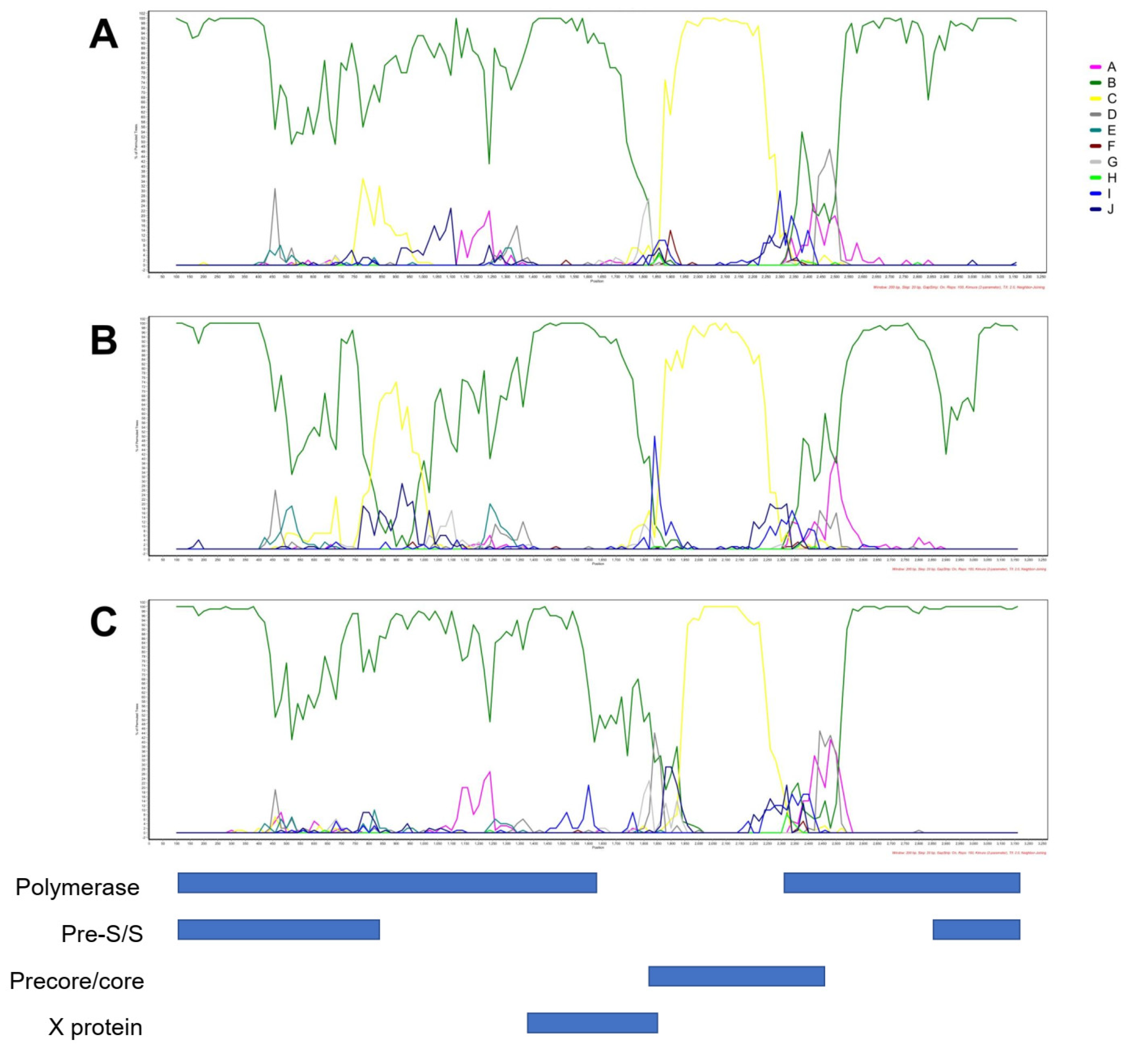
3.4. Genotyping by Phylogenetic Analysis
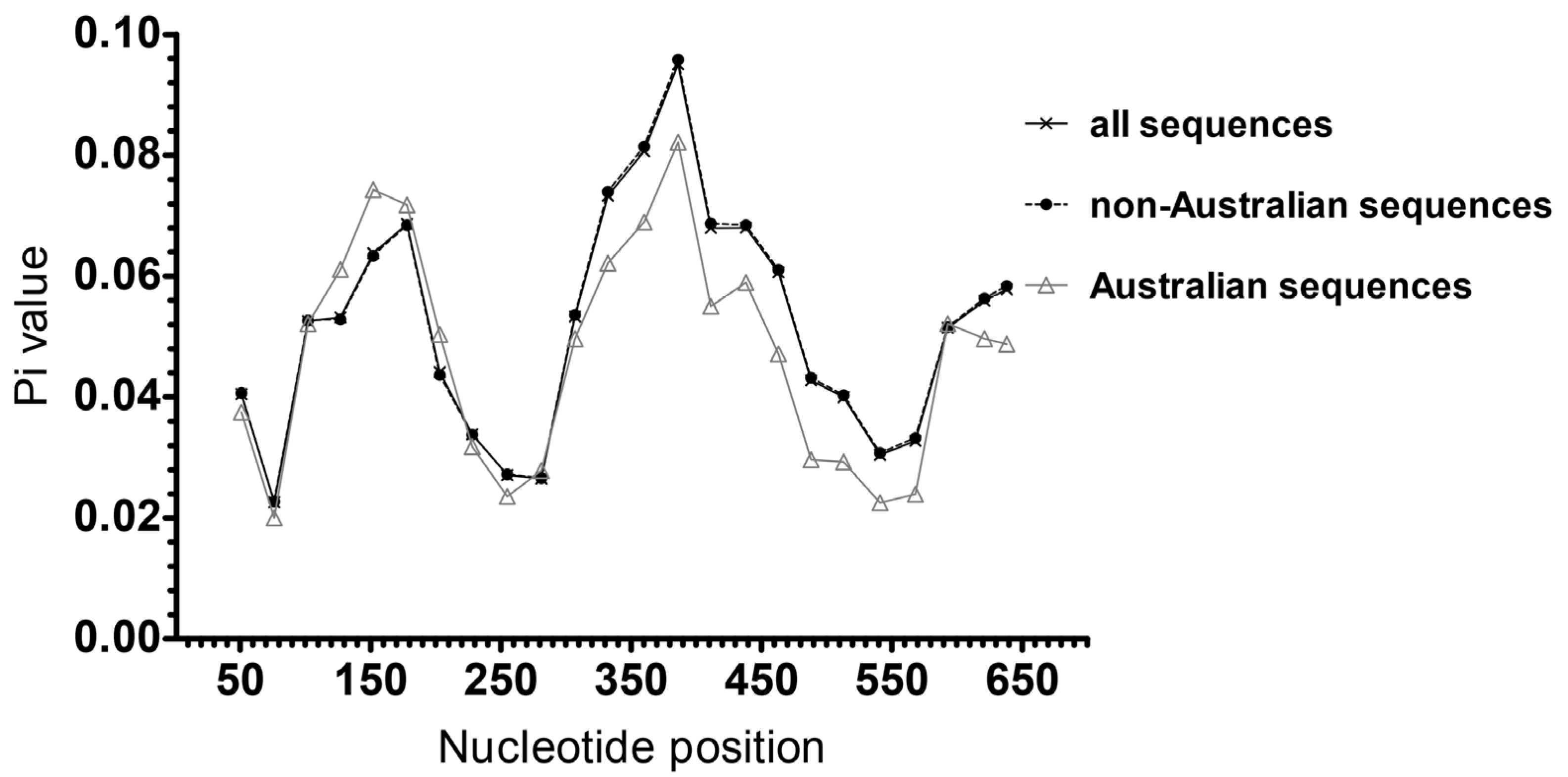
3.5. Analysis of HBsAg Nucleotide and AA Diversity
3.6. Variant Calling
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vos, T.; Barber, R.M.; Bell, B.; Bertozzi-Villa, A.; Biryukov, S.; Bolliger, I.; Charlson, F.; Davis, A.; Degenhardt, L.; Dicker, D. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar] [CrossRef] [Green Version]
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Wang, Z.; Zeng, J.; Li, T.; Zheng, X.; Xu, X.; Ye, X.; Lu, L.; Zhu, W.; Yang, B.; Allain, J.P.; et al. Prevalence of hepatitis B surface antigen (HBsAg) in a blood donor population born prior to and after implementation of universal HBV vaccination in Shenzhen, China. BMC Infect. Dis. 2016, 16, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osei, E.; Lokpo, S.Y.; Agboli, E. Sero-prevalence of hepatitis B infection among blood donors in a secondary care hospital, Ghana (2014): A retrospective analysis. BMC Res. Notes 2017, 10, 391. [Google Scholar] [CrossRef] [Green Version]
- Babanejad, M.; Izadi, N.; Najafi, F.; Alavian, S.M. The HBsAg prevalence among blood donors from eastern Mediterranean and middle eastern countries: A systematic review and meta-analysis. Hepat. Mon. 2016, 16, e35664. [Google Scholar] [CrossRef] [Green Version]
- Romanò, L.; Velati, C.; Cambiè, G.; Fomiatti, L.; Galli, C.; Zanetti, A.R.; SIMTI study group for HBV infection among first-time blood donors. Hepatitis B virus infection among first-time blood donors in Italy: Prevalence and correlates between serological patterns and occult infection. Blood Transfus. 2013, 11, 281–288. [Google Scholar]
- Clive, S.; Laila, K.; Veronica, H.; Rebecca, G. (Eds.) Transfusion-Transmissible Infections in Australia: 2017 Surveillance Report; Australian Red Cross Blood Service, University of New South Wales, and Kirby Institute: Macquarie Park, Australia, 2017. [Google Scholar]
- McNaughton, A.L.; D’Arienzo, V.; Ansari, M.A.; Lumley, S.F.; Littlejohn, M.; Revill, P.; McKeating, J.A.; Matthews, P.C. Insights from deep sequencing of the HBV genome—Unique, tiny, and misunderstood. Gastroenterology 2019, 156, 384–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, A.; Zoulim, F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007, 127, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Hudu, S.A.; Malik, Y.A.; Niazlin, M.T.; Harmal, N.S.; Sekawi, Z. An overview of hepatitis B virus surface antigen mutant in the Asia Pacific. Curr. Issues Mol. Biol. 2013, 16, 69–78. [Google Scholar]
- Norder, H.; Couroucé, A.M.; Coursaget, P.; Echevarria, J.M.; Lee, S.D.; Mushahwar, I.K.; Robertson, B.H.; Locarnini, S.; Magnius, L.O. Genetic diversity of hepatitis B virus strains derived worldwide: Genotypes, subgenotypes, and HBsAg subtypes. Intervirology 2004, 47, 289–309. [Google Scholar] [CrossRef]
- McNaughton, A.L.; Revill, P.A.; Littlejohn, M.; Matthews, P.C.; Ansari, M.A. Analysis of genomic-length HBV sequences to determine genotype and subgenotype reference sequences. J. Gen. Virol. 2020, 101, 271. [Google Scholar] [CrossRef]
- Wang, B.; Feng, Y.; Li, Z.; Duan, H.; Zhao, T.; Zhang, A.; Liu, L.; Baloch, Z.; Xia, X. Distribution and diversity of hepatitis B virus genotypes in Yunnan, China. J. Med. Virol. 2014, 86, 1675–1682. [Google Scholar] [CrossRef] [Green Version]
- SHARI, Z.; Yari, F.; Gharebaghiyan, A. Sequence analysis of the polymerase gene in hepatitis B virus infected blood donors in Iran. Arch. Iran Med. 2012, 15, 88–90. [Google Scholar]
- Olinger, C.M.; Venard, V.; Njayou, M.; Oyefolu, A.O.B.; Maïga, I.; Kemp, A.J.; Omilabu, S.A.; Le Faou, A.; Muller, C.P. Phylogenetic analysis of the precore/core gene of hepatitis B virus genotypes E and A in West Africa: New subtypes, mixed infections and recombinations. J. Gen. Virol. 2006, 87, 1163–1173. [Google Scholar] [CrossRef]
- Dumpis, U.; Holmes, E.; Mendy, M.; Hill, A.; Thursz, M.; Hall, A.; Whittle, H.; Karayiannis, P. Transmission of hepatitis B virus infection in Gambian families revealed by phylogenetic analysis. J. Hepatol. 2001, 35, 99–104. [Google Scholar] [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes 2018, 9, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Frias, F. Quasispecies structure, cornerstone of hepatitis B virus infection: Mass sequencing approach. World J. Gastroenterol. 2013, 19, 6995–7023. [Google Scholar] [CrossRef]
- Karayiannis, P. Hepatitis B virus: Virology, molecular biology, life cycle and intrahepatic spread. Hepatol. Int. 2017, 11, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Kim, Y.; Park, E.; Ryu, H.M.; Jung, G. Fidelity of hepatitis B virus polymerase. JBIC J. Biol. Inorg. Chem. 2003, 270, 2929–2936. [Google Scholar] [CrossRef] [Green Version]
- Yin, F.; Wu, Z.; Fang, W.; Wu, C.; Rayner, S.; Han, M.; Deng, F.; Du, R.; Liu, J.; Wang, M.; et al. Resistant mutations and quasispecies complexity of hepatitis B virus during telbivudine treatment. J. Gen. Virol. 2015, 96, 3302–3312. [Google Scholar] [CrossRef]
- Hatazawa, Y.; Yano, Y.; Okada, R.; Tanahashi, T.; Hayashi, H.; Hirano, H.; Minami, A.; Kawano, Y.; Tanaka, M.; Fukumoto, T.; et al. Quasispecies variant of pre-S/S gene in HBV-related hepatocellular carcinoma with HBs antigen positive and occult infection. Infect. Agents Cancer 2018, 13, 7. [Google Scholar] [CrossRef] [Green Version]
- Lazarevic, I.; Banko, A.; Miljanovic, D.; Cupic, M. Immune-Escape Hepatitis B Virus Mutations Associated with Viral Reactivation upon Immunosuppression. Viruses 2019, 11, 778. [Google Scholar] [CrossRef] [Green Version]
- Candotti, D.; Allain, J.-P. Transfusion-transmitted hepatitis B virus infection. J. Hepatol. 2009, 51, 798–809. [Google Scholar] [CrossRef] [Green Version]
- Pollicino, T.; Amaddeo, G.; Restuccia, A.; Raffa, G.; Alibrandi, A.; Cutroneo, G.; Favaloro, A.; Maimone, S.; Squadrito, G.; Raimondo, G. Impact of hepatitis B virus (HBV) preS/S genomic variability on HBV surface antigen and HBV DNA serum levels. Hepatology 2012, 56, 434–443. [Google Scholar] [CrossRef]
- Rezaee, R.; Poorebrahim, M.; Najafi, S.; Sadeghi, S.; Pourdast, A.; Alavian, S.M.; Poortahmasebi, V.; Alavian, S.E. Impacts of the G145R Mutation on the Structure and Immunogenic Activity of the Hepatitis B Surface Antigen: A Computational Analysis. Zahedan J. Res. Med. Sci. 2016, 16, e39097. [Google Scholar] [CrossRef] [Green Version]
- Mello, F.C.; Martel, N.; Gomes, S.A.; Araujo, N.M. Expression of hepatitis B virus surface antigen containing Y100C variant frequently detected in occult HBV infection. Hepat. Res. Treat. 2011, 2011, 695859. [Google Scholar] [CrossRef]
- Allison, K.M.; Faddy, H.M.; Margaritis, A.; Ismay, S.; Marks, D.C. The impact on blood donor screening for human immunodeficiency virus, hepatitis C virus, and hepatitis B virus using plasma from frozen-thawed plasma preparation tubes. Transfusion 2015, 56, 449–456. [Google Scholar] [CrossRef]
- QIAGEN. For Simultaneous Purification of Viral RNA and DNA from Plasma, Serum, and Cell-Free Body Fluids. QIAamp® MinElute® Virus Spin Handbook. 2010. Available online: https://www.qiagen.com/us/resources/resourcedetail?id=8798cda6-4c55-4c0e-a302-966521c81aec&lang=en (accessed on 22 March 2019).
- Primerdesign™ Ltd. Hepatitis B Virus Core Protein Region Genesig® Standard kit. Available online: https://www.genesig.com/assets/files/hbv_std.pdf (accessed on 10 January 2020).
- Chook, J.B.; Teo, W.L.; Ngeow, Y.F.; Tee, K.K.; Ng, K.P.; Mohamed, R. Universal Primers for Detection and Sequencing of Hepatitis B Virus Genomes across Genotypes A to G. J. Clin. Microbiol. 2015, 53, 1831–1835. [Google Scholar] [CrossRef] [Green Version]
- Invitrogen by ThermoFisher Scientific, User Guide: Invitrogen™ Platinum™ SuperFi™ PCR Master Mix (Pub. No. MAN0014883). Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0014883_Platinum_SuperFi_PCR_MM_UG_not_interactive.pdf (accessed on 1 June 2017).
- Biotium, Product Information: GelRed™ Nucleic Acid Gel Stain, 10,000X. Available online: https://biotium.com/wp-content/uploads/2015/02/PI-41002-41003.pdf (accessed on 15 July 2015).
- Rio, D.C.; Ares, M.; Hannon, G.J.; Nilsen, T.W. Ethanol Precipitation of RNA and the Use of Carriers. Cold Spring Harb. Protoc. 2010, 2010, 5440. [Google Scholar] [CrossRef]
- Illumina, Reference Guide: NexteraTM DNA Flex Library Prep (Document # 1000000025416v07). Available online: https://sapac.support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/illumina_prep/illumina-dna-prep-reference-guide-1000000025416-09.pdf (accessed on 8 August 2019).
- Rozanov, M.; Plikat, U.; Chappey, C.; Kochergin, A.; Tatusova, T. A web-based genotyping resource for viral sequences. Nucleic Acids Res. 2004, 32, W654–W659. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Misawa, K.I.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Dhanda, S.K.; Mahajan, S.; Paul, S.; Yan, Z.; Kim, H.; Jespersen, M.C.; Jurtz, V.; Andreatta, M.; Greenbaum, J.A.; Marcatili, P.; et al. IEDB-AR: Immune epitope database—analysis resource in 2019. Nucleic Acids Res. 2019, 47, W502–W506. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, P.; Kim, Y.; Haste-Andersen, P.; Beaver, J.; Bourne, P.E.; Bui, H.-H.; Buus, S.; Frankild, S.; Greenbaum, J.; et al. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 2008, 36, W513–W518. [Google Scholar] [CrossRef] [Green Version]
- Neumann-Fraune, M.; Beggel, B.; Kaiser, R.; Obermeier, M. Hepatitis B Virus Drug Resistance Tools: One Sequence, Two Predictions. Intervirology 2014, 57, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.L.; Li, X.; Li, J.; Zhang, Z.H. Genetic variation of occult hepatitis B virus infection. World J. Gastroenterol. 2016, 22, 3531–3546. [Google Scholar] [CrossRef]
- Phan, N.M.H.; Faddy, H.; Flower, R.; Spann, K.; Roulis, E. In Silico Analysis of Genetic Diversity of Human Hepatitis B Virus in Southeast Asia, Australia and New Zealand. Viruses 2020, 12, 427. [Google Scholar] [CrossRef] [Green Version]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [Green Version]
- Torresi, J.; Earnest-Silveira, L.; Deliyannisd, G.; Edgttond, K.; Zhuange, H.; Locarnini, S.A.; Fyfec, J.; Sozzic, T.; Jackson, D.C. Reduced Antigenicity of the Hepatitis B Virus HBsAg Protein Arising as a Consequence of Sequence Changes in the Overlapping Polymerase Gene That Are Selected by Lamivudine Therapy. Virology 2002, 293, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Inoue, T.; Tanaka, Y. Cross-Protection of Hepatitis B Vaccination among Different Genotypes. Vaccines 2020, 8, 456. [Google Scholar] [CrossRef]
- Kleinman, S.H.; Lelie, N.; Busch, M.P. Infectivity of human immunodeficiency virus-1, hepatitis C virus, and hepatitis B virus and risk of transmission by transfusion. Transfusion 2009, 49, 2454–2489. [Google Scholar] [CrossRef] [PubMed]
- Putri, W.A.; Yano, Y.; Yamani, L.N.; Liang, Y.; Mardian, Y.; Utsumi, T.; Lusida, M.I.; Hayashi, Y. Association between quasispecies variants of hepatitis B virus, as detected by high-throughput sequencing, and progression of advanced liver disease in Indonesian patients. Mol. Med. Rep. 2019, 20, 16–24. [Google Scholar]
- HIV, Viral Hepatitis and Sexually Transmissible Infections in Australia: Annual Surveillance Report 2018; Kirby Institute and UNSW Sydney: Sydney, Australia, 2018.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Date Collected | HIV-1/HCV/HBV Multiplex NAT | HBV Discriminatory NAT | HCV Discriminatory NAT | HIV-1 Discriminatory NAT | Total Anti-HBc | Anti-HBc IgM | Anti-HBe | HbeAg | Total Anti-HBs | HBsAg |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HBV 1 | 12-Dec-97 | REAC | REAC | NONREAC | NONREAC | POS | NEG | POS | NEG | NEG | POS |
| HBV 2 | 12-Dec-97 | REAC | REAC | NONREAC | NONREAC | POS | NA | NA | NA | NA | POS |
| HBV 3 | 06-Apr-98 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 4 | 31-May-02 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 5 | 25-Sep-02 | REAC | REAC | REAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 6 | 11-Oct-02 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 7 | 29-Jan-03 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 8 | 07-Dec-03 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 9 | 09-Feb-04 | REAC | REAC | NONREAC | NONREAC | POS | NEG | POS | NEG | NEG | POS |
| HBV 10 | 05-Mar-04 | REAC | REAC | NONREAC | NONREAC | POS | NEG | POS | NEG | NEG | POS |
| HBV 11 | 11-Mar-04 | REAC | REAC | NONREAC | NONREAC | POS | NEG | POS | NEG | NEG | POS |
| HBV 12 | 01-Apr-04 | REAC | REAC | NONREAC | INVALID | NA | NA | NA | NA | NA | POS |
| HBV 13 | 20-May-04 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 14 | 05-Jul-04 | REAC | REAC | NONREAC | NONREAC | NA | NA | NA | NA | NA | POS |
| HBV 15 | 14-Apr-11 | REAC | REAC | NONREAC | NONREAC | POS | NA | NA | NA | NEG | POS |
| HBV 16 | 26-May-11 | REAC | REAC | NONREAC | NONREAC | POS | NA | NA | NA | NEG | POS |
| Genotype | Determined by the Phylogeny of Core Genes | Determined by the Phylogeny of Polymerase/Large Surface Genes | Determined by Phylogeny of Whole Genomes | Consensus Genotype | |
|---|---|---|---|---|---|
| Sample | |||||
| HBV 1 | D | N/A * | N/A * | D + | |
| HBV 2 | C | N/A * | N/A * | C + | |
| HBV 3 | C | B | B | B | |
| HBV 4 | D | D | N/A * | D | |
| HBV 5 | N/A * | N/A * | N/A * | N/A * | |
| HBV 6 | C | N/A * | N/A * | C + | |
| HBV 7 | C | B | B | B | |
| HBV 8 | N/A * | N/A * | N/A * | N/A * | |
| HBV 9 | A | A | N/A * | A | |
| HBV 10 | C | C | N/A * | C | |
| HBV 11 | C | N/A * | N/A * | C + | |
| HBV 12 | C | C | C | C | |
| HBV 13 | C | B | B | B | |
| HBV 14 | D | N/A * | N/A * | D + | |
| HBV 15 | A | A | N/A * | A | |
| HBV 16 | C | N/A * | N/A * | C + | |
| Sample | Sequence Length (bp)/Genotype | Polymerase Sequences | Large Surface Sequences | Middle Surface Sequences | Small Surface Sequences | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total SNV/Length (bp) | sSNV | nsSNV | Total SNV/Length (bp) | sSNV | nsSNV | Total SNV/Length (bp) | sSNV | nsSNV | Total SNV/Length (bp) | sSNV | nsSNV | ||
| HBV 1 | 945/D | N/A | N/A | N/A | N/A | ||||||||
| HBV 2 | 1910/C | N/A | N/A | N/A | N/A | ||||||||
| HBV 3 | 3182/B * | 198/2499 | 112 | 86 | 87/1170 | 49 | 38 | 52/846 | 24 | 28 | 32/681 | 13 | 20 |
| HBV 4 | 3089/D | 93/2499 | 60 | 33 | 30/1170 | 15 | 15 | 17/846 | 7 | 10 | 11/681 | 6 | 5 |
| HBV 5 | 1530 | N/A | N/A | N/A | N/A | ||||||||
| HBV 6 | 947/C | N/A | N/A | N/A | N/A | ||||||||
| HBV 7 | 3182/B * | 201/2499 | 118 | 83 | 93/1170 | 52 | 41 | 57/846 | 26 | 31 | 40/681 | 17 | 23 |
| HBV 8 | N/A | N/A | N/A | N/A | N/A | ||||||||
| HBV 9 | 3100/A | 198/2511 | 104 | 94 | 79/1170 | 39 | 40 | 48/846 | 19 | 29 | 31/681 | 13 | 18 |
| HBV 10 | 3080/C | 122/1710 | 57 | 65 | 36/420 | 26 | 10 | 11/96 | 5 | 6 | 25/681 | 10 | 15 |
| HBV 11 | 947/C | N/A | N/A | N/A | N/A | ||||||||
| HBV 12 | 3181/C * | 133/1710 | 59 | 11 | 67/1170 | 38 | 29 | 41/846 | 18 | 23 | 26/681 | 9 | 17 |
| HBV 13 | 3197/B * | 208/2514 | 116 | 92 | 98/1170 | 55 | 43 | 61/846 | 27 | 34 | 37/681 | 16 | 21 |
| HBV 14 | 1910/D | N/A | N/A | N/A | N/A | ||||||||
| HBV 15 | 3093/A | 196/2505 | 108 | 88 | 77/1170 | 44 | 10 | 44/846 | 19 | 25 | 29/681 | 13 | 16 |
| HBV 16 | 939/C | N/A | N/A | N/A | N/A | ||||||||
| Variant Frequency | Polymerase Gene | Large Surface Gene | Middle Surface Gene | Small Surface Gene | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total | sSNV | nsSNV | Total | sSNV | nsSNV | Total | sSNV | nsSNV | Total | sSNV | nsSNV | |
| >20–80% | 20 (17%) | 5 | 15 | 15 (37%) | 6 | 9 | 12 (86%) | 4 | 8 | 1 (5%) | 0 | 1 |
| >80–100% | 97 (83%) | 51 | 46 | 26 (63%) | 22 | 4 | 2 (14%) | 2 | 0 | 20 (95%) | 9 | 11 |
| Total | 117 | 56 | 61 | 41 | 37 | 30 | 14 | 12 | 15 | 21 | 9 | 15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, N.M.H.; Faddy, H.M.; Flower, R.L.; Dimech, W.J.; Spann, K.M.; Roulis, E.V. Low Genetic Diversity of Hepatitis B Virus Surface Gene amongst Australian Blood Donors. Viruses 2021, 13, 1275. https://doi.org/10.3390/v13071275
Phan NMH, Faddy HM, Flower RL, Dimech WJ, Spann KM, Roulis EV. Low Genetic Diversity of Hepatitis B Virus Surface Gene amongst Australian Blood Donors. Viruses. 2021; 13(7):1275. https://doi.org/10.3390/v13071275
Chicago/Turabian StylePhan, Ngoc Minh Hien, Helen M. Faddy, Robert L. Flower, Wayne J. Dimech, Kirsten M. Spann, and Eileen V. Roulis. 2021. "Low Genetic Diversity of Hepatitis B Virus Surface Gene amongst Australian Blood Donors" Viruses 13, no. 7: 1275. https://doi.org/10.3390/v13071275
APA StylePhan, N. M. H., Faddy, H. M., Flower, R. L., Dimech, W. J., Spann, K. M., & Roulis, E. V. (2021). Low Genetic Diversity of Hepatitis B Virus Surface Gene amongst Australian Blood Donors. Viruses, 13(7), 1275. https://doi.org/10.3390/v13071275