
Viral Control of Glioblastoma


{kind=link}
{kind=link}
{kind=link}
Abstract
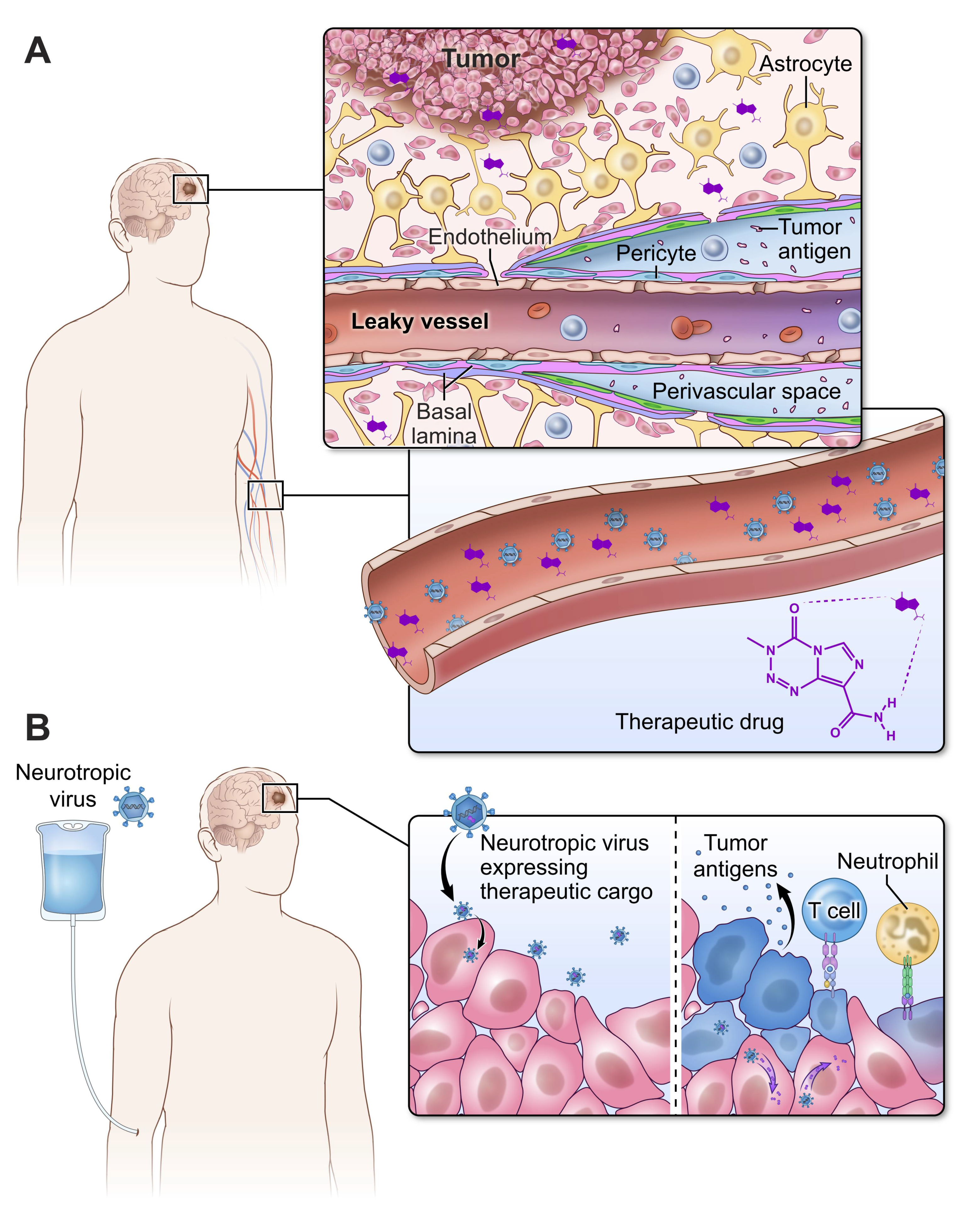
:1. Introduction
2. Endogenous Viruses Found in GBM
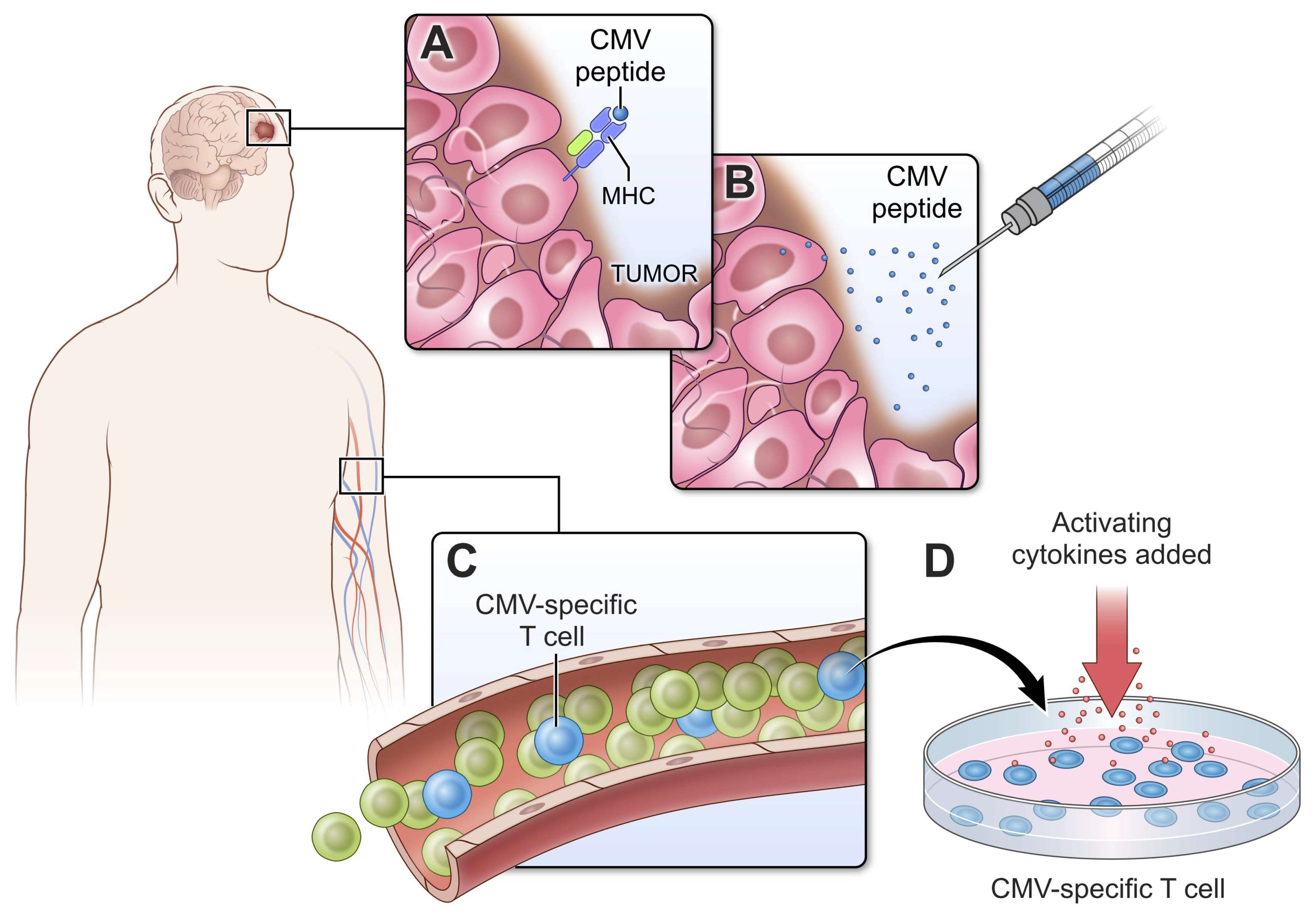
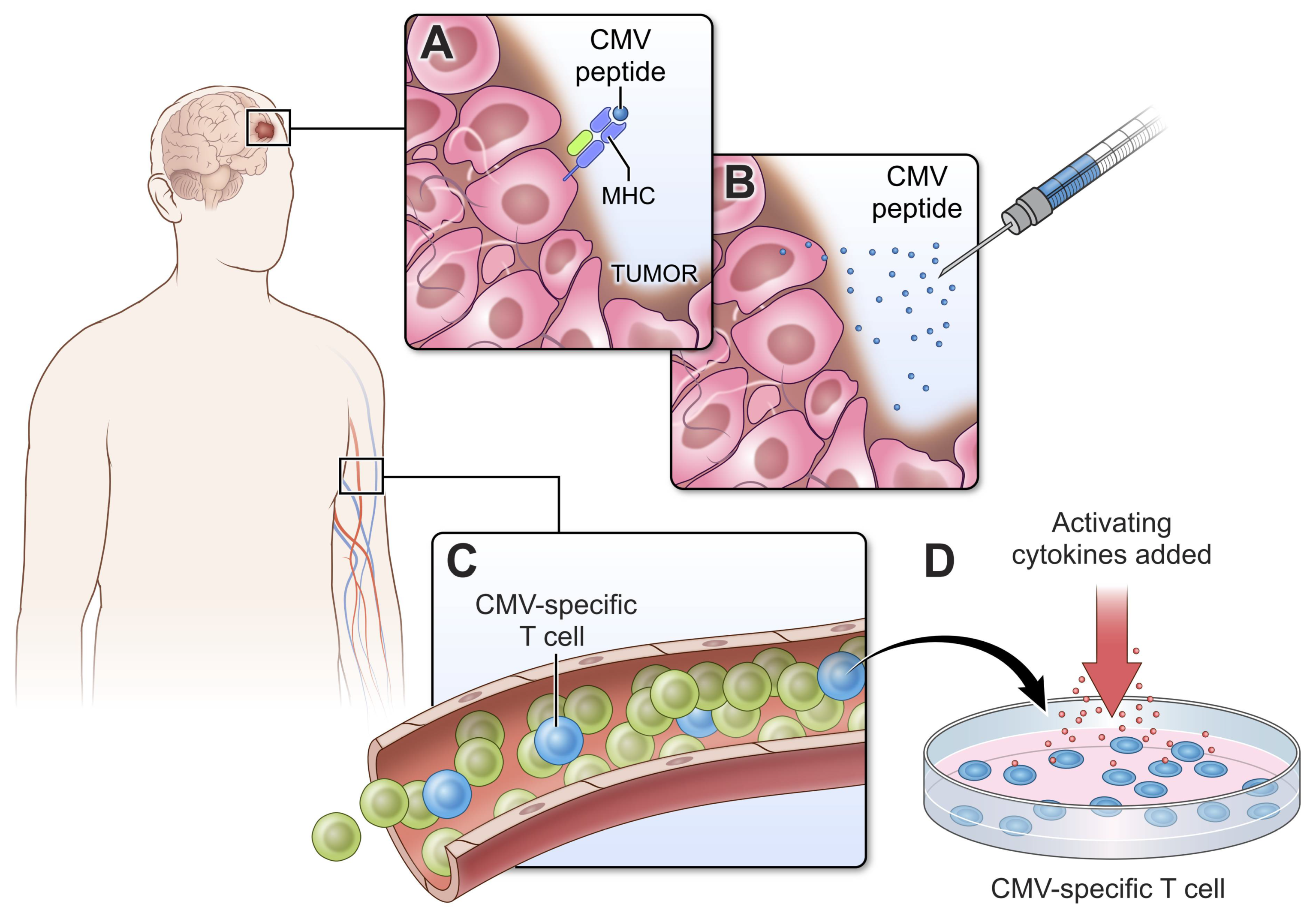
2.1. Cytomegalovirus
2.2. Epstein–Barr Virus
2.3. Human Endogenous Retroviruses
2.4. Exploiting Endogenous Viruses in GBM for Treatment
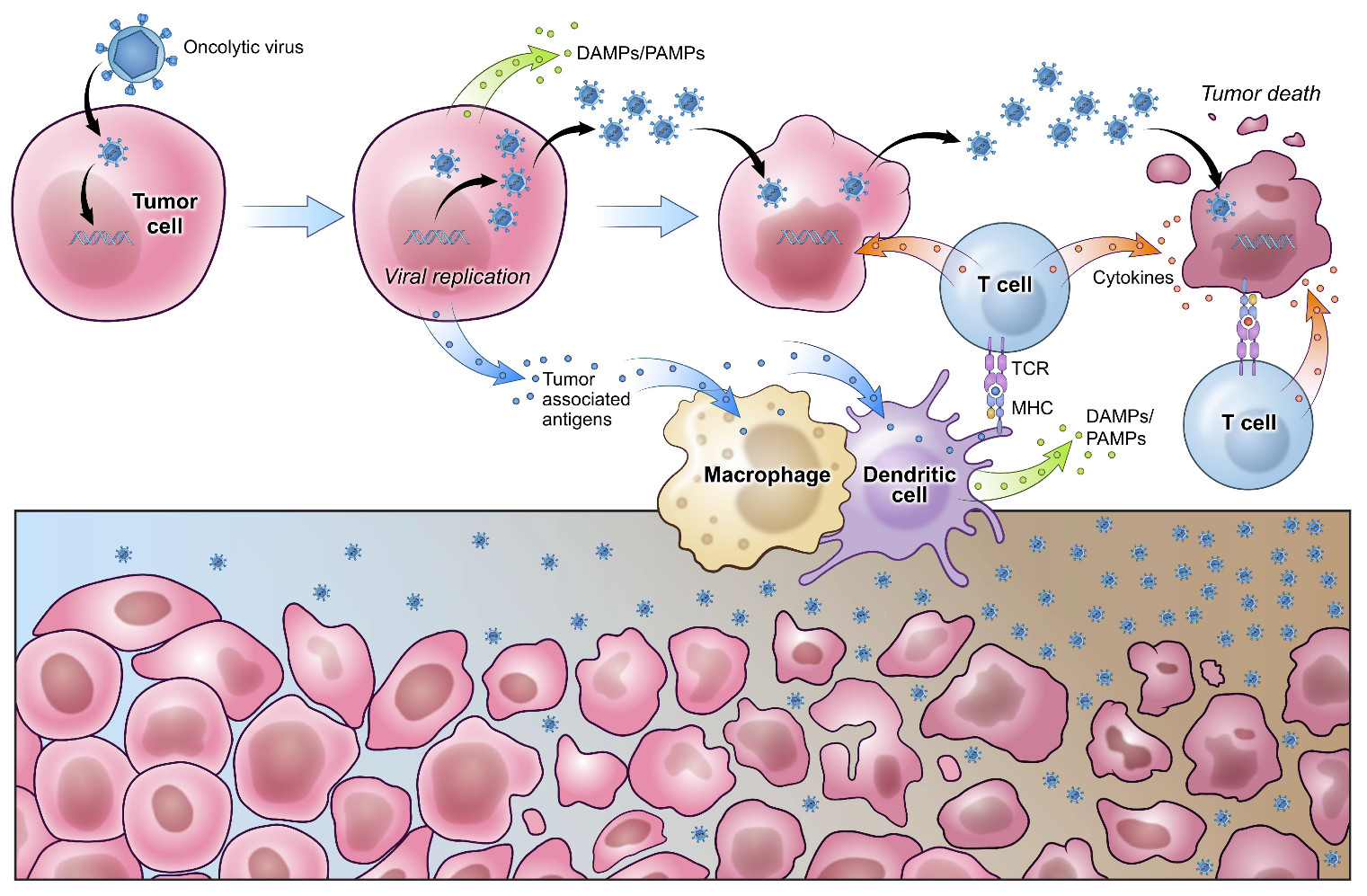
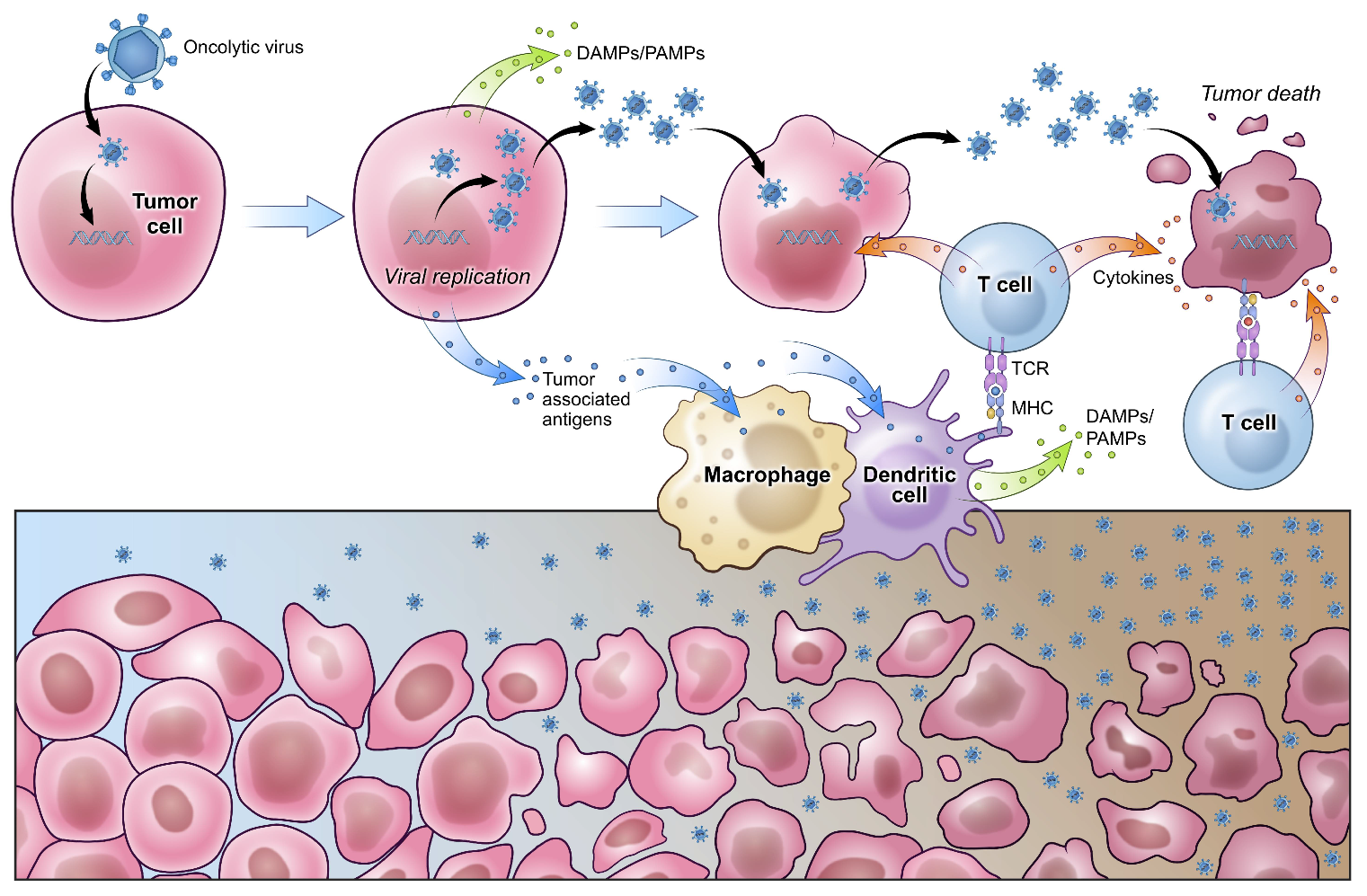
3. Oncolytic Viral Therapy
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gimple, R.C.; Bhargava, S.; Dixit, D.; Rich, J.N. Glioblastoma stem cells: Lessons from the tumor hierarchy in a lethal cancer. Genes Dev. 2019, 33, 591–609. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Fulop, J.; Liu, M.; Blanda, R.; Kromer, C.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-Oncology 2015, 17 (Suppl. 4), iv1–iv62. [Google Scholar] [CrossRef]
- Lee, J.H.; Lee, J.E.; Kahng, J.Y.; Kim, S.H.; Park, J.S.; Yoon, S.J.; Um, J.Y.; Kim, W.K.; Lee, J.K.; Park, J.; et al. Human glioblastoma arises from subventricular zone cells with low-level driver mutations. Nature 2018, 560, 243–247. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (≥ 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef]
- Fukushima, S.; Narita, Y.; Miyakita, Y.; Ohno, M.; Takizawa, T.; Takusagawa, Y.; Mori, M.; Ichimura, K.; Tsuda, H.; Shibui, S. A case of more than 20 years survival with glioblastoma, and development of cavernous angioma as a delayed complication of radiotherapy. Neuropathology 2013, 33, 576–581. [Google Scholar] [CrossRef]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs Maintenance Temozolomide Alone on Survival in Patients With Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306–2316. [Google Scholar] [CrossRef] [Green Version]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E., 2nd; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- De Bonis, P.; Albanese, A.; Lofrese, G.; de Waure, C.; Mangiola, A.; Pettorini, B.L.; Pompucci, A.; Balducci, M.; Fiorentino, A.; Lauriola, L.; et al. Postoperative infection may influence survival in patients with glioblastoma: Simply a myth? Neurosurgery 2011, 69, 864–868. [Google Scholar] [CrossRef]
- Eakin, E. Bacteria on the Brain. The New Yorker, 7 December 2015. [Google Scholar]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar] [PubMed]
- Scheurer, M.E.; Bondy, M.L.; Aldape, K.D.; Albrecht, T.; El-Zein, R. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008, 116, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poltermann, S.; Schlehofer, B.; Steindorf, K.; Schnitzler, P.; Geletneky, K.; Schlehofer, J.R. Lack of association of herpesviruses with brain tumors. J. Neurovirol. 2006, 12, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro-Oncology 2008, 10, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatier, J.; Uro-Coste, E.; Pommepuy, I.; Labrousse, F.; Allart, S.; Trémoulet, M.; Delisle, M.B.; Brousset, P. Detection of human cytomegalovirus genome and gene products in central nervous system tumours. Br. J. Cancer 2005, 92, 747–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; McGregor Dallas, S.R.; Smit, M.; Soroceanu, L.; Cobbs, C.S. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro-Oncology 2012, 14, 246–255. [Google Scholar] [CrossRef]
- Ranganathan, P.; Clark, P.A.; Kuo, J.S.; Salamat, M.S.; Kalejta, R.F. Significant association of multiple human cytomegalovirus genomic Loci with glioblastoma multiforme samples. J. Virol. 2012, 86, 854–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spector, D.H. Human cytomegalovirus riding the cell cycle. Med. Microbiol. Immunol. 2015, 204, 409–419. [Google Scholar] [CrossRef]
- Stragliotto, G.; Pantalone, M.R.; Rahbar, A.; Bartek, J.; Söderberg-Naucler, C. Valganciclovir as Add-on to Standard Therapy in Glioblastoma Patients. Clin. Cancer Res. 2020, 26, 4031–4039. [Google Scholar] [CrossRef] [PubMed]
- Krenzlin, H.; Behera, P.; Lorenz, V.; Passaro, C.; Zdioruk, M.; Nowicki, M.O.; Grauwet, K.; Zhang, H.; Skubal, M.; Ito, H.; et al. Cytomegalovirus promotes murine glioblastoma growth via pericyte recruitment and angiogenesis. J. Clin. Investig. 2019, 129, 1671–1683. [Google Scholar] [CrossRef] [Green Version]
- Münz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salahuddin, S.; Fath, E.K.; Biel, N.; Ray, A.; Moss, C.R.; Patel, A.; Patel, S.; Hilding, L.; Varn, M.; Ross, T.; et al. Epstein-Barr Virus Latent Membrane Protein-1 Induces the Expression of SUMO-1 and SUMO-2/3 in LMP1-positive Lymphomas and Cells. Sci. Rep. 2019, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.; Begue-Pastor, N.; Chaganti, S.; Croom-Carter, D.; Shannon-Lowe, C.; Kube, D.; Feederle, R.; Delecluse, H.J.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus infection of naïve B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: Implications for virus biology. PLoS Pathog. 2012, 8, e1002697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between Epstein-Barr virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Menet, A.; Speth, C.; Larcher, C.; Prodinger, W.M.; Schwendinger, M.G.; Chan, P.; Jäger, M.; Schwarzmann, F.; Recheis, H.; Fontaine, M.; et al. Epstein-Barr virus infection of human astrocyte cell lines. J. Virol. 1999, 73, 7722–7733. [Google Scholar] [CrossRef] [Green Version]
- Słońska, A.; Cymerys, J.; Chodkowski, M.; Bąska, P.; Krzyżowska, M.; Bańbura, M.W. Human herpesvirus type 2 infection of primary murine astrocytes causes disruption of the mitochondrial network and remodeling of the actin cytoskeleton: An in vitro morphological study. Arch. Virol. 2021, 166, 1371–1383. [Google Scholar] [CrossRef]
- Fujimoto, H.; Asaoka, K.; Imaizumi, T.; Ayabe, M.; Shoji, H.; Kaji, M. Epstein-Barr virus infections of the central nervous system. Intern. Med. 2003, 42, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soldan, S.S.; Lieberman, P.M. Epstein-Barr Virus Infection in the Development of Neurological Disorders. Drug Discov. Today Dis. Models 2020, 32 Pt A, 35–52. [Google Scholar] [CrossRef]
- Strong, M.J.; Blanchard, E.t.; Lin, Z.; Morris, C.A.; Baddoo, M.; Taylor, C.M.; Ware, M.L.; Flemington, E.K. A comprehensive next generation sequencing-based virome assessment in brain tissue suggests no major virus–tumor association. Acta Neuropathol. Commun. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markovitz, D.M. “Reverse genomics” and human endogenous retroviruses. Trans. Am. Clin. Climatol. Assoc. 2014, 125, 57–62. [Google Scholar] [PubMed]
- Grow, E.J.; Flynn, R.A.; Chavez, S.L.; Bayless, N.L.; Wossidlo, M.; Wesche, D.J.; Martin, L.; Ware, C.B.; Blish, C.A.; Chang, H.Y.; et al. Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 2015, 522, 221–225. [Google Scholar] [CrossRef] [Green Version]
- Mayer, J.; Sauter, M.; Rácz, A.; Scherer, D.; Mueller-Lantzsch, N.; Meese, E. An almost-intact human endogenous retrovirus K on human chromosome 7. Nat. Genet. 1999, 21, 257–258. [Google Scholar] [CrossRef]
- Serafino, A.; Balestrieri, E.; Pierimarchi, P.; Matteucci, C.; Moroni, G.; Oricchio, E.; Rasi, G.; Mastino, A.; Spadafora, C.; Garaci, E.; et al. The activation of human endogenous retrovirus K (HERV-K) is implicated in melanoma cell malignant transformation. Exp. Cell Res. 2009, 315, 849–862. [Google Scholar] [CrossRef]
- Kleiman, A.; Senyuta, N.; Tryakin, A.; Sauter, M.; Karseladze, A.; Tjulandin, S.; Gurtsevitch, V.; Mueller-Lantzsch, N. HERV-K(HML-2) GAG/ENV antibodies as indicator for therapy effect in patients with germ cell tumors. Int. J. Cancer 2004, 110, 459–461. [Google Scholar] [CrossRef]
- Ma, W.; Hong, Z.; Liu, H.; Chen, X.; Ding, L.; Liu, Z.; Zhou, F.; Yuan, Y. Human Endogenous Retroviruses-K (HML-2) Expression Is Correlated with Prognosis and Progress of Hepatocellular Carcinoma. Biomed. Res. Int. 2016, 2016, 8201642. [Google Scholar] [CrossRef]
- Ishida, T.; Obata, Y.; Ohara, N.; Matsushita, H.; Sato, S.; Uenaka, A.; Saika, T.; Miyamura, T.; Chayama, K.; Nakamura, Y.; et al. Identification of the HERV-K gag antigen in prostate cancer by SEREX using autologous patient serum and its immunogenicity. Cancer Immun. 2008, 8, 15. [Google Scholar]
- Kahyo, T.; Tao, H.; Shinmura, K.; Yamada, H.; Mori, H.; Funai, K.; Kurabe, N.; Suzuki, M.; Tanahashi, M.; Niwa, H.; et al. Identification and association study with lung cancer for novel insertion polymorphisms of human endogenous retrovirus. Carcinogenesis 2013, 34, 2531–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang-Johanning, F.; Radvanyi, L.; Rycaj, K.; Plummer, J.B.; Yan, P.; Sastry, K.J.; Piyathilake, C.J.; Hunt, K.K.; Johanning, G.L. Human endogenous retrovirus K triggers an antigen-specific immune response in breast cancer patients. Cancer Res. 2008, 68, 5869–5877. [Google Scholar] [CrossRef] [Green Version]
- Wang-Johanning, F.; Liu, J.; Rycaj, K.; Huang, M.; Tsai, K.; Rosen, D.G.; Chen, D.T.; Lu, D.W.; Barnhart, K.F.; Johanning, G.L. Expression of multiple human endogenous retrovirus surface envelope proteins in ovarian cancer. Int. J. Cancer 2007, 120, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Yang, Y.; Zhang, N.; Soto, C.; Jiang, X.; An, Z.; Zheng, W.J. Human Endogenous Retroviruses in Glioblastoma Multiforme. Microorganisms 2021, 9, 764. [Google Scholar] [CrossRef] [PubMed]
- Kessler, A.F.; Wiesner, M.; Denner, J.; Kämmerer, U.; Vince, G.H.; Linsenmann, T.; Löhr, M.; Ernestus, R.I.; Hagemann, C. Expression-analysis of the human endogenous retrovirus HERV-K in human astrocytic tumors. BMC Res. Notes 2014, 7, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, E.; Mao, T.; Dong, H.; Boisserand, L.S.B.; Antila, S.; Bosenberg, M.; Alitalo, K.; Thomas, J.L.; Iwasaki, A. VEGF-C-driven lymphatic drainage enables immunosurveillance of brain tumours. Nature 2020, 577, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ozga, A.J.; Chow, M.T.; Luster, A.D. Chemokines and the immune response to cancer. Immunity 2021, 54, 859–874. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e21. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Carare, R.O.; Bernardes-Silva, M.; Newman, T.A.; Page, A.M.; Nicoll, J.A.; Perry, V.H.; Weller, R.O. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: Significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol. Appl. Neurobiol. 2008, 34, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Ineichen, B.V.; Detmar, M.; Proulx, S.T. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat. Commun. 2017, 8, 1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kida, S.; Pantazis, A.; Weller, R.O. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol. Appl. Neurobiol. 1993, 19, 480–488. [Google Scholar] [CrossRef]
- Ahn, J.H.; Cho, H.; Kim, J.H.; Kim, S.H.; Ham, J.S.; Park, I.; Suh, S.H.; Hong, S.P.; Song, J.H.; Hong, Y.K.; et al. Meningeal lymphatic vessels at the skull base drain cerebrospinal fluid. Nature 2019, 572, 62–66. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012, 33, 579–589. [Google Scholar] [CrossRef]
- Mastorakos, P.; McGavern, D. The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 2019, 4, eaav0492. [Google Scholar] [CrossRef]
- Greter, M.; Heppner, F.L.; Lemos, M.P.; Odermatt, B.M.; Goebels, N.; Laufer, T.; Noelle, R.J.; Becher, B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat. Med. 2005, 11, 328–334. [Google Scholar] [CrossRef]
- Lodygin, D.; Odoardi, F.; Schläger, C.; Körner, H.; Kitz, A.; Nosov, M.; van den Brandt, J.; Reichardt, H.M.; Haberl, M.; Flügel, A. A combination of fluorescent NFAT and H2B sensors uncovers dynamics of T cell activation in real time during CNS autoimmunity. Nat. Med. 2013, 19, 784–790. [Google Scholar] [CrossRef]
- Kawakami, N.; Nägerl, U.V.; Odoardi, F.; Bonhoeffer, T.; Wekerle, H.; Flügel, A. Live imaging of effector cell trafficking and autoantigen recognition within the unfolding autoimmune encephalomyelitis lesion. J. Exp. Med. 2005, 201, 1805–1814. [Google Scholar] [CrossRef]
- Khan, N.; Shariff, N.; Cobbold, M.; Bruton, R.; Ainsworth, J.A.; Sinclair, A.J.; Nayak, L.; Moss, P.A. Cytomegalovirus seropositivity drives the CD8 T cell repertoire toward greater clonality in healthy elderly individuals. J. Immunol. 2002, 169, 1984–1992. [Google Scholar] [CrossRef] [Green Version]
- Alejenef, A.; Pachnio, A.; Halawi, M.; Christmas, S.E.; Moss, P.A.; Khan, N. Cytomegalovirus drives Vδ2neg γδ T cell inflation in many healthy virus carriers with increasing age. Clin. Exp. Immunol. 2014, 176, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, A.R.; Shin, E.C. Cytomegalovirus Infection and Memory T Cell Inflation. Immune Netw. 2015, 15, 186–190. [Google Scholar] [CrossRef] [Green Version]
- Facoetti, A.; Nano, R.; Zelini, P.; Morbini, P.; Benericetti, E.; Ceroni, M.; Campoli, M.; Ferrone, S. Human leukocyte antigen and antigen processing machinery component defects in astrocytic tumors. Clin. Cancer Res. 2005, 11, 8304–8311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, M.; Sipos, B.; Pieper, N.; Biskup, S. Major histocompatibility complex class 1 (MHC1) loss among patients with glioblastoma (GBM). J. Clin. Oncol. 2020, 38, e14523. [Google Scholar] [CrossRef]
- Yeung, J.T.; Hamilton, R.L.; Ohnishi, K.; Ikeura, M.; Potter, D.M.; Nikiforova, M.N.; Ferrone, S.; Jakacki, R.I.; Pollack, I.F.; Okada, H. LOH in the HLA class I region at 6p21 is associated with shorter survival in newly diagnosed adult glioblastoma. Clin. Cancer Res. 2013, 19, 1816–1826. [Google Scholar] [CrossRef] [Green Version]
- Satoh, E.; Mabuchi, T.; Satoh, H.; Asahara, T.; Nukui, H.; Naganuma, H. Reduced expression of the transporter associated with antigen processing 1 molecule in malignant glioma cells, and its restoration by interferon-gamma and -beta. J. Neurosurg. 2006, 104, 264–271. [Google Scholar] [CrossRef]
- Cuburu, N.; Bialkowski, L.; Pontejo, S.M.; Bell, A.; Kim, R.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T. Abstract 5563: Harnessing pre-existing viral immunity for development of a broadly applicable tumor immunotherapy. Cancer Res. 2020, 80, 5563. [Google Scholar]
- Thompson, E.; Landi, D.; Thompson, E.; Lipp, E.; Balajonda, B.; Herndon, J., II; Buckley, E.; Flahiff, C.; Jaggers, D.; Schroeder, K.; et al. CTIM-21. Peptide vaccine directed to CMV pp65 for treatment of recurrent malignant glioma and medulloblastoma in children and young adults: Preliminary results of a phase I trial. Neuro-Oncology 2020, 22 (Suppl. 2), ii37. [Google Scholar] [CrossRef]
- Batich, K.A.; Mitchell, D.A.; Healy, P.; Herndon, J.E.; Sampson, J.H. Once, Twice, Three Times a Finding: Reproducibility of Dendritic Cell Vaccine Trials Targeting Cytomegalovirus in Glioblastoma. Clin. Cancer Res. 2020, 26, 5297. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, D.A.; Batich, K.A.; Gunn, M.D.; Huang, M.N.; Sanchez-Perez, L.; Nair, S.K.; Congdon, K.L.; Reap, E.A.; Archer, G.E.; Desjardins, A.; et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 2015, 519, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Lineburg, K.E.; Martins, J.P.; Ambalathingal, G.R.; Neller, M.A.; Morrison, B.; Matthews, K.K.; Rehan, S.; Crooks, P.; Panikkar, A.; et al. Autologous CMV-specific T cells are a safe adjuvant immunotherapy for primary glioblastoma multiforme. J. Clin. Investig. 2020, 130, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Schuessler, A.; Smith, C.; Beagley, L.; Boyle, G.M.; Rehan, S.; Matthews, K.; Jones, L.; Crough, T.; Dasari, V.; Klein, K.; et al. Autologous T-cell therapy for cytomegalovirus as a consolidative treatment for recurrent glioblastoma. Cancer Res. 2014, 74, 3466–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghazi, A.; Ashoori, A.; Hanley, P.J.; Brawley, V.S.; Shaffer, D.R.; Kew, Y.; Powell, S.Z.; Grossman, R.; Grada, Z.; Scheurer, M.E.; et al. Generation of polyclonal CMV-specific T cells for the adoptive immunotherapy of glioblastoma. J. Immunother. 2012, 35, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Reap, E.A.; Suryadevara, C.M.; Batich, K.A.; Sanchez-Perez, L.; Archer, G.E.; Schmittling, R.J.; Norberg, P.K.; Herndon, J.E.; Healy, P.; Congdon, K.L.; et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018, 78, 256. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, M. Viral Xenogenization of Intact Tumor Cells. In Advances in Cancer Research; Klein, G., Weinhouse, S., Eds.; Academic Press: Cambridge, MA, USA, 1979; Volume 30, pp. 279–299. [Google Scholar]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662. [Google Scholar] [CrossRef] [PubMed]
- Dobrikova, E.Y.; Goetz, C.; Walters, R.W.; Lawson, S.K.; Peggins, J.O.; Muszynski, K.; Ruppel, S.; Poole, K.; Giardina, S.L.; Vela, E.M.; et al. Attenuation of neurovirulence, biodistribution, and shedding of a poliovirus:rhinovirus chimera after intrathalamic inoculation in Macaca fascicularis. J. Virol. 2012, 86, 2750–2759. [Google Scholar] [CrossRef] [Green Version]
- Chandramohan, V.; Bryant, J.D.; Piao, H.; Keir, S.T.; Lipp, E.S.; Lefaivre, M.; Perkinson, K.; Bigner, D.D.; Gromeier, M.; McLendon, R.E. Validation of an Immunohistochemistry Assay for Detection of CD155, the Poliovirus Receptor, in Malignant Gliomas. Arch. Pathol. Lab. Med. 2017, 141, 1697–1704. [Google Scholar] [CrossRef]
- Brown, M.C.; Holl, E.K.; Boczkowski, D.; Dobrikova, E.; Mosaheb, M.; Chandramohan, V.; Bigner, D.D.; Gromeier, M.; Nair, S.K. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci. Transl. Med. 2017, 9, eaan4220. [Google Scholar] [CrossRef] [Green Version]
- Mosaheb, M.M.; Dobrikova, E.Y.; Brown, M.C.; Yang, Y.; Cable, J.; Okada, H.; Nair, S.K.; Bigner, D.D.; Ashley, D.M.; Gromeier, M. Genetically stable poliovirus vectors activate dendritic cells and prime antitumor CD8 T cell immunity. Nat. Commun. 2020, 11, 524. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Todo, T. ATIM-14. Results of phase II clinical trial of oncolytic herpes virus G47Δ in patients with glioblastoma. Neuro-Oncology 2019, 21 (Suppl. 6), vi4. [Google Scholar] [CrossRef]
- Agarwalla, P.K.; Aghi, M.K. Oncolytic herpes simplex virus engineering and preparation. Methods Mol. Biol. 2012, 797, 1–19. [Google Scholar]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Zhu, Z.; Gorman, M.J.; McKenzie, L.D.; Chai, J.N.; Hubert, C.G.; Prager, B.C.; Fernandez, E.; Richner, J.M.; Zhang, R.; Shan, C.; et al. Zika virus has oncolytic activity against glioblastoma stem cells. J. Exp. Med. 2017, 214, 2843–2857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Mesci, P.; Bernatchez, J.A.; Gimple, R.C.; Wang, X.; Schafer, S.T.; Wettersten, H.I.; Beck, S.; Clark, A.E.; Wu, Q.; et al. Zika Virus Targets Glioblastoma Stem Cells through a SOX2-Integrin αvβ5 Axis. Cell Stem Cell 2020, 26, 187–204.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Guo, P.; Kruh, G.D.; Vicini, P.; Wang, X.; Gallo, J.M. Predicting human tumor drug concentrations from a preclinical pharmacokinetic model of temozolomide brain disposition. Clin. Cancer Res. 2007, 13, 4271–4279. [Google Scholar] [CrossRef] [Green Version]
- Alessandrini, F.; Menotti, L.; Avitabile, E.; Appolloni, I.; Ceresa, D.; Marubbi, D.; Campadelli-Fiume, G.; Malatesta, P. Eradication of glioblastoma by immuno-virotherapy with a retargeted oncolytic HSV in a preclinical model. Oncogene 2019, 38, 4467–4479. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihelson, N.; McGavern, D.B. Viral Control of Glioblastoma. Viruses 2021, 13, 1264. https://doi.org/10.3390/v13071264
Mihelson N, McGavern DB. Viral Control of Glioblastoma. Viruses. 2021; 13(7):1264. https://doi.org/10.3390/v13071264
Chicago/Turabian StyleMihelson, Nicole, and Dorian B. McGavern. 2021. "Viral Control of Glioblastoma" Viruses 13, no. 7: 1264. https://doi.org/10.3390/v13071264
APA StyleMihelson, N., & McGavern, D. B. (2021). Viral Control of Glioblastoma. Viruses, 13(7), 1264. https://doi.org/10.3390/v13071264