
From Recoding to Peptides for MHC Class I Immune Display: Enriching Viral Expression, Virus Vulnerability and Virus Evasion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pox Virus, Alphavirus and Retrovirus Dynamic Codon Redefinition
3. Retroviral Frameshifting
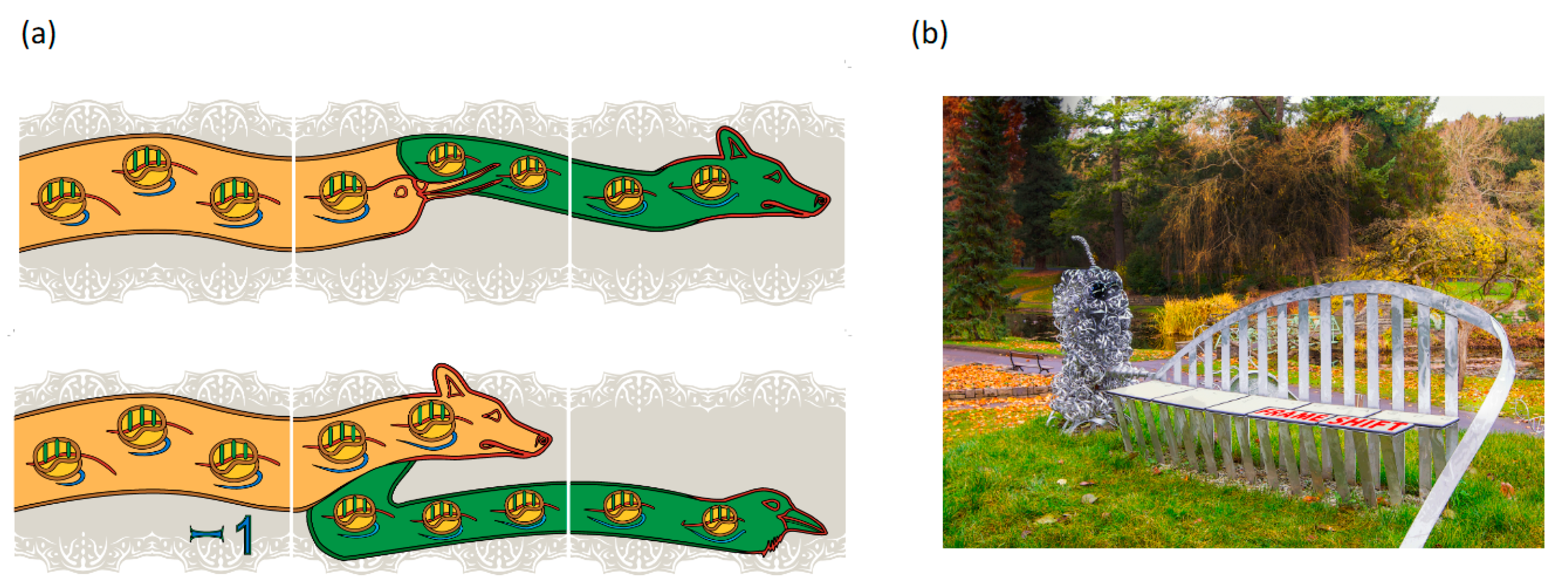
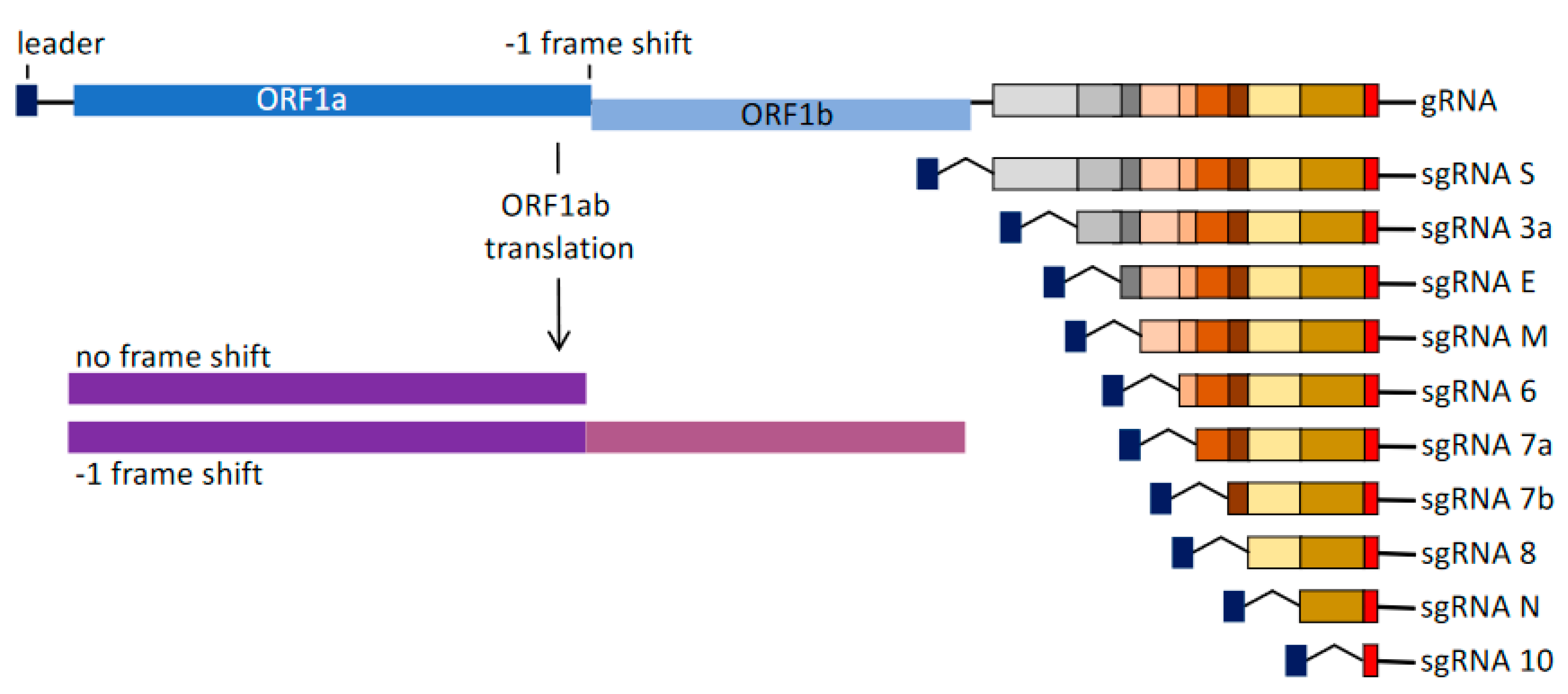
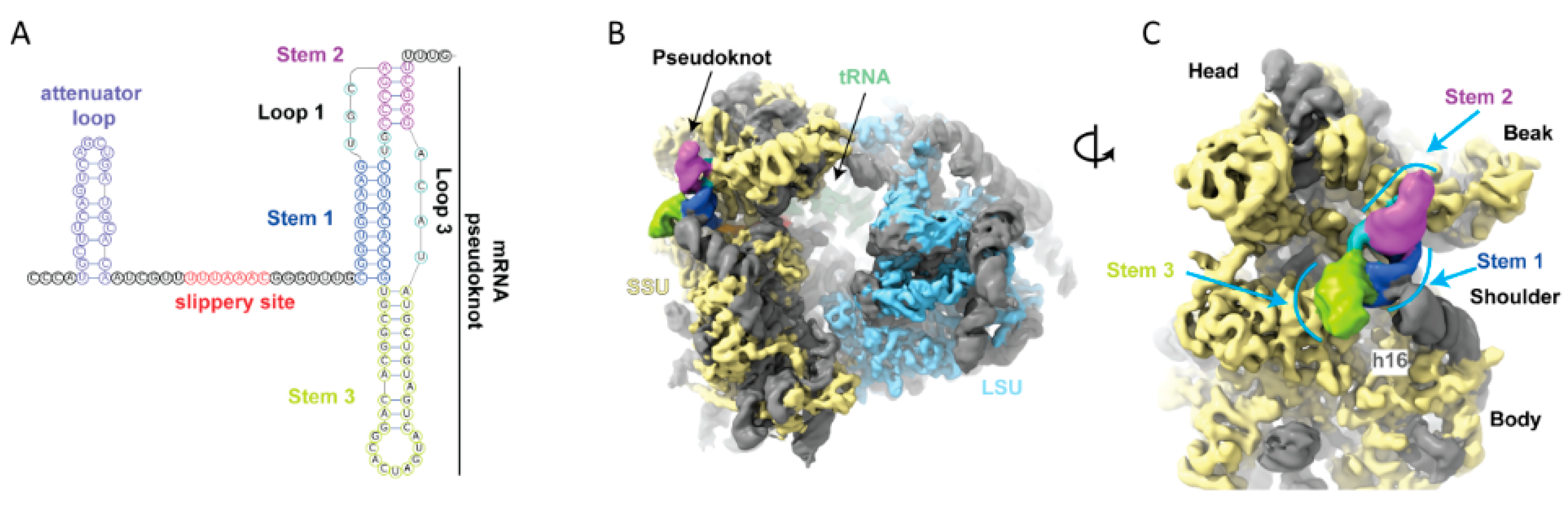
4. −1 Frameshifting Shift Sites Leading to Coronavirus Frameshifting
5. More Recently Identified Viral Recoding Points to Greater Mechanistic Diversity
6. Releasing a Newly Synthesized Peptide without Terminating Decoding: StopGo and Its Use to Determine the Degree of Importance of Retroviral Recoding
7. Implications for Synthetic Manipulation of Frameshifting, Readthrough and StopGo for the Successful Delivery of Nanoparticle-Complexed Nucleoside-Modified RNA and of DNA Vaccines
8. Finding Framing Imperfection, or Rather Its ‘Trade-Off’ Occurrence, Despite Crick’s ‘Half-Right’ Reason for Thinking It Would Not Exist
9. Utilizing Imperfections, Including of Framing, to Inhibit Viruses (and Cancer): DRiPS
10. Herpes Virus: DNA Viruses Can Use Adventitious Frameshifting to Evade Drugs and Programmed Frameshifting to Perhaps Facilitate Avoidance of Immune Detection
11. Immune Evasion
12. Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gesteland, R.F.; Weiss, R.B.; Atkins, J.F. Recoding: Reprogrammed Genetic Decoding. Science 1992, 257, 1640–1641. [Google Scholar] [CrossRef]
- Atkins, J.F.; Gesteland, R.F. Recoding: Expansion of Decoding Rules Enriches Gene Expression; Springer: New York, NY, USA, 2010; p. 466. [Google Scholar]
- Choi, J.; Grosely, R.; Prabhakar, A.; Lapointe, C.P.; Wang, J.; Puglisi, J.D. How Messenger RNA and Nascent Chain Sequences Regulate Translation Elongation. Annu. Rev. Biochem. 2018, 87, 421–449. [Google Scholar] [CrossRef]
- Rodnina, M.V.; Korniy, N.; Klimova, M.; Karki, P.; Peng, B.Z.; Senyushkina, T.; Belardinelli, R.; Maracci, C.; Wohlgemuth, I.; Samatova, E.; et al. Translational Recoding: Canonical Translation Mechanisms Reinterpreted. Nucleic Acids Res. 2020, 48, 1056–1067. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; Toptan, T.; Da Silva, S.R.; Atkins, J.F.; Moore, P.S.; Chang, Y. Human DNA Tumor Viruses Generate Alternative Reading Frame Proteins through Repeat Sequence Recoding. Proc. Natl. Acad. Sci. USA 2014, 111, E4342–E4349. [Google Scholar] [CrossRef] [Green Version]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal Frameshifting and Transcriptional Slippage: From Genetic Steganography and Cryptography to Adventitious Use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef] [Green Version]
- Weiner, A.M.; Weber, K. A Single UGA Codon Functions as a Natural Termination Signal in the Coliphage Qβ Coat Protein Cistron. J. Mol. Biol. 1973, 80, 837–855. [Google Scholar] [CrossRef]
- Barrell, B.G.; Air, G.M.; Hutchison, C.A. Overlapping Genes in Bacteriophage ΦX174. Nature 1976, 264, 34–41. [Google Scholar] [CrossRef]
- Atkins, J.F.; Steitz, J.A.; Anderson, C.W.; Model, P. Binding of Mammalian Ribosomes to MS2 Phage RNA Reveals an Overlapping Gene Encoding a Lysis Function. Cell 1979, 18, 247–256. [Google Scholar] [CrossRef]
- Gamarnik, A.V.; Andino, R. Switch from Translation to RNA Replication in a Positive-Stranded RNA Virus. Genes Dev. 1998, 12, 2293–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, W.A.; Jackson, J.; Feng, Y. Cis- and Trans-Regulation of Luteovirus Gene Expression by the 3′ End of the Viral Genome. Virus Res. 2015, 206, 37–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Franz, C.J.; Wu, G.; Renshaw, H.; Zhao, G.; Firth, A.E.; Wang, D. Orsay Virus Utilizes Ribosomal Frameshifting to Express a Novel Protein That Is Incorporated into Virions. Virology 2014, 450, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Molteni, C.G.; Principi, N.; Esposito, S. Reactive Oxygen and Nitrogen Species during Viral Infections. Free Radic. Res. 2014, 48, 1163–1169. [Google Scholar] [CrossRef]
- Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. [Google Scholar] [CrossRef] [Green Version]
- Guillin, O.M.; Vindry, C.; Ohlmann, T.; Chavatte, L. Selenium, Selenoproteins and Viral Infection. Nutrients 2019, 11, 2101. [Google Scholar] [CrossRef] [Green Version]
- Brigelius-Flohé, R.; Flohé, L. Regulatory Phenomena in the Glutathione Peroxidase Superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, Biology and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Avery, J.C.; Hoffmann, P.R. Selenium, Selenoproteins, and Immunity. Nutrients 2018, 10, 1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korwar, A.M.; Hossain, A.; Lee, T.-J.; Shay, A.E.; Basrur, V.; Conlon, K.; Smith, P.B.; Carlson, B.A.; Salis, H.M.; Patterson, A.D.; et al. Selenium-Dependent Metabolic Reprogramming during Inflammation and Resolution. J. Biol. Chem. 2021, 296, 100410. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Zhong, N.; Carlson, B.A.; Perella, C.M.; Hatfield, D.L.; Beck, M.A. Decreased Selenoprotein Expression Alters the Immune Response during Influenza Virus Infection in Mice. J. Nutr. 2007, 137, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Shisler, J.L.; Senkevich, T.G.; Berry, M.J.; Moss, B. Ultraviolet-Induced Cell Death Blocked by a Selenoprotein from a Human Dermatotropic Poxvirus. Science 1998, 279, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Mix, H.; Lobanov, A.V.; Gladyshev, V.N. SECIS Elements in the Coding Regions of Selenoprotein Transcripts Are Functional in Higher Eukaryotes. Nucleic Acids Res. 2007, 35, 414–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McFadden, G. Even Viruses Can Learn to Cope with Stress. Science 1998, 279, 40. [Google Scholar] [CrossRef] [PubMed]
- Fradejas-Villar, N.; Seeher, S.; Anderson, C.B.; Doengi, M.; Carlson, B.A.; Hatfield, D.L.; Schweizer, U.; Howard, M.T. The RNA-Binding Protein Secisbp2 Differentially Modulates UGA Codon Reassignment and RNA Decay. Nucleic Acids Res. 2017, 45, 4094–4107. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.C.; Ho, S.C.; Chen, Y.Y.; Khoo, K.H.; Hsu, P.H.; Yen, H.C.S. CRL2 Aids Elimination of Truncated Selenoproteins Produced by Failed UGA/Sec Decoding. Science 2015, 349, 91–95. [Google Scholar] [CrossRef] [Green Version]
- Baclaocos, J.; Santesmasses, D.; Mariotti, M.; Bierła, K.; Vetick, M.B.; Lynch, S.; McAllen, R.; Mackrill, J.J.; Loughran, G.; Guigó, R.; et al. Processive Recoding and Metazoan Evolution of Selenoprotein P: Up to 132 UGAs in Molluscs. J. Mol. Biol. 2019, 431, 4381–4407. [Google Scholar] [CrossRef]
- Santesmasses, D.; Mariotti, M.; Gladyshev, V.N. Bioinformatics of Selenoproteins. Antioxid. Redox Signal. 2020, 33, 525–536. [Google Scholar] [CrossRef]
- Berry, M.J.; Banu, L.; Chen, Y.; Mandel, S.J.; Kieffer, J.D.; Harney, J.W.; Larsen, P.R. Recognition of UGA as a Selenocysteine Codon in Type I Deiodinase Requires Sequences in the 3′ Untranslated Region. Nature 1991, 353, 273–276. [Google Scholar] [CrossRef]
- Wu, S.; Mariotti, M.; Santesmasses, D.; Hill, K.E.; Baclaocos, J.; Aparicio-Prat, E.; Li, S.; Mackrill, J.; Wu, Y.; Howard, M.T.; et al. Human Selenoprotein P and S Variant mRNAs with Different Numbers of SECIS Elements and Inferences from Mutant Mice of the Roles of Multiple SECIS Elements. Open Biol. 2016, 6, 160241. [Google Scholar] [CrossRef] [PubMed]
- Budiman, M.E.; Bubenik, J.L.; Miniard, A.C.; Middleton, L.M.; Gerber, C.A.; Cash, A.; Driscoll, D.M. Eukaryotic Initiation Factor 4a3 Is a Selenium-Regulated RNA-Binding Protein That Selectively Inhibits Selenocysteine Incorporation. Mol. Cell 2009, 35, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Howard, M.T.; Copeland, P.R. New Directions for Understanding the Codon Redefinition Required for Selenocysteine Incorporation. Biol. Trace Elem. Res. 2019, 192, 18–25. [Google Scholar] [CrossRef]
- Jedrychowski, M.P.; Lu, G.Z.; Szpyt, J.; Mariotti, M.; Garrity, R.; Paulo, J.A.; Schweppe, D.K.; Laznik-Bogoslavski, D.; Kazak, L.; Murphy, M.P.; et al. Facultative Protein Selenation Regulates Redox Sensitivity, Adipose Tissue Thermogenesis, and Obesity. Proc. Natl. Acad. Sci. USA 2020, 117, 10789–10796. [Google Scholar] [CrossRef]
- Firth, A.E.; Wills, N.M.; Gesteland, R.F.; Atkins, J.F. Stimulation of Stop Codon Readthrough: Frequent Presence of an Extended 3′ RNA Structural Element. Nucleic Acids Res. 2011, 39, 6679–6691. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Shao, S.; Murray, J.; Hegde, R.S.; Ramakrishnan, V. Structural Basis for Stop Codon Recognition in Eukaryotes. Nature 2015, 524, 493–496. [Google Scholar] [CrossRef]
- Matheisl, S.; Berninghausen, O.; Becker, T.; Beckmann, R. Structure of a Human Translation Termination Complex. Nucleic Acids Res. 2015, 43, 8615–8626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philipson, L.; Andersson, P.; Olshevsky, U.; Weinberg, R.; Baltimore, D.; Gesteland, R. Translation of MuLV and MSV RNAs in Nuclease-Treated Reticulocyte Extracts: Enhancement of the gag-pol Polypeptide with Yeast Suppressor tRNA. Cell 1978, 13, 189–199. [Google Scholar] [CrossRef]
- Yoshinaka, Y.; Katoh, I.; Copeland, T.D.; Oroszlan, S. Murine Leukemia Virus Protease Is Encoded by the gag-pol Gene and Is Synthesized through Suppression of an Amber Termination Codon. Proc. Natl. Acad. Sci. USA 1985, 82, 1618–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.X.; Hatfield, D.L.; Rein, A.; Levin, J.G. Translational Readthrough of the Murine Leukemia Virus gag Gene Amber Codon Does Not Require Virus-Induced Alteration of tRNA. J. Virol. 1989, 63, 2405–2410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wills, N.M.; Gesteland, R.F.; Atkins, J.F. Evidence That a Downstream Pseudoknot Is Required for Translational Read-through of the Moloney Murine Leukemia Virus gag Stop Codon. Proc. Natl. Acad. Sci. USA 1991, 88, 6991–6995. [Google Scholar] [CrossRef] [Green Version]
- Wills, N.M.; Gesteland, R.F.; Atkins, J.F. Pseudoknot-Dependent Read-through of Retroviral gag Termination Codons: Importance of Sequences in the Spacer and Loop 2. EMBO J. 1994, 13, 4137–4144. [Google Scholar] [CrossRef]
- Feng, Y.X.; Yuan, H.; Rein, A.; Levin, J.G. Bipartite Signal for Read-through Suppression in Murine Leukemia Virus mRNA: An Eight-Nucleotide Purine-Rich Sequence Immediately Downstream of the gag Termination Codon Followed by an RNA Pseudoknot. J. Virol. 1992, 66, 5127–5132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felsenstein, K.M.; Goff, S.P. Mutational Analysis of the gag-pol Junction of Moloney Murine Leukemia Virus: Requirements for Expression of the gag-pol Fusion Protein. J. Virol. 1992, 66, 6601–6608. [Google Scholar] [CrossRef] [Green Version]
- Alam, S.L.; Wills, N.M.; Ingram, J.A.; Atkins, J.F.; Gesteland, R.F. Structural Studies of the RNA Pseudoknot Required for Readthrough of the gag-Termination Codon of Murine Leukemia Virus. J. Mol. Biol. 1999, 288, 837–852. [Google Scholar] [CrossRef]
- Houck-Loomis, B.; Durney, M.A.; Salguero, C.; Shankar, N.; Nagle, J.M.; Goff, S.P.; D’Souza, V.M. An Equilibrium-Dependent Retroviral mRNA Switch Regulates Translational Recoding. Nature 2011, 480, 561–564. [Google Scholar] [CrossRef]
- Orlova, M.; Yueh, A.; Leung, J.; Goff, S.P. Reverse Transcriptase of Moloney Murine Leukemia Virus Binds to Eukaryotic Release Factor 1 to Modulate Suppression of Translational Termination. Cell 2003, 115, 319–331. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zhu, Y.; Baker, S.L.; Bowler, M.W.; Chen, B.J.; Chen, C.; Hogg, J.R.; Goff, S.P.; Song, H. Structural Basis of Suppression of Host Translation Termination by Moloney Murine Leukemia Virus. Nat. Commun. 2016, 7, 12070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irigoyen, N.; Dinan, A.M.; Brierley, I.; Firth, A.E. Ribosome Profiling of the Retrovirus Murine Leukemia Virus. Retrovirology 2018, 15, 10. [Google Scholar] [CrossRef] [Green Version]
- Lyon, K.; Aguilera, L.U.; Morisaki, T.; Munsky, B.; Stasevich, T.J. Live-Cell Single RNA Imaging Reveals Bursts of Translational Frameshifting. Mol. Cell 2019, 75, 172–183.e9. [Google Scholar] [CrossRef]
- Jacks, T.; Varmus, H.E. Expression of the Rous Sarcoma Virus pol Gene by Ribosomal Frameshifting. Science 1985, 230, 1237–1242. [Google Scholar] [CrossRef]
- Jacks, T.; Townsley, K.; Varmus, H.E.; Majors, J. Two Efficient Ribosomal Frameshifting Events Are Required for Synthesis of Mouse Mammary Tumor Virus gag-Related Polyproteins. Proc. Natl. Acad. Sci. USA 1987, 84, 4298–4302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacks, T.; Power, M.D.; Masiarz, F.R.; Luciw, P.A.; Barr, P.J.; Varmus, H.E. Characterization of Ribosomal Frameshifting in HIV-1 gag-pol Expression. Nature 1988, 331, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Madhani, H.D.; Masiarz, F.R.; Varmus, H.E. Signals for Ribosomal Frameshifting in the Rous Sarcoma Virus gag-pol Region. Cell 1988, 55, 447–458. [Google Scholar] [CrossRef]
- Hizi, A.; Henderson, L.E.; Copeland, T.D.; Sowder, R.C.; Hixson, C.V.; Oroszlan, S. Characterization of Mouse Mammary Tumor Virus gag-pro Gene Products and the Ribosomal Frameshift Site by Protein Sequencing. Proc. Natl. Acad. Sci. USA 1987, 84, 7041–7045. [Google Scholar] [CrossRef] [Green Version]
- Yoshinaka, Y.; Katoh, I.; Copeland, T.D.; Smythers, G.W.; Oroszlan, S. Bovine Leukemia Virus Protease: Purification, Chemical Analysis, and in Vitro Processing of gag Precursor Polyproteins. J. Virol. 1986, 57, 826–832. [Google Scholar] [CrossRef] [Green Version]
- Marczinke, B.; Fisher, R.; Vidakovic, M.; Bloys, A.J.; Brierley, I. Secondary Structure and Mutational Analysis of the Ribosomal Frameshift Signal of Rous Sarcoma Virus. J. Mol. Biol. 1998, 284, 205–225. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.B.; Dunn, D.M.; Shuh, M.; Atkins, J.F.; Gesteland, R.F. E. Coli Ribosomes Re-Phase on Retroviral Frameshift Signals at Rates Ranging from 2 to 50 Percent. New Biol. 1989, 1, 159–169. [Google Scholar]
- Yelverton, E.; Lindsley, D.; Yamauchi, P.; Gallant, J.A. The Function of a Ribosomal Frameshifting Signal from Human Immunodeficiency Virus-1 in Escherichia coli. Mol. Microbiol. 1994, 11, 303–313. [Google Scholar] [CrossRef]
- Korniy, N.; Goyal, A.; Hoffmann, M.; Samatova, E.; Peske, F.; Pöhlmann, S.; Rodnina, M.V. Modulation of HIV-1 gag/gag-pol Frameshifting by tRNA Abundance. Nucleic Acids Res. 2019, 47, 5210–5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korniy, N.; Samatova, E.; Anokhina, M.M.; Peske, F.; Rodnina, M.V. Mechanisms and Biomedical Implications of –1 Programmed Ribosome Frameshifting on Viral and Bacterial mRNAs. FEBS Lett. 2019, 593, 1468–1482. [Google Scholar] [CrossRef]
- Charbonneau, J.; Gendron, K.; Ferbeyre, G.; Brakier-Gingras, L. The 5′ UTR of HIV-1 Full-Length mRNA and the Tat Viral Protein Modulate the Programmed -1 Ribosomal Frameshift That Generates HIV-1 Enzymes. RNA 2012, 18, 519–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Miranda, P.; Becker, J.T.; Benner, B.E.; Blume, A.; Sherer, N.M.; Butcher, S.E. Stability of HIV Frameshift Site RNA Correlates with Frameshift Efficiency and Decreased Virus Infectivity. J. Virol. 2016, 90, 6906–6917. [Google Scholar] [CrossRef] [Green Version]
- Penno, C.; Kumari, R.; Baranov, P.V.; Van Sinderen, D.; Atkins, J.F. Specific Reverse Transcriptase Slippage at the HIV Ribosomal Frameshift Sequence: Potential Implications for Modulation of GagPol Synthesis. Nucleic Acids Res. 2017, 45, 10156–10167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penno, C.; Sharma, V.; Coakley, A.; Motherway, M.O.C.; Van Sinderen, D.; Lubkowska, L.; Kireeva, M.L.; Kashlev, M.; Baranov, P.V.; Atkins, J.F. Productive mRNA Stem Loop-Mediated Transcriptional Slippage: Crucial Features in Common with Intrinsic Terminators. Proc. Natl. Acad. Sci. USA 2015, 112, 1984–1993. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.Y.W.; Miller, W.A.; Atkins, J.F.; Firth, A.E. An Overlapping Essential Gene in the Potyviridae. Proc. Natl. Acad. Sci. USA 2008, 105, 5897–5902. [Google Scholar] [CrossRef] [Green Version]
- Olspert, A.; Chung, B.Y.; Atkins, J.F.; Carr, J.P.; Firth, A.E. Transcriptional Slippage in the Positive-sense RNA Virus Family Potyviridae. EMBO Rep. 2015, 16, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Rodamilans, B.; Valli, A.; Mingot, A.; San León, D.; Baulcombe, D.; López-Moya, J.J.; García, J.A. RNA Polymerase Slippage as a Mechanism for the Production of Frameshift Gene Products in Plant Viruses of the Potyviridae Family. J. Virol. 2015, 89, 6965–6967. [Google Scholar] [CrossRef] [Green Version]
- Weiss, R.; Gallant, J. Mechanism of Ribosome Frameshifting during Translation of the Genetic Code. Nature 1983, 302, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.F.; Nichols, B.P.; Thompson, S. The Nucleotide Sequence of the First Externally Suppressible-1 Frameshift Mutant, and of Some Nearby Leaky Frameshift Mutants. EMBO J. 1983, 2, 1345–1350. [Google Scholar] [CrossRef]
- Weiss, R.B.; Dunn, D.M.; Atkins, J.F.; Gesteland, R.F. Slippery Runs, Shifty Stops, Backward Steps, and Forward Hops: -2, -1, +1, +2, +5, and +6 Ribosomal Frameshifting. Cold Spring Harb. Symp. Quant. Biol. 1987, 52, 687–693. [Google Scholar] [CrossRef]
- Dunn, J.J.; Studier, F.W.; Gottesman, M. Complete Nucleotide Sequence of Bacteriophage T7 DNA and the Locations of T7 Genetic Elements. J. Mol. Biol. 1983, 166, 477–535. [Google Scholar] [CrossRef]
- Condron, B.G.; Gesteland, R.F.; Atkins, J.F. An Analysis of Sequences Stimulating Frameshifting in the Decoding of Gene 10 of Bacteriophage T7. Nucleic Acids Res. 1991, 19, 5607–5612. [Google Scholar] [CrossRef]
- Atkins, J.F.; Gesteland, R.F.; Reid, B.R.; Anderson, C.W. Normal tRNAs Promote Ribosomal Frameshifting. Cell 1979, 18, 1119–1131. [Google Scholar] [CrossRef]
- Dayhuff, T.J.; Atkins, J.F.; Gesteland, R.F. Characterization of Ribosomal Frameshift Events by Protein Sequence Analysis. J. Biol. Chem. 1986, 261, 7491–7500. [Google Scholar] [CrossRef]
- Atkins, J.F.; Herr, A.J.; Massire, C.; O’Connor, M.; Ivanov, I.; Gesteland, R.F. Poking a Hole in the Sanctity of the Triplet Code: Inferences for Framing. In The Ribosome. Structure, Function, Antibiotics and Cellular Interactions; Garret, R.A., Douthwaite, S.R., Liljas, A., Matheson, A.T., Moore, P.B., Noller, H.F., Eds.; ASM Press: Washington, DC, USA, 2000; pp. 367–383. [Google Scholar]
- Caulfield, T.; Coban, M.; Tek, A.; Flores, S.C. Molecular Dynamics Simulations Suggest a Non-Doublet Decoding Model of –1 Frameshifting by tRNAser3. Biomolecules 2019, 9, 745. [Google Scholar] [CrossRef] [Green Version]
- Brierley, I.; Boursnell, M.E.; Binns, M.M.; Bilimoria, B.; Blok, V.C.; Brown, T.D.; Inglis, S.C. An Efficient Ribosomal Frame-Shifting Signal in the Polymerase-Encoding Region of the Coronavirus IBV. EMBO J. 1987, 6, 3779–3785. [Google Scholar] [CrossRef] [PubMed]
- Brierley, I.; Jenner, A.J.; Inglis, S.C. Mutational Analysis of the “Slippery-Sequence” Component of a Coronavirus Ribosomal Frameshifting Signal. J. Mol. Biol. 1992, 227, 463–479. [Google Scholar] [CrossRef]
- Dinan, A.M.; Keep, S.; Bickerton, E.; Britton, P.; Firth, A.E.; Brierley, I. Comparative Analysis of Gene Expression in Virulent and Attenuated Strains of Infectious Bronchitis Virus at Subcodon Resolution. J. Virol. 2019, 93, 714–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licznar, P.; Mejlhede, N.; Prère, M.F.; Wills, N.; Gesteland, R.F.; Atkins, J.F.; Fayet, O. Programmed Translational -1 Frameshifting on Hexanucleotide Motifs and the Wobble Properties of tRNAs. EMBO J. 2003, 22, 4770–4778. [Google Scholar] [CrossRef] [Green Version]
- Pleij, C.W.A.; Rietveld, K.; Bosch, L. A New Principle of RNA Folding Based on Pseudoknotting. Nucleic Acids Res. 1985, 13, 1717–1731. [Google Scholar] [CrossRef]
- ten Dam, E.B.; Pleij, C.W.A.; Bosch, L. RNA Pseudoknots: Translational Frameshifting and Readthrough on Viral RNAs. Virus Genes 1990, 4, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Brierley, I.; Digard, P.; Inglis, S.C. Characterization of an Efficient Coronavirus Ribosomal Frameshifting Signal: Requirement for an RNA Pseudoknot. Cell 1989, 57, 537–547. [Google Scholar] [CrossRef]
- Brierley, I.; Rolley, N.J.; Jenner, A.J.; Inglis, S.C. Mutational Analysis of the RNA Pseudoknot Component of a Coronavirus Ribosomal Frameshifting Signal. J. Mol. Biol. 1991, 220, 889–902. [Google Scholar] [CrossRef]
- Napthine, S.; Liphardt, J.; Bloys, A.; Routledge, S.; Brierley, I. The Role of RNA Pseudoknot Stem 1 Length in the Promotion of Efficient -1 Ribosomal Frameshifting. J. Mol. Biol. 1999, 288, 305–320. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 Gene Content and COVID-19 Mutation Impact by Comparing 44 Sarbecovirus Genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef] [PubMed]
- Ziv, O.; Price, J.; Shalamova, L.; Kamenova, T.; Goodfellow, I.; Weber, F.; Miska, E.A. The Short- and Long-Range RNA-RNA Interactome of SARS-CoV-2. Mol. Cell 2020, 80, 1067–1077. [Google Scholar] [CrossRef] [PubMed]
- Baranov, P.V.; Henderson, C.M.; Anderson, C.B.; Gesteland, R.F.; Atkins, J.F.; Howard, M.T. Programmed Ribosomal Frameshifting in Decoding the SARS-CoV Genome. Virology 2005, 332, 498–510. [Google Scholar] [CrossRef] [Green Version]
- Plant, E.P.; Pérez-Alvarado, G.C.; Jacobs, J.L.; Mukhopadhyay, B.; Hennig, M.; Dinman, J.D. A Three-Stemmed mRNA Pseudoknot in the SARS Coronavirus Frameshift Signal. PLoS Biol. 2005, 3, 1012–1023. [Google Scholar] [CrossRef] [Green Version]
- Su, M.C.; Chang, C.T.; Chu, C.H.; Tsai, C.H.; Chang, K.Y. An Atypical RNA Pseudoknot Stimulator and an Upstream Attenuation Signal for -1 Ribosomal Frameshifting of SARS Coronavirus. Nucleic Acids Res. 2005, 33, 4265–4275. [Google Scholar] [CrossRef] [Green Version]
- Dos Ramos, F.; Carrasco, M.; Doyle, T.; Brierley, I. Programmed -1 Ribosomal Frameshifting in the SARS Coronavirus. Biochem Soc. Trans. 2004, 32, 1081–1083. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, D.; Plant, E.P.; Sims, A.C.; Yount, B.L.; Roth, B.M.; Eldho, N.V.; Pérez-Alvarado, G.C.; Armbruster, D.W.; Baric, R.S.; Dinman, J.D.; et al. RNA Dimerization Plays a Role in Ribosomal Frameshifting of the SARS Coronavirus. Nucleic Acids Res. 2013, 41, 2594–2608. [Google Scholar] [CrossRef]
- Plant, E.P.; Sims, A.C.; Baric, R.S.; Dinman, J.D.; Taylor, D.R. Altering SARS Coronavirus Frameshift Efficiency Affects Genomic and Subgenomic RNA Production. Viruses 2013, 5, 279–294. [Google Scholar] [CrossRef] [Green Version]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol Ratio Is Important for Human Immunodeficiency Virus Type 1 RNA Dimerization and Viral Infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Chin, W.X.; Han, Q.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016, 12, e1005357. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yang, X.; Yao, Z.; Dong, X.; Zhang, D.; Hu, Y.; Zhang, S.; Lin, J.; Chen, J.; An, S.; et al. C19orf66 Interrupts Zika Virus Replication by Inducing Lysosomal Degradation of Viral NS3. PLoS Negl. Trop. Dis. 2020, 14, e0008083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinast, V.; Plociennikowska, A.; Anggakusuma; Bracht, T.; Todt, D.; Brown, R.J.P.; Boldanova, T.; Zhang, Y.; Brüggemann, Y.; Friesland, M.; et al. C19orf66 Is an Interferon-Induced Inhibitor of HCV Replication That Restricts Formation of the Viral Replication Organelle. J. Hepatol. 2020, 73, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 2019, 176, 625–635.e14. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural Basis of Ribosomal Frameshifting during Translation of the SARS-CoV-2 RNA Genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Baranov, P.V.; Atkins, J.F.; Yordanova, M.M. Augmented Genetic Decoding: Global, Local and Temporal Alterations of Decoding Processes and Codon Meaning. Nat. Rev. Genet. 2015, 16, 517–529. [Google Scholar] [CrossRef]
- Meydan, S.; Klepacki, D.; Karthikeyan, S.; Margus, T.; Thomas, P.; Jones, J.E.; Khan, Y.; Briggs, J.; Dinman, J.D.; Vázquez-Laslop, N.; et al. Programmed Ribosomal Frameshifting Generates a Copper Transporter and a Copper Chaperone from the Same Gene. Mol. Cell 2017, 65, 207–219. [Google Scholar] [CrossRef] [Green Version]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An Overlapping Protein-Coding Region in Influenza A Virus Segment 3 Modulates the Host Response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.E.; Jagger, B.W.; Wise, H.M.; Nelson, C.C.; Parsawar, K.; Wills, N.M.; Napthine, S.; Taubenberger, J.K.; Digard, P.; Atkins, J.F. Ribosomal Frameshifting Used in Influenza A Virus Expression Occurs within the Sequence UCC-UUU-CGU and Is in the +1 Direction. Open Biol. 2012, 2, 120109. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; MacDonald, L.A.; Takimoto, T. Influenza A Virus Protein PA-X Contributes to Viral Growth and Suppression of the Host Antiviral and Immune Responses. J. Virol. 2015, 89, 6442–6452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Identification of Amino Acid Residues in Influenza A Virus PA-X That Contribute to Enhanced Shutoff Activity. Front. Microbiol. 2019, 10, 432. [Google Scholar] [CrossRef] [Green Version]
- Oishi, K.; Yamayoshi, S.; Kawaoka, Y. Mapping of a Region of the PA-X Protein of Influenza A Virus That Is Important for Its Shutoff Activity. J. Virol. 2015, 89, 8661–8665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinan, A.M.; Atkins, J.F.; Firth, A.E. ASXL Gain-of-Function Truncation Mutants: Defective and Dysregulated Forms of a Natural Ribosomal Frameshifting Product? Biol. Direct 2017, 12, 24. [Google Scholar] [CrossRef]
- Bock, L.V.; Caliskan, N.; Korniy, N.; Peske, F.; Rodnina, M.V.; Grubmüller, H. Thermodynamic Control of −1 Programmed Ribosomal Frameshifting. Nat. Commun. 2019, 10, 4598. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; O’Loughlin, S.; Atkins, J.F.; Puglisi, J.D. The Energy Landscape of −1 Ribosomal Frameshifting. Sci. Adv. 2020, 6, eaax6969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firth, A.E.; Atkins, J.F. A Conserved Predicted Pseudoknot in the NS2A-Encoding Sequence of West Nile and Japanese Encephalitis Flaviviruses Suggests NS1′ May Derive from Ribosomal Frameshifting. Virol. J. 2009, 6, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melian, E.B.; Hinzman, E.; Nagasaki, T.; Firth, A.E.; Wills, N.M.; Nouwens, A.S.; Blitvich, B.J.; Leung, J.; Funk, A.; Atkins, J.F.; et al. NS1′ of Flaviviruses in the Japanese Encephalitis Virus Serogroup Is a Product of Ribosomal Frameshifting and Plays a Role in Viral Neuroinvasiveness. J. Virol. 2010, 84, 1641–1647. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Li, Q.; Jia, F.; Zhang, L.; Wan, S.; Li, Y.; Song, Y.; Chen, H.; Cao, S.; Ye, J. The Japanese Encephalitis Virus NS1′ Protein Inhibits Type I IFN Production by Targeting MAVS. J. Immunol. 2020, 204, 1287–1298. [Google Scholar] [CrossRef]
- Sun, J.; Yu, Y.; Deubel, V. Japanese Encephalitis Virus NS1′ Protein Depends on Pseudoknot Secondary Structure and Is Cleaved by Caspase during Virus Infection and Cell Apoptosis. Microbes Infect. 2012, 14, 930–940. [Google Scholar] [CrossRef]
- Ye, Q.; Li, X.F.; Zhao, H.; Li, S.H.; Deng, Y.Q.; Cao, R.Y.; Song, K.Y.; Wang, H.J.; Hua, R.H.; Yu, Y.X.; et al. A Single Nucleotide Mutation in NS2A of Japanese Encephalitis-Live Vaccine Virus (SA14-14-2) Ablates NS1′ Formation and Contributes to Attenuation. J. Gen. Virol. 2012, 93, 1959–1964. [Google Scholar] [CrossRef]
- Wang, J.; Li, X.; Gu, J.; Fan, Y.; Zhao, P.; Cao, R.; Chen, P. The A66G Back Mutation in NS2A of JEV SA14-14-2 Strain Contributes to Production of NS1′ Protein and the Secreted NS1′ Can Be Used for Diagnostic Biomarker for Virulent Virus Infection. Infect. Genet. Evol. 2015, 36, 116–125. [Google Scholar] [CrossRef]
- Halma, M.T.J.; Ritchie, D.B.; Cappellano, T.R.; Neupane, K.; Woodside, M.T. Complex Dynamics under Tension in a High-Efficiency Frameshift Stimulatory Structure. Proc. Natl. Acad. Sci. USA 2019, 116, 19500–19505. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Blitvich, B.J.; Wills, N.M.; Miller, C.L.; Atkins, J.F. Evidence for Ribosomal Frameshifting and a Novel Overlapping Gene in the Genomes of Insect-Specific Flaviviruses. Virology 2010, 399, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Boon, J.A.; Snijder, E.J.; Chirnside, E.D.; de Vries, A.A.; Horzinek, M.C.; Spaan, W.J. Equine Arteritis Virus Is Not a Togavirus but Belongs to the Coronaviruslike Superfamily. J. Virol. 1991, 65, 2910–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Snijder, E.J. The PRRSV Replicase: Exploring the Multifunctionality of an Intriguing Set of Nonstructural Proteins. Virus Res. 2010, 154, 61–76. [Google Scholar] [CrossRef]
- Fang, Y.; Treffers, E.E.; Li, Y.; Tas, A.; Sun, Z.; Van Der Meer, Y.; De Ru, A.H.; Van Veelen, P.A.; Atkins, J.F.; Snijder, E.J.; et al. Efficient—2 Frameshifting by Mammalian Ribosomes to Synthesize an Additional Arterivirus Protein. Proc. Natl. Acad. Sci. USA 2012, 109, 2920–2928. [Google Scholar] [CrossRef] [Green Version]
- Guo, R.; Yan, X.; Li, Y.; Cui, J.; Misra, S.; Firth, A.E.; Snijder, E.J.; Fang, Y. A Swine Arterivirus Deubiquitinase Stabilizes Two Major Envelope Proteins and Promotes Production of Viral Progeny. PLoS Pathog. 2021, 17, e1009403. [Google Scholar] [CrossRef]
- Li, Y.; Treffers, E.E.; Napthine, S.; Tas, A.; Zhu, L.; Sun, Z.; Bell, S.; Mark, B.L.; Van Veelen, P.A.; Van Hemert, M.J.; et al. Transactivation of Programmed Ribosomal Frameshifting by a Viral Protein. Proc. Natl. Acad. Sci. USA 2014, 111, 2172–2181. [Google Scholar] [CrossRef] [Green Version]
- Napthine, S.; Treffers, E.E.; Bell, S.; Goodfellow, I.; Fang, Y.; Firth, A.E.; Snijder, E.J.; Brierley, I. A Novel Role for Poly(C) Binding Proteins in Programmed Ribosomal Frameshifting. Nucleic Acids Res. 2016, 44, 5491–5503. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Treffers, E.E.; Meier, M.; Patel, T.R.; Stetefeld, J.; Snijder, E.J.; Mark, B.L. Molecular Characterization of the RNA-Protein Complex Directing 22/21 Programmed Ribosomal Frameshifting during Arterivirus Replicase Expression. J. Biol. Chem. 2020, 295, 17904–17921. [Google Scholar] [CrossRef]
- Loughran, G.; Firth, A.E.; Atkins, J.F. Ribosomal Frameshifting into an Overlapping Gene in the 2B-Encoding Region of the Cardiovirus Genome. Proc. Natl. Acad. Sci. USA 2011, 108, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finch, L.K.; Ling, R.; Napthine, S.; Olspert, A.; Michiels, T.; Lardinois, C.; Bell, S.; Loughran, G.; Brierley, I.; Firth, A.E. Characterization of Ribosomal Frameshifting in Theiler’s Murine Encephalomyelitis Virus. J. Virol. 2015, 89, 8580–8589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napthine, S.; Ling, R.; Finch, L.K.; Jones, J.D.; Bell, S.; Brierley, I.; Firth, A.E. Protein-Directed Ribosomal Frameshifting Temporally Regulates Gene Expression. Nat. Commun. 2017, 8, 15582. [Google Scholar] [CrossRef] [Green Version]
- Napthine, S.; Bell, S.; Hil, C.H.; Brierley, I.; Firt, A.E. Characterization of the Stimulators of Protein-Directed Ribosomal Frameshifting in Theiler’s Murine Encephalomyelitis Virus. Nucleic Acids Res. 2019, 47, 8207–8223. [Google Scholar] [CrossRef]
- Firth, A.E.; Chung, B.Y.W.; Fleeton, M.N.; Atkins, J.F. Discovery of Frameshifting in Alphavirus 6K Resolves a 20-Year Enigma. Virol. J. 2008, 5, 108. [Google Scholar] [CrossRef] [Green Version]
- Chung, B.Y.W.; Firth, A.E.; Atkins, J.F. Frameshifting in Alphaviruses: A Diversity of 3′ Stimulatory Structures. J. Mol. Biol. 2010, 397, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.G.; Maas, S.; Rich, A. Comparative Mutational Analysis of Cis-Acting RNA Signals for Translational Frameshifting in HIV-1 and HTLV-2. Nucleic Acids Res. 2001, 29, 1125–1131. [Google Scholar] [CrossRef] [Green Version]
- Harrington, H.R.; Zimmer, M.H.; Chamness, L.M.; Nash, V.; Penn, W.D.; Miller, T.F.; Mukhopadhyay, S.; Schlebach, J.P. Cotranslational Folding Stimulates Programmed Ribosomal Frameshifting in the Alphavirus Structural Polyprotein. J. Biol. Chem. 2020, 295, 6798–6808. [Google Scholar] [CrossRef] [Green Version]
- Larsen, B.; Wills, N.M.; Gesteland, R.F.; Atkins, J.F. rRNA-mRNA Base Pairing Stimulates a Programmed -1 Ribosomal Frameshift. J. Bacteriol. 1994, 176, 6842–6851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.T.; Cho, C.P.; Lin, Y.H.; Chang, K.Y. A General Strategy to Inhibiting Viral -1 Frameshifting Based on Upstream Attenuation Duplex Formation. Nucleic Acids Res. 2016, 44, 256–266. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.P.; Cho, C.P.; Chang, K.Y. mRNA-Mediated Duplexes Play Dual Roles in the Regulation of Bidirectional Ribosomal Frameshifting. Int. J. Mol. Sci. 2018, 19, 3867. [Google Scholar] [CrossRef] [Green Version]
- Firth, A.E.; Wang, Q.S.; Jan, E.; Atkins, J.F. Bioinformatic Evidence for a Stem-Loop Structure 5′-Adjacent to the IGR-IRES and for an Overlapping Gene in the Bee Paralysis Dicistroviruses. Virol. J. 2009, 6, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q.; Wang, Q.S.; Firth, A.E.; Chan, M.M.Y.; Gouw, J.W.; Guarna, M.M.; Foster, L.J.; Atkins, J.F.; Jan, E. Alternative Reading Frame Selection Mediated by a tRNA-like Domain of an Internal Ribosome Entry Site. Proc. Natl. Acad. Sci. USA 2012, 109, 630–639. [Google Scholar] [CrossRef] [Green Version]
- Kerr, C.H.; Wang, Q.S.; Moon, K.M.; Keatings, K.; Allan, D.W.; Foster, L.J.; Jan, E. IRES-Dependent Ribosome Repositioning Directs Translation of a +1 Overlapping ORF That Enhances Viral Infection. Nucleic Acids Res. 2018, 46, 11952–11967. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, M.L.L.; Luke, G.; Mehrotra, A.; Li, X.; Hughes, L.E.; Gani, D.; Ryan, M.D. Analysis of the Aphthovirus 2A/2B Polyprotein “cleavage” Mechanism Indicates Not a Proteolytic Reaction, but a Novel Translational Effect: A Putative Ribosomal “Skip”. J. Gen. Virol. 2001, 82, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.F.; Wills, N.M.; Loughran, G.; Wu, C.-Y.; Parsawar, K.; Ryan, M.D.; Wang, C.-H.; Nelson, C.C. A Case for “StopGo”: Reprogramming Translation to Augment Codon Meaning of GGN by Promoting Unconventional Termination (Stop) after Addition of Glycine and Then Allowing Continued Translation (Go). RNA 2007, 13, 803–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doronina, V.A.; Wu, C.; de Felipe, P.; Sachs, M.S.; Ryan, M.D.; Brown, J.D. Site-Specific Release of Nascent Chains from Ribosomes at a Sense Codon. Mol. Cell. Biol. 2008, 28, 4227–4239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.D.; Ryan, M.D. Ribosome “Skipping”: “Stop-Carry On” or “StopGo” Translation. In Recoding: Expansion of Decoding Rules Enriches Gene Expression; Atkins, J.F., Gesteland, R.F., Eds.; Springer: New York, NY, USA, 2010; pp. 101–121. [Google Scholar]
- Luke, G.A.; de Felipe, P.; Lukashev, A.; Kallioinen, S.E.; Bruno, E.A.; Ryan, M.D. Occurrence, Function and Evolutionary Origins of “2A-like” Sequences in Virus Genomes. J. Gen. Virol. 2008, 89, 1036–1042. [Google Scholar] [CrossRef]
- Loughran, G.; Libbey, J.E.; Uddowla, S.; Scallan, M.F.; Ryan, M.D.; Fujinami, R.S.; Rieder, E.; Atkins, J.F. Theiler’s Murine Encephalomyelitis Virus Contrasts with Encephalomyocarditis and Foot-and-Mouth Disease Viruses in Its Functional Utilization of the StopGo Non-Standard Translation Mechanism. J. Gen. Virol. 2013, 94, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjær, J.; Belsham, G.J. Modifications to the Foot-and-Mouth Disease Virus 2A Peptide: Influence on Polyprotein Processing and Virus Replication. J. Virol. 2018, 92, e02218-17. [Google Scholar] [CrossRef] [Green Version]
- Kjær, J.; Belsham, G.J. Selection of Functional 2A Sequences within Foot-and-Mouth Disease Virus; Requirements for the NPGP Motif with a Distinct Codon Bias. RNA 2018, 24, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Luke, G.A.; Ryan, M.D. Therapeutic Applications of the ‘NPGP’ Family of Viral 2As. Rev. Med. Virol. 2018, 28, 2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loughran, G.; Howard, M.T.; Firth, A.E.; Atkins, J.F. Avoidance of Reporter Assay Distortions from Fused Dual Reporters. Rna 2017, 23, 1285–1289. [Google Scholar] [CrossRef]
- Khan, Y.A.; Loughran, G.; Steckelberg, A.L.; Brown, K.; Kiniry, S.J.; Stewart, H.; Baranov, P.V.; Kieft, J.S.; Firth, A.E.; Atkins, J.F. Evaluating Programmed Frameshifting in CCR5 mRNA Decoding. Nature 2021, in press. [Google Scholar]
- Yu, F.H.; Huang, K.J.; Wang, C.T. HIV-1 Mutant Assembly, Processing and Infectivity Expresses Pol Independent of Gag. Viruses 2020, 12, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, O.N.; Baltimore, D. Relationship of Retrovirus Polyprotein Cleavages to Virion Maturation Studied with Temperature-Sensitive Murine Leukemia Virus Mutants. J. Virol. 1978, 26, 750–761. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.S.; He, C. Pseudouridine in a New Era of RNA Modifications. Cell Res. 2015, 25, 153–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression Kinetics of Nucleoside-Modified mRNA Delivered in Lipid Nanoparticles to Mice by Various Routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Mauger, D.M.; Joseph Cabral, B.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K. In Vitro-Transcribed mRNA Therapeutics: Out of the Shadows and Into the Spotlight. Mol. Ther. 2019, 27, 691–692. [Google Scholar] [CrossRef] [Green Version]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-Engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollner, C.J.; Richner, J.M. mRNA Vaccines against Flaviviruses. Vaccines 2021, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, F.; Everton, E.; Smith, A.R.; Liu, H.; Osota, E.; Beattie, M.; Tam, Y.; Pardi, N.; Weissman, D.; Gouon-Evans, V. Murine Liver Repair via Transient Activation of Regenerative Pathways in Hepatocytes Using Lipid Nanoparticle-Complexed Nucleoside-Modified mRNA. Nat. Commun. 2021, 12, 613. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.T.; Gesteland, R.F.; Atkins, J.F. Efficient Stimulation of Site-Specific Ribosome Frameshifting by Antisense Oligonucleotides. RNA 2004, 10, 1653–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsthoorn, R.C.L.; Laurs, M.; Sohet, F.; Hilbers, C.W.; Heus, H.A.; Pleij, C.W.A. Novel Application of sRNA: Stimulation of Ribosomal Frameshifting. RNA 2004, 10, 1702–1703. [Google Scholar] [CrossRef] [Green Version]
- Henderson, C.M.; Anderson, C.B.; Howard, M.T. Antisense-Induced Ribosomal Frameshifting. Nucleic Acids Res. 2006, 34, 4302–4310. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Noteborn, M.H.M.; Olsthoorn, R.C.L. Stimulation of Ribosomal Frameshifting by Antisense LNA. Nucleic Acids Res. 2010, 38, 8277–8283. [Google Scholar] [CrossRef]
- Yewdell, J.W. Individuals Cannot Rely on COVID-19 Herd Immunity: Durable Immunity to Viral Disease Is Limited to Viruses with Obligate Viremic Spread. PLoS Pathog. 2021, 17, e1009509. [Google Scholar] [CrossRef]
- Qiao, J.; Li, Y.-S.; Zeng, R.; Liu, F.-L.; Luo, R.-H.; Huang, C.; Wang, Y.-F.; Zhang, J.; Quan, B.; Shen, C.; et al. SARS-CoV-2 M pro Inhibitors with Antiviral Activity in a Transgenic Mouse Model. Science 2021, 371, 1374–1378. [Google Scholar] [CrossRef]
- Günther, S.; Reinke, P.Y.A.; Fernández-Garciá, Y.; Lieske, J.; Lane, T.J.; Ginn, H.M.; Koua, F.H.M.; Ehrt, C.; Ewert, W.; Oberthuer, D.; et al. X-Ray Screening Identifies Active Site and Allosteric Inhibitors of SARS-CoV-2 Main Protease. Science 2021, 372, 642–646. [Google Scholar] [CrossRef]
- Sun, Y.; Abriola, L.; Surovtseva, Y.V.; Lindenbach, B.D.; Guo, J.U. Restriction of SARS-CoV-2 Replication by Targeting Programmed −1 Ribosomal Frameshifting In Vitro. Proc. Natl. Acad. Sci. USA 2021, in press. [Google Scholar] [CrossRef]
- Atkins, J.F.; Ryan, M.D. Foot and Mouth’s Achilles’ Heel? Nat. Biotechnol. 2008, 26, 1335–1336. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, H.J.; Martin, R.G.; Ames, B.N. Classification of Aminotransferase (C Gene) Mutants in the Histidine Operon. J. Mol. Biol. 1966, 21, 335–355. [Google Scholar] [CrossRef]
- Crick, F.H. What Mad Pursuit: A Personal View of Scientific Discovery; Basic Books: New York, NY, USA, 1988; p. 208. [Google Scholar]
- Newton, A. Isolation and Characterization of Frameshift Mutations in the lac Operon. J. Mol. Biol. 1970, 49, 589–601. [Google Scholar] [CrossRef]
- Atkins, J.F.; Elseviers, D.; Gorini, L. Low Activity of β-Galactosidase in Frameshift Mutants of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 1192–1195. [Google Scholar] [CrossRef] [Green Version]
- Atkins, J.F.; Björk, G.R. A Gripping Tale of Ribosomal Frameshifting: Extragenic Suppressors of Frameshift Mutations Spotlight P-Site Realignment. Microbiol. Mol. Biol. Rev. 2009, 73, 178–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bretscher, M.S. Translocation in Protein Synthesis: A Hybrid Structure Model. Nature 1968, 218, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Moazed, D.; Noller, H.F. Intermediate States in the Movement of Transfer RNA in the Ribosome. Nature 1989, 342, 142–148. [Google Scholar] [CrossRef]
- Atkins, J.F.; Gesteland, R.F. mRNA Readout at 40. Nature 2001, 414, 693. [Google Scholar] [CrossRef]
- Crick, F.H.C.; Barnett, L.; Brenner, S.; Watts-Tobin, R.J. General Nature of the Genetic Code for Proteins. Nature 1961, 192, 1227–1232. [Google Scholar] [CrossRef]
- Ibba, M.; Becker, H.D.; Stathopoulos, C.; Tumbula, D.L.; Söll, D. The Adaptor Hypothesis Revisited. Trends Biochem. Sci. 2000, 25, 311–316. [Google Scholar] [CrossRef]
- Spirin, A.S. Ribosomal Translocation: Facts and Models. Prog. Nucleic Acid Res. Mol. Biol. 1985, 32, 75–114. [Google Scholar] [PubMed]
- Southworth, D.R.; Brunelle, J.L.; Green, R. EFG-Independent Translocation of the mRNA:tRNA Complex Is Promoted by Modification of the Ribosome with Thiol-Specific Reagents. J. Mol. Biol. 2002, 324, 611–623. [Google Scholar] [CrossRef]
- Geggier, P.; Dave, R.; Feldman, M.B.; Terry, D.S.; Altman, R.B.; Munro, J.B.; Blanchard, S.C. Conformational Sampling of Aminoacyl-tRNA during Selection on the Bacterial Ribosome. J. Mol. Biol. 2010, 399, 576–595. [Google Scholar] [CrossRef] [Green Version]
- Rodnina, M.V.; Peske, F.; Peng, B.Z.; Belardinelli, R.; Wintermeyer, W. Converting GTP Hydrolysis into Motion: Versatile Translational Elongation Factor G. Biol. Chem. 2019, 401, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.Z.; Bock, L.V.; Belardinelli, R.; Peske, F.; Grubmüller, H.; Rodnina, M.V. Active Role of Elongation Factor G in Maintaining the mRNA Reading Frame during Translation. Sci. Adv. 2019, 5, eaax8030. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Lancaster, L.; Donohue, J.P.; Noller, H.F. Spontaneous Ribosomal Translocation of mRNA and tRNAs into a Chimeric Hybrid State. Proc. Natl. Acad. Sci. USA 2019, 116, 7813–7818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niblett, D.; Nelson, C.; Leung, C.S.; Rexroad, G.; Cosy, J.; Zhou, J.; Lancaster, L.; Noller, H.F. Mutations in Domain IV of Elongation Factor EF-G Confer -1 Frameshifting. RNA 2021, 27, 40–53. [Google Scholar] [CrossRef]
- Atkins, J.F.; Böck, A.; Matsufuji, S.; Gesteland, R.F. Dynamics of the genetic code. In The RNA World: The Nature of Modern RNA Suggest a Prebiotic RNA World; Gesteland, R.F., Cech, T.R., Atkins, J.F., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1999; pp. 637–673. [Google Scholar]
- Yewdell, J.W. Hide and Seek in the Peptidome. Science 2003, 301, 1334–1335. [Google Scholar] [CrossRef]
- Bourdetsky, D.; Schmelzer, C.E.H.; Admon, A. The Nature and Extent of Contributions by Defective Ribosome Products to the HLA Peptidome. Proc. Natl. Acad. Sci. USA 2014, 111, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Qian, S.-B.; Reits, E.; Neefjes, J.; Deslich, J.M.; Bennink, J.R.; Yewdell, J.W. Tight Linkage between Translation and MHC Class I Peptide Ligand Generation Implies Specialized Antigen Processing for Defective Ribosomal Products. J. Immunol. 2006, 177, 227–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dersh, D.; Hollý, J.; Yewdell, J.W. A Few Good Peptides: MHC Class I-Based Cancer Immunosurveillance and Immunoevasion. Nat. Rev. Immunol. 2021, 21, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.; Müller, M.; Pak, H.S.; Harnett, D.; Huber, F.; Grun, D.; Leleu, M.; Auger, A.; Arnaud, M.; Stevenson, B.J.; et al. Integrated Proteogenomic Deep Sequencing and Analytics Accurately Identify Non-Canonical Peptides in Tumor Immunopeptidomes. Nat. Commun. 2020, 11, 1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, E.; Vincent, K.; Fortier, M.H.; Laverdure, J.P.; Bramoullé, A.; Hardy, M.P.; Voisin, G.; Roux, P.P.; Lemieux, S.; Thibault, P.; et al. The MHC I Immunopeptidome Conveys to the Cell Surface an Integrative View of Cellular Regulation. Mol. Syst. Biol. 2011, 7, 533. [Google Scholar] [CrossRef] [PubMed]
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, É.; Bonneil, É.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Côté, C.; et al. Noncoding Regions Are the Main Source of Targetable Tumor-Specific Antigens. Sci. Transl. Med. 2018, 10, eaau5516. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Kishton, R.J.; Angel, M.; Conn, C.S.; Dalla-Venezia, N.; Marcel, V.; Vincent, A.; Catez, F.; Ferré, S.; Ayadi, L.; et al. Ribosomal Proteins Regulate MHC Class I Peptide Generation for Immunosurveillance. Mol. Cell 2019, 73, 1162–1173.e5. [Google Scholar] [CrossRef] [Green Version]
- Bartok, O.; Pataskar, A.; Nagel, R.; Laos, M.; Goldfarb, E.; Hayoun, D.; Levy, R.; Körner, P.R.; Kreuger, I.Z.M.; Champagne, J.; et al. Anti-Tumour Immunity Induces Aberrant Peptide Presentation in Melanoma. Nature 2020, 590, 332–337. [Google Scholar] [CrossRef]
- De Crécy-Lagard, V.; Boccaletto, P.; Mangleburg, C.G.; Sharma, P.; Lowe, T.M.; Leidel, S.A.; Bujnicki, J.M. Matching tRNA Modifications in Humans to Their Known and Predicted Enzymes. Nucleic Acids Res. 2019, 47, 2143–2159. [Google Scholar] [CrossRef]
- Nagayoshi, Y.; Chujo, T.; Hirata, S.; Nakatsuka, H.; Chen, C.W.; Takakura, M.; Miyauchi, K.; Ikeuchi, Y.; Carlyle, B.C.; Kitchen, R.R.; et al. Loss of Ftsj1 Perturbs Codon-Specific Translation Efficiency in the Brain and Is Associated with X-Linked Intellectual Disability. Sci. Adv. 2021, 7, eabf3072. [Google Scholar] [CrossRef]
- Girstmair, H.; Saffert, P.; Rode, S.; Czech, A.; Holland, G.; Bannert, N.; Ignatova, Z. Depletion of Cognate Charged Transfer RNA Causes Translational Frameshifting within the Expanded CAG Stretch in Huntingtin. Cell Rep. 2013, 3, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Brunner, A.D.; Cogan, J.Z.; Nunez, J.K.; Fields, A.P.; Adamson, B.; Itzhak, D.N.; Li, J.Y.; Mann, M.; Leonetti, M.D.; et al. Pervasive Functional Translation Of Noncanonical Human Open Reading Frames. Science 2020, 367, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260.e29. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, I.P.; Wei, J.; Caster, S.Z.; Smith, K.M.; Michel, A.M.; Zhang, Y.; Firth, A.E.; Freitag, M.; Dunlap, J.C.; Bell-Pedersen, D.; et al. Translation Initiation from Conserved Non-AUG Codons Provides Additional Layers of Regulation and Coding Capacity. MBio 2017, 8, e00844-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzani, I.; Ivanov, I.P.; Andreev, D.E.; Dmitriev, R.I.; Dean, K.A.; Baranov, P.V.; Atkins, J.F.; Loughran, G. Systematic Analysis of the PTEN 5′ Leader Identifies a Major AUU Initiated Proteoform. Open Biol. 2016, 6, 150203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starck, S.R.; Shastri, N. Nowhere to Hide: Unconventional Translation Yields Cryptic Peptides for Immune Surveillance. Immunol. Rev. 2016, 272, 8–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Damme, P.; Gawron, D.; Van Criekinge, W.; Menschaert, G. N-Terminal Proteomics and Ribosome Profiling Provide a Comprehensive View of the Alternative Translation Initiation Landscape in Mice and Men. Mol. Cell. Proteomics 2014, 13, 1245–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.P.; Firth, A.E.; Michel, A.M.; Atkins, J.F.; Baranov, P.V. Identification of Evolutionarily Conserved Non-AUG-Initiated N-Terminal Extensions in Human Coding Sequences. Nucleic Acids Res. 2011, 39, 4220–4234. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, A.R.; Higdon, A.L.; Hollerer, I.; Fields, A.P.; Jungreis, I.; Diamond, P.D.; Kellis, M.; Jovanovic, M.; Brar, G.A. Translation Initiation Site Profiling Reveals Widespread Synthesis of Non-AUG-Initiated Protein Isoforms in Yeast. Cell Syst. 2020, 11, 145–160.e5. [Google Scholar] [CrossRef]
- Khan, Y.A.; Jungreis, I.; Wright, J.C.; Mudge, J.M.; Choudhary, J.S.; Firth, A.E.; Kellis, M. Evidence for a Novel Overlapping Coding Sequence in POLG Initiated at a CUG Start Codon. BMC Genet. 2020, 21, 25. [Google Scholar] [CrossRef] [Green Version]
- Loughran, G.; Zhdanov, A.V.; Mikhaylova, M.S.; Rozov, F.N.; Datskevich, P.N.; Kovalchuk, S.I.; Serebryakova, M.V.; Kiniry, S.J.; Michel, A.M.; O’Connor, P.B.F.; et al. Unusually Efficient CUG Initiation of an Overlapping Reading Frame in POLG mRNA Yields Novel Protein POLGARF. Proc. Natl. Acad. Sci. USA 2020, 117, 24936–24946. [Google Scholar] [CrossRef]
- Erhard, F.; Dölken, L.; Schilling, B.; Schlosser, A. Identification of the Cryptic HLA-I Immunopeptidome. Cancer Immunol. Res. 2020, 8, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Zhang, J.; Lian, X.; Sun, L.; Meng, K.; Chen, Y.; Sun, Z.; Yin, X.; Li, Y.; Zhao, J.; et al. A Hidden Human Proteome Encoded by “non-Coding” Genes. Nucleic Acids Res. 2019, 47, 8111–8125. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.A.; Brunelle, M.; Lucier, J.F.; Delcourt, V.; Levesque, M.; Grenier, F.; Samandi, S.; Leblanc, S.; Aguilar, J.D.; Dufour, P.; et al. OpenProt: A More Comprehensive Guide to Explore Eukaryotic Coding Potential and Proteomes. Nucleic Acids Res. 2019, 47, 403–410. [Google Scholar] [CrossRef] [Green Version]
- Na, C.H.; Barbhuiya, M.A.; Kim, M.S.; Verbruggen, S.; Eacker, S.M.; Pletnikova, O.; Troncoso, J.C.; Halushka, M.K.; Menschaert, G.; Overall, C.M.; et al. Discovery of Noncanonical Translation Initiation Sites through Mass Spectrometric Analysis of Protein N Termini. Genome Res. 2018, 28, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, I.P.; Shin, B.S.; Loughran, G.; Tzani, I.; Young-Baird, S.K.; Cao, C.; Atkins, J.F.; Dever, T.E. Polyamine Control of Translation Elongation Regulates Start Site Selection on Antizyme Inhibitor mRNA via Ribosome Queuing. Mol. Cell 2018, 70, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Prasad, S.; Starck, S.R.; Shastri, N. Presentation of Cryptic Peptides by MHC Class I Is Enhanced by Inflammatory Stimuli. J. Immunol. 2016, 197, 2981–2991. [Google Scholar] [CrossRef] [Green Version]
- Yewdell, J.W.; Hollý, J. DRiPs Get Molecular. Curr. Opin. Immunol. 2020, 64, 130–136. [Google Scholar] [CrossRef]
- Ruiz Cuevas, M.V.; Hardy, M.P.; Hollý, J.; Bonneil, É.; Durette, C.; Courcelles, M.; Lanoix, J.; Côté, C.; Staudt, L.M.; Lemieux, S.; et al. Most Non-Canonical Proteins Uniquely Populate the Proteome or Immunopeptidome. Cell Rep. 2021, 34, 108815. [Google Scholar] [CrossRef]
- Hukelmann, J.L.; Anderson, K.E.; Sinclair, L.V.; Grzes, K.M.; Murillo, A.B.; Hawkins, P.T.; Stephens, L.R.; Lamond, A.I.; Cantrell, D.A. The Cytotoxic T Cell Proteome and Its Shaping by the Kinase mTOR. Nat. Immunol. 2016, 17, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Yewdell, J.W.; Dersh, D.; Fåhraeus, R. Peptide Channeling: The Key to MHC Class I Immunosurveillance? Trends Cell Biol. 2019, 29, 929–939. [Google Scholar] [CrossRef]
- Goodenough, E.; Robinson, T.M.; Zook, M.B.; Flanigan, K.M.; Atkins, J.F.; Howard, M.T.; Eisenlohr, L.C. Cryptic MHC Class I-Binding Peptides Are Revealed by Aminoglycoside-Induced Stop Codon Read-through into the 3′ UTR. Proc. Natl. Acad. Sci. USA 2014, 111, 5670–5675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.J.; Sanjana, N.E.; Kishton, R.J.; Eidizadeh, A.; Vodnala, S.K.; Cam, M.; Gartner, J.J.; Jia, L.; Steinberg, S.M.; Yamamoto, T.N.; et al. Identification of Essential Genes for Cancer Immunotherapy. Nature 2017, 548, 537–542. [Google Scholar] [CrossRef] [Green Version]
- Ferretti, M.B.; Karbstein, K. Does Functional Specialization of Ribosomes Really Exist? RNA 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Horsburgh, B.C.; Kollmus, H.; Hauser, H.; Coen, D.M. Translational Recoding Induced by G-Rich mRNA Sequences That Form Unusual Structures. Cell 1996, 86, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, A.; Chen, S.-H.; Horsburgh, B.C.; Coen, D.M. Translational Compensation of a Frameshift Mutation Affecting Herpes Simplex Virus Thymidine Kinase Is Sufficient To Permit Reactivation from Latency. J. Virol. 2003, 77, 4703–4709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zook, M.B.; Howard, M.T.; Sinnathamby, G.; Atkins, J.F.; Eisenlohr, L.C. Epitopes Derived by Incidental Translational Frameshifting Give Rise to a Protective CTL Response. J. Immunol. 2006, 176, 6928–6934. [Google Scholar] [CrossRef] [Green Version]
- Grentzmann, G.; Ingram, J.A.; Kelly, P.J.; Gesteland, R.F.; Atkins, J.F. A Dual-Luciferase Reporter System for Studying Recoding Signals. RNA 1998, 4, 479–486. [Google Scholar]
- Pan, D.; Coen, D.M. Net -1 Frameshifting on a Noncanonical Sequence in a Herpes Simplex Virus Drug-Resistant Mutant Is Stimulated by Nonstop mRNA. Proc. Natl. Acad. Sci. USA 2012, 109, 14852–14857. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekaran, V.; Juszkiewicz, S.; Choi, J.; Puglisi, J.D.; Brown, A.; Shao, S.; Ramakrishnan, V.; Hegde, R.S. Mechanism of Ribosome Stalling during Translation of a Poly(A) Tail. Nat. Struct. Mol. Biol. 2019, 26, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Tesina, P.; Lessen, L.N.; Buschauer, R.; Cheng, J.; Wu, C.C.; Berninghausen, O.; Buskirk, A.R.; Becker, T.; Beckmann, R.; Green, R. Molecular Mechanism of Translational Stalling by Inhibitory Codon Combinations and Poly(A) Tracts. EMBO J. 2020, 39, e103365. [Google Scholar] [CrossRef]
- Smith, A.M.; Costello, M.S.; Kettring, A.H.; Wingo, R.J.; Moore, S.D. Ribosome Collisions Alter Frameshifting at Translational Reprogramming Motifs in Bacterial mRNAs. Proc. Natl. Acad. Sci. USA 2019, 116, 21769–21779. [Google Scholar] [CrossRef] [Green Version]
- Rocha, E.P.C.; Matic, I.; Taddei, F. Over-Representation of Repeats in Stress Response Genes: A Strategy to Increase Versatility under Stressful Conditions? Nucleic Acids Res. 2002, 30, 1886–1894. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, J.P.; Ronson, C.W. Silencing Quorum Sensing and Ice Mobility through Antiactivation and Ribosomal Frameshifting. Mob. Genet. Elements 2015, 5, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Loving, C.L.; Osorio, F.A.; Murtaugh, M.P.; Zuckermann, F.A. Innate and Adaptive Immunity against Porcine Reproductive and Respiratory Syndrome Virus. Vet. Immunol. Immunopathol. 2015, 167, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shang, P.; Shyu, D.; Carrillo, C.; Naraghi-Arani, P.; Jaing, C.J.; Renukaradhya, G.J.; Firth, A.E.; Snijder, E.J.; Fang, Y. Nonstructural Proteins Nsp2TF and Nsp2N of Porcine Reproductive and Respiratory Syndrome Virus (PRRSV) Play Important Roles in Suppressing Host Innate Immune Responses. Virology 2018, 517, 164–176. [Google Scholar] [CrossRef]
- Sun, Z.; Chen, Z.; Lawson, S.R.; Fang, Y. The Cysteine Protease Domain of Porcine Reproductive and Respiratory Syndrome Virus Nonstructural Protein 2 Possesses Deubiquitinating and Interferon Antagonism Functions. J. Virol. 2010, 84, 7832–7846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Li, Y.; Ransburgh, R.; Snijder, E.J.; Fang, Y. Nonstructural Protein 2 of Porcine Reproductive and Respiratory Syndrome Virus Inhibits the Antiviral Function of Interferon-Stimulated Gene 15. J. Virol. 2012, 86, 3839–3850. [Google Scholar] [CrossRef] [Green Version]
- Van Kasteren, P.B.; Bailey-Elkin, B.A.; James, T.W.; Ninaber, D.K.; Beugeling, C.; Khajehpour, M.; Snijder, E.J.; Mark, B.L.; Kikkert, M. Deubiquitinase Function of Arterivirus Papain-like Protease 2 Suppresses the Innate Immune Response in Infected Host Cells. Proc. Natl. Acad. Sci. USA 2013, 110, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Barcena, M.; Limpens, R.W.A.L.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. Ultrastructural Characterization of Arterivirus Replication Structures: Reshaping the Endoplasmic Reticulum To Accommodate Viral RNA Synthesis. J. Virol. 2012, 86, 2474–2487. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Dojcinovic, D.; Guillaume, P.; Luescher, I. Analysis, Isolation, and Activation of Antigen-Specific CD4+ and CD8+ T Cells by Soluble MHC-Peptide Complexes. Front. Immunol. 2013, 4, 218. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.M.; Subramaniam, S.; Ni, Y.Y.; Cao, D.; Meng, X.J. The Non-Structural Protein Nsp2TF of Porcine Reproductive and Respiratory Syndrome Virus down-Regulates the Expression of Swine Leukocyte Antigen Class I. Virology 2016, 491, 115–124. [Google Scholar] [CrossRef]
- Randow, F.; Lehner, P.J. Viral Avoidance and Exploitation of the Ubiquitin System. Nat. Cell Biol. 2009, 11, 527–534. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of Antigen Processing by the Internal Repeat Region of the Epstein-Barr Virus Nuclear Antigen-1. Nature 1995, 375, 685–688. [Google Scholar] [CrossRef]
- Yin, Y.; Manoury, B.; Fåhraeus, R. Self-Inhibition of Synthesis and Antigen Presentation by Epstein-Barr Virus-Encoded EBNA1. Science 2003, 301, 1371–1374. [Google Scholar] [CrossRef]
- Kwun, H.J.; da Silva, S.R.; Shah, I.M.; Blake, N.; Moore, P.S.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 Mimics Epstein-Barr Virus EBNA1 Immune Evasion through Central Repeat Domain Effects on Protein Processing. J. Virol. 2007, 81, 8225–8235. [Google Scholar] [CrossRef] [Green Version]
- Kwun, H.J.; da Silva, S.R.; Qin, H.; Ferris, R.L.; Tan, R.; Chang, Y.; Moore, P.S. The Central Repeat Domain 1 of Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Latency Associated-Nuclear Antigen 1 (LANA1) Prevents Cis MHC Class I Peptide Presentation. Virology 2011, 412, 357–365. [Google Scholar] [CrossRef]
- Stochmanski, S.J.; Therrien, M.; Laganière, J.; Rochefort, D.; Laurent, S.; Karemera, L.; Gaudet, R.; Vyboh, K.; Van Meyel, D.J.; Di Cristo, G.; et al. Expanded ATXN3 Frameshifting Events Are Toxic in Drosophila and Mammalian Neuron Models. Hum. Mol. Genet. 2012, 21, 2211–2218. [Google Scholar] [CrossRef] [Green Version]
- Saffert, P.; Adamla, F.; Schieweck, R.; Atkins, J.F.; Ignatova, Z. An Expanded CAG Repeat in Huntingtin Causes +1 Frameshifting. J. Biol. Chem. 2016, 291, 18505–18513. [Google Scholar] [CrossRef] [Green Version]
- Tabet, R.; Schaeffer, L.; Freyermuth, F.; Jambeau, M.; Workman, M.; Lee, C.Z.; Lin, C.C.; Jiang, J.; Jansen-West, K.; Abou-Hamdan, H.; et al. CUG Initiation and Frameshifting Enable Production of Dipeptide Repeat Proteins from ALS/FTD C9ORF72 Transcripts. Nat. Commun. 2018, 9, 152. [Google Scholar] [CrossRef]
- Murat, P.; Zhong, J.; Lekieffre, L.; Cowieson, N.P.; Clancy, J.L.; Preiss, T.; Balasubramanian, S.; Khanna, R.; Tellam, J. G-Quadruplexes Regulate Epstein-Barr Virus-Encoded Nuclear Antigen 1 mRNA Translation. Nat. Chem. Biol. 2014, 10, 358–364. [Google Scholar] [CrossRef]
- Dabral, P.; Babu, J.; Zareie, A.; Verma, S.C. LANA and HnRNP A1 Regulate the Translation of LANA mRNA through G-Quadruplexes. J. Virol. 2019, 94, e01508–e01519. [Google Scholar] [CrossRef]
- Lista, M.J.; Martins, R.P.; Billant, O.; Contesse, M.A.; Findakly, S.; Pochard, P.; Daskalogianni, C.; Beauvineau, C.; Guetta, C.; Jamin, C.; et al. Nucleolin Directly Mediates Epstein-Barr Virus Immune Evasion through Binding to G-Quadruplexes of EBNA1 mRNA. Nat. Commun. 2017, 8, 16043. [Google Scholar] [CrossRef] [PubMed]
- Martins, R.P.; Malbert-Colas, L.; Lista, M.J.; Daskalogianni, C.; Apcher, S.; Pla, M.; Findakly, S.; Blondel, M.; Fåhraeus, R. Nuclear Processing of Nascent Transcripts Determines Synthesis of Full-Length Proteins and Antigenic Peptides. Nucleic Acids Res. 2019, 47, 3086–3100. [Google Scholar] [CrossRef]
- Penno, C.; Kumari, R.; Baranov, P.V.; Van Sinderen, D.; Atkins, J.F. Stimulation of Reverse Transcriptase Generated CDNAs with Specific Indels by Template RNA Structure: Retrotransposon, DNTP Balance, RT-Reagent Usage. Nucleic Acids Res. 2017, 45, 10143–10155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sun, T.; Bi, Z.; Ni, J.Q.; Pastor-Pareja, J.C.; Javid, B. Premature Termination Codon Readthrough in Drosophila Varies in a Developmental and Tissue-Specific Manner. Sci. Rep. 2020, 10, 8485. [Google Scholar] [CrossRef]
- Hudson, A.M.; Szabo, N.L.; Loughran, G.; Wills, N.M.; Atkins, J.F.; Cooley, L. Tissue-Specific Dynamic Codon Redefinition in Drosophila. Proc. Natl. Acad. Sci. USA 2021, 118, 5. [Google Scholar] [CrossRef] [PubMed]
- Karki, P.; Carney, T.D.; Maracci, C.; Yatsenko, A.S.; Shcherbata, H.R.; Rodnina, M.V. Tissue-Specific Regulation of Translational Readthrough Tunes Functions of the Traffic Jam Transcription Factor. bioRxiv 2020. [Google Scholar] [CrossRef]
- Swart, E.C.; Serra, V.; Petroni, G.; Nowacki, M. Genetic Codes with No Dedicated Stop Codon: Context-Dependent Translation Termination. Cell 2016, 166, 691–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaphy, S.M.; Mariotti, M.; Gladyshev, V.N.; Atkins, J.F.; Baranov, P.V. Novel Ciliate Genetic Code Variants Including the Reassignment of All Three Stop Codons to Sense Codons in Condylostoma magnum. Mol. Biol. Evol. 2016, 33, 2885–2889. [Google Scholar] [CrossRef] [Green Version]
- Záhonová, K.; Kostygov, A.Y.; Ševčíková, T.; Yurchenko, V.; Eliáš, M. An Unprecedented Non-Canonical Nuclear Genetic Code with All Three Termination Codons Reassigned as Sense Codons. Curr. Biol. 2016, 26, 2364–2369. [Google Scholar] [CrossRef] [Green Version]
- Bachvaroffi, T.R. A Precedented Nuclear Genetic Code with All Three Termination Codons Reassigned as Sense Codons in the Syndinean Amoebophrya Sp. Ex Karlodinium veneficum. PLoS ONE 2019, 14, e0212912. [Google Scholar] [CrossRef]
- Lobanov, A.V.; Heaphy, S.M.; Turanov, A.A.; Gerashchenko, M.V.; Pucciarelli, S.; Devaraj, R.R.; Xie, F.; Petyuk, V.A.; Smith, R.D.; Klobutcher, L.A.; et al. Position-Dependent Termination and Widespread Obligatory Frameshifting in Euplotes Translation. Nat. Struct. Mol. Biol. 2017, 24, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Westergren Jakobsson, A.; Segerman, B.; Wallerman, O.; Bergström Lind, S.; Zhao, H.; Rubin, C.-J.; Pettersson, U.; Akusjärvi, G. The Human Adenovirus 2 Transcriptome: An Amazing Complexity of Alternatively Spliced mRNAs. J. Virol. 2020, 95, e01869-20. [Google Scholar]
- Lewis, J.B.; Anderson, C.W.; Atkins, J.F. Further Mapping of Late Adenovirus Genes by Cell-Free Translation of RNA Selected by Hybridization to Specific DNA Fragments. Cell 1977, 12, 37–44. [Google Scholar] [CrossRef]
- Atkins, J.F.; Weiss, R.B.; Gesteland, R.F. Ribosome Gymnastics-Degree of Difficulty 9.5, Style 10.0. Cell 1990, 62, 413–423. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atkins, J.F.; O’Connor, K.M.; Bhatt, P.R.; Loughran, G. From Recoding to Peptides for MHC Class I Immune Display: Enriching Viral Expression, Virus Vulnerability and Virus Evasion. Viruses 2021, 13, 1251. https://doi.org/10.3390/v13071251
Atkins JF, O’Connor KM, Bhatt PR, Loughran G. From Recoding to Peptides for MHC Class I Immune Display: Enriching Viral Expression, Virus Vulnerability and Virus Evasion. Viruses. 2021; 13(7):1251. https://doi.org/10.3390/v13071251
Chicago/Turabian StyleAtkins, John F., Kate M. O’Connor, Pramod R. Bhatt, and Gary Loughran. 2021. "From Recoding to Peptides for MHC Class I Immune Display: Enriching Viral Expression, Virus Vulnerability and Virus Evasion" Viruses 13, no. 7: 1251. https://doi.org/10.3390/v13071251
APA StyleAtkins, J. F., O’Connor, K. M., Bhatt, P. R., & Loughran, G. (2021). From Recoding to Peptides for MHC Class I Immune Display: Enriching Viral Expression, Virus Vulnerability and Virus Evasion. Viruses, 13(7), 1251. https://doi.org/10.3390/v13071251