
Leveraging Novel Integrated Single-Cell Analyses to Define HIV-1 Latency Reversal


Abstract
:1. Introduction
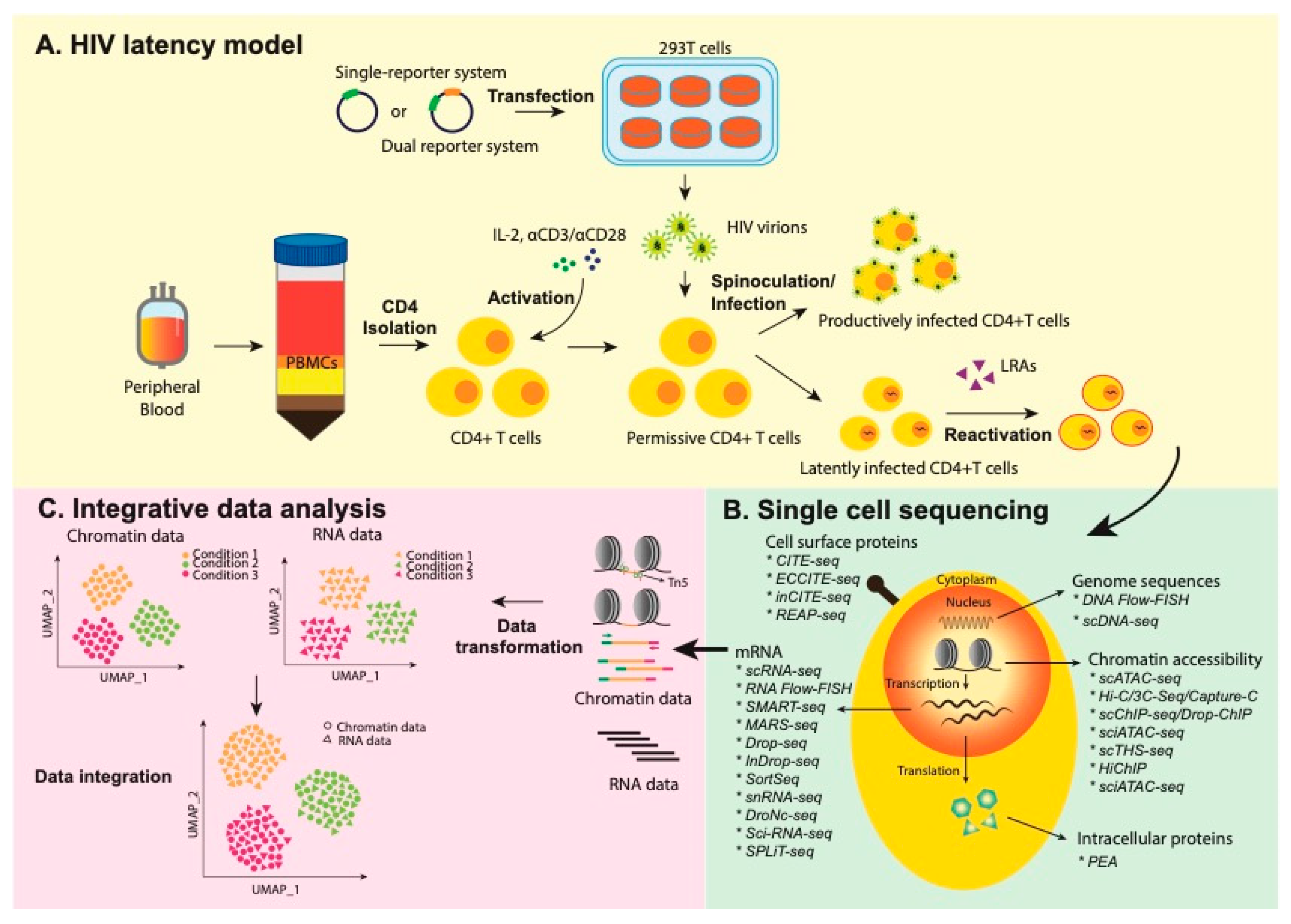
2. Single-Cell Studies of HIV-1 Latency
3. Multimodal Profiling of HIV-1 Latency
3.1. Epigenomics from Single Cells
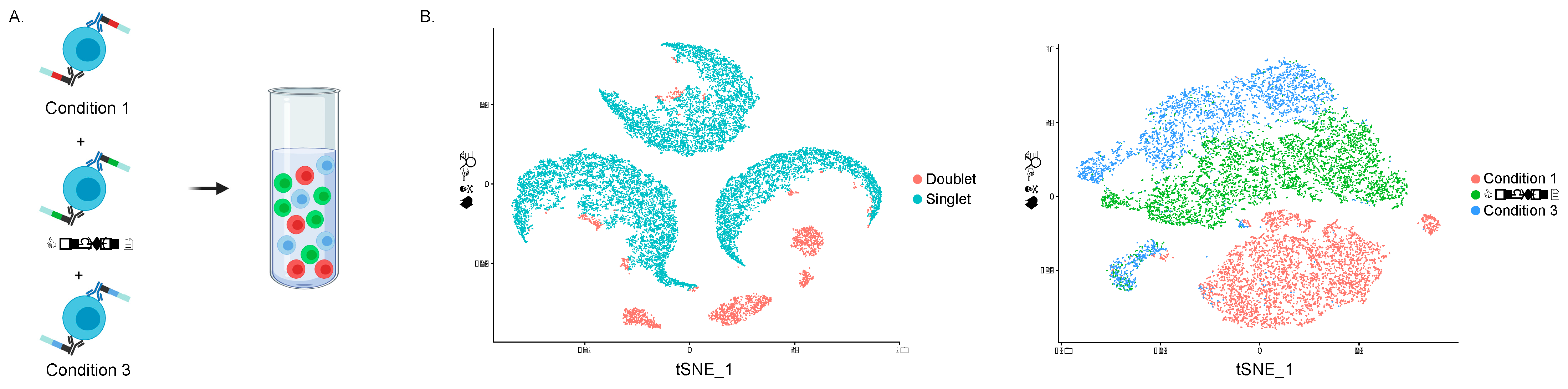
3.2. Integrated Protein and RNA Measurements
3.3. Other Frontier Single-Cell Technologies

{kind=link}
{kind=link}
| Single-Cell Method | Acronym | Target | Reference |
|---|---|---|---|
| Single-cell RNA sequencing | scRNA-seq | mRNA | [107] |
| Switch mechanism at the 5′ End of RNA templates single-cell sequencing | SMART-seq | Full-length capture of RNA | [108] |
| Massively parallel single-cell RNA-sequencing | MARS-seq | 3′-end only | [109] |
| Drop-seq | Drop-seq | 3′-end only | [110] |
| Indexing droplets RNA sequencing | InDrop | 3′-end only | [111] |
| Single-nucleus RNA sequencing | snRNA-seq | RNA | [112] |
| Massively parallel single-nucleus RNA-seq | DroNc-seq | 3′-end only | [103] |
| Single-cell combinatorial indexing RNA sequencing | Sci-RNA-seq | 3′-end only | [113] |
| Split-pool ligation-based transcriptome sequencing | SPLiT-seq | 3′-end only | [101] |
| single-cell assay for transposase-accessible chromatin sequencing | scATAC-seq | Chromatin accessibility | [79] |
| Chromosome conformation capture (3C) coupled with sequencing | Hi-C/3C-Seq/Capture-C | Chromatin structure | [114] |
| Droplet-based single-cell chromatin immune-precipitation sequencing | scChIP-seq/Drop-ChIP | Chromatin fragments | [115] |
| Single-cell transposome hypersensitive site sequencing | scTHS-seq | Chromatin accessibility | [116] |
| Chromosome conformation capture sequencing combining chromatin immunoprecipitation | HiChIP | Chromasome capture | [117] |
| Single-cell combinatorial indexing ATAC-seq | sciATAC-seq | Chromatin accessibility | [118] |
| Cellular indexing of transcriptomes and epitopes by sequencing | CITE-seq | Multiomic | [74] |
| RNA expression and protein sequencing assay | REAP-seq | Multiomic | [75] |
| Expanded CRISPR-compatible cellular indexing of transcriptomes and epitopes by sequencing | ECCITE-seq | Multiomic | [97] |
| Intranuclear cellular indexing of transcriptomes and epitopes | inCITE-seq | Intranuclear protein and transcriptome | [100] |
4. Integration of Single-Cell Datasets
4.1. M3S
4.2. Mixscape
4.3. Seurat Toolkits
4.4. LIGER
4.5. Harmony
4.6. BindSC
4.7. MAESTRO
4.8. ScAI
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siliciano, R.F.; Greene, W.C. HIV Latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Laskey, S.B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Hoh, R.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiener, B.; Horsburgh, B.A.; Eden, J.-S.; Barton, K.; Schlub, T.E.; Lee, E.; von Stockenstrom, S.; Odevall, L.; Milush, J.M.; Liegler, T.; et al. Identification of Genetically Intact HIV-1 Proviruses in Specific CD4 + T Cells from Effectively Treated Participants. Cell Rep. 2017, 21, 813–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-Competent Noninduced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.Q.; Orlova-Fink, N.; Einkauf, K.; Chowdhury, F.Z.; Sun, X.; Harrington, S.; Kuo, H.-H.; Hua, S.; Chen, H.-R.; Ouyang, Z.; et al. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Investig. 2017, 127, 2689–2696. [Google Scholar] [CrossRef]
- Imamichi, H.; Dewar, R.L.; Adelsberger, J.W.; Rehm, C.A.; O’Doherty, U.; Paxinos, E.E.; Fauci, A.S.; Lane, H.C. Defective HIV-1 proviruses produce novel protein-coding RNA species in HIV-infected patients on combination antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2016, 113, 8783–8788. [Google Scholar] [CrossRef] [Green Version]
- Pollack, R.A.; Jones, R.B.; Pertea, M.; Bruner, K.M.; Martin, A.R.; Thomas, A.; Capoferri, A.A.; Beg, S.A.; Huang, S.-H.; Karandish, S.; et al. Defective HIV-1 Proviruses Are Expressed and Can Be Recognized by Cytotoxic T Lymphocytes, which Shape the Proviral Landscape. Cell Host Microbe 2017, 21, 494–506.e4. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.T.; McMahon, D.K.; Bosch, R.J.; Lalama, C.M.; Cyktor, J.C.; Macatangay, B.J.; Rinaldo, C.R.; Riddler, S.A.; Hogg, E.; Godfrey, C.; et al. Levels of HIV-1 persistence on antiretroviral therapy are not associated with markers of inflammation or activation. PLoS Pathog. 2017, 13, e1006285. [Google Scholar] [CrossRef]
- Bachmann, N.; the Swiss HIV Cohort Study; Von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; et al. Determinants of HIV-1 reservoir size and long-term dynamics during suppressive ART. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ramratnam, B.; Mittler, J.E.; Zhang, L.; Boden, D.; Hurley, A.; Fang, F.; Macken, C.A.; Perelson, A.S.; Markowitz, M.; Ho, D.D. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 2000, 6, 82–85. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nat. Cell Biol. 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Bertagnolli, L.N.; White, J.A.; Simonetti, F.R.; Beg, S.A.; Lai, J.; Tomescu, C.; Murray, A.J.; Antar, A.; Zhang, H.; Margolick, J.B.; et al. The role of CD32 during HIV-1 infection. Nat. Cell Biol. 2018, 561, E17–E19. [Google Scholar] [CrossRef] [PubMed]
- Osuna, C.E.; Lim, S.Y.; Kublin, J.L.; Apps, R.; Chen, E.; Mota, T.M.; Huang, S.H.; Ren, Y.; Bachtel, N.D.; Tsibris, A.M.; et al. Evidence that CD32a does not mark the HIV-1 latent reservoir. Nature 2018, 561, E20–E28. [Google Scholar] [CrossRef] [PubMed]
- Pérez, L.; Anderson, J.; Chipman, J.; Thorkelson, A.; Chun, T.-W.; Moir, S.; Haase, A.T.; Douek, D.C.; Schacker, T.W.; Boritz, E.A. Conflicting evidence for HIV enrichment in CD32+ CD4 T cells. Nat. Cell Biol. 2018, 561, E9–E16. [Google Scholar] [CrossRef]
- Jiang, C.; Lian, X.; Gao, C.; Sun, X.; Einkauf, K.B.; Chevalier, J.M.; Chen, S.M.Y.; Hua, S.; Rhee, B.; Chang, K.; et al. Distinct viral reservoirs in individuals with spontaneous control of HIV-1. Nat. Cell Biol. 2020, 585, 261–267. [Google Scholar] [CrossRef]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Collora, J.A.; Liu, R.; Albrecht, K.; Ho, Y.-C. The single-cell landscape of immunological responses of CD4+ T cells in HIV versus severe acute respiratory syndrome coronavirus 2. Curr. Opin. HIV AIDS 2021, 16, 36–47. [Google Scholar] [CrossRef]
- Plantin, J.; Massanella, M.; Chomont, N. Inducible HIV RNA transcription assays to measure HIV persistence: Pros and cons of a compromise. Retrovirology 2018, 15, 1–11. [Google Scholar] [CrossRef]
- Yukl, S.A.; Kaiser, P.; Kim, P.; Telwatte, S.; Joshi, S.K.; Vu, M.; Lampiris, H.; Wong, J.K. HIV latency in isolated patient CD4+T cells may be due to blocks in HIV transcriptional elongation, completion, and splicing. Sci. Transl. Med. 2018, 10, eaap9927. [Google Scholar] [CrossRef] [Green Version]
- Procopio, F.A.; Fromentin, R.; Kulpa, D.A.; Brehm, J.H.; Bebin, A.-G.; Strain, M.C.; Richman, D.D.; O’Doherty, U.; Palmer, S.; Hecht, F.; et al. A Novel Assay to Measure the Magnitude of the Inducible Viral Reservoir in HIV-infected Individuals. EBioMedicine 2015, 2, 874–883. [Google Scholar] [CrossRef] [Green Version]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Günthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of Replication-Competent HIV Despite Prolonged Suppression of Plasma Viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.-W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nat. Cell Biol. 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Laird, G.M.; Eisele, E.E.; Rabi, S.A.; Lai, J.; Chioma, S.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Rapid Quantification of the Latent Reservoir for HIV-1 Using a Viral Outgrowth Assay. PLoS Pathog. 2013, 9, e1003398. [Google Scholar] [CrossRef] [Green Version]
- Wonderlich, E.R.; Subramanian, K.; Cox, B.; Wiegand, A.; Lackman-Smith, C.; Bale, M.; Stone, M.; Hoh, R.; Kearney, M.F.; Maldarelli, F.; et al. Effector memory differentiation increases detection of replication-competent HIV-l in resting CD4+ T cells from virally suppressed individuals. PLoS Pathog. 2019, 15, e1008074. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, F.R.; White, J.A.; Tumiotto, C.; Ritter, K.D.; Cai, M.; Gandhi, R.T.; Deeks, S.G.; Howell, B.J.; Montaner, L.J.; Blankson, J.N.; et al. Intact proviral DNA assay analysis of large cohorts of people with HIV provides a benchmark for the frequency and composition of persistent proviral DNA. Proc. Natl. Acad. Sci. USA 2020, 117, 18692–18700. [Google Scholar] [CrossRef]
- Gadol, N.; Crutcher, G.J.; Busch, M.P. Detection of intracellular HIV in lymphocytes by flow cytometry. Cytometry 1994, 15, 359–370. [Google Scholar] [CrossRef]
- Kux, A.; Bertram, S.; Hufert, F.T.; Schmitz, H.; von Laer, D. Antibodies to p24 antigen do not specifically detect HIV-infected lymphocytes in AIDS patients. J. Immunol. Methods 1996, 191, 179–186. [Google Scholar] [CrossRef]
- Cameron, P.U.; Hunter, S.D.; Jolley, D.; Sonza, S.; Mijch, A.; Crowe, S.M. Specificity of binding of HIV-1 anti-p24 antibodies to CD4+ lymphocytes from HIV-infected subjects. Cytometry 1998, 33, 83–88. [Google Scholar] [CrossRef]
- Lassen, K.G.; Hebbeler, A.M.; Bhattacharyya, D.; Lobritz, M.A.; Greene, W.C. A Flexible Model of HIV-1 Latency Permitting Evaluation of Many Primary CD4 T-Cell Reservoirs. PLoS ONE 2012, 7, e30176. [Google Scholar] [CrossRef] [Green Version]
- Chun, T.-W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Zerbato, J.M.; Serrao, E.; Lenzi, G.; Kim, B.; Ambrose, Z.; Watkins, S.C.; Engelman, A.N.; Sluis-Cremer, N. Establishment and Reversal of HIV-1 Latency in Naive and Central Memory CD4 + T Cells In Vitro. J. Virol. 2016, 90, 8059–8073. [Google Scholar] [CrossRef] [Green Version]
- Dobrowolski, C.; Valadkhan, S.; Graham, A.C.; Shukla, M.; Ciuffi, A.; Telenti, A.; Karn, J. Entry of Polarized Effector Cells into Quiescence Forces HIV Latency. mBio 2019, 10, e00337-19. [Google Scholar] [CrossRef] [Green Version]
- Agosto, L.M.; Henderson, A.J. CD4+T Cell Subsets and Pathways to HIV Latency. AIDS Res. Hum. Retrovir. 2018, 34, 780–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agosto, L.M.; Herring, M.B.; Mothes, W.; Henderson, A.J. HIV-1-Infected CD4+ T Cells Facilitate Latent Infection of Resting CD4+ T Cells through Cell-Cell Contact. Cell Rep. 2018, 24, 2088–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finzi, D.; Blankson, J.N.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.C.; Smith, K.A.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Chun, T.-W.; Engel, D.; Berrey, M.M.; Shea, T.; Corey, L.; Fauci, A.S. Early establishment of a pool of latently infected, resting CD4+ T cells during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 1998, 95, 8869–8873. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.-R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Archin, N.M.; Keedy, K.S.; Espeseth, A.; Dang, H.; Hazuda, D.J.; Margolis, D.M. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 2009, 23, 1799–1806. [Google Scholar] [CrossRef] [Green Version]
- Archin, N.M.; Kirchherr, J.L.; Sung, J.A.; Clutton, G.; Sholtis, K.; Xu, Y.; Allard, B.; Stuelke, E.; Kashuba, A.D.; Kuruc, J.D.; et al. Interval dosing with the HDAC inhibitor vorinostat effectively reverses HIV latency. J. Clin. Investig. 2017, 127, 3126–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Brinkmann, C.R.; Olesen, R.; Erikstrup, C.; Solomon, A.; Winckelmann, A.; Palmer, S.; Dinarello, C.; Buzon, M.; et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: A phase 1/2, single group, clinical trial. Lancet. HIV 2014, 1, 13–21. [Google Scholar] [CrossRef]
- Søgaard, O.S.; Graversen, M.E.; Leth, S.; Olesen, R.; Brinkmann, C.R.; Nissen, S.K.; Kjaer, A.S.; Schleimann, M.H.; Denton, P.W.; Hey-Cunningham, W.J.; et al. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11, e1005142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, C.; Serrano-Villar, S.; Madrid-Elena, N.; Elias, M.J.P.; Martín, M.E.; Barbas, C.; Ruipérez, J.; Muñoz, E.; Muñoz-Fernández, M.A.; Castor, T.; et al. Bryostatin-1 for latent virus reactivation in HIV-infected patients on antiretroviral therapy. AIDS 2016, 30, 1385–1392. [Google Scholar] [CrossRef]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-free HIV-1 remission and viral rebound after allogeneic stem cell transplantation: Report of 2 cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef]
- Hill, A.L.; Rosenbloom, D.I.S.; Fu, F.; Nowak, M.A.; Siliciano, R.F. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc. Natl. Acad. Sci. USA 2014, 111, 13475–13480. [Google Scholar] [CrossRef] [Green Version]
- Cillo, A.R.; Sobolewski, M.D.; Bosch, R.J.; Fyne, E.; Piatak, M.; Coffin, J.M.; Mellors, J.W. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2014, 111, 7078–7083. [Google Scholar] [CrossRef] [Green Version]
- Hosmane, N.N.; Kwon, K.J.; Bruner, K.M.; Capoferri, A.A.; Beg, S.; Rosenbloom, D.I.; Keele, B.F.; Ho, Y.-C.; Siliciano, J.D.; Siliciano, R.F. Proliferation of latently infected CD4+ T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef]
- Dar, R.D.; Hosmane, N.N.; Arkin, M.R.; Siliciano, R.F.; Weinberger, L.S. Screening for noise in gene expression identifies drug synergies. Science 2014, 344, 1392–1396. [Google Scholar] [CrossRef] [Green Version]
- Bradley, T.; Ferrari, G.; Haynes, B.F.; Margolis, D.M.; Browne, E.P. Single-Cell Analysis of Quiescent HIV Infection Reveals Host Transcriptional Profiles that Regulate Proviral Latency. Cell Rep. 2018, 25, 107–117.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerbato, J.M.; McMahon, D.K.; Sobolewski, M.D.; Mellors, J.W.; Sluis-Cremer, N. Naive CD4+ T Cells Harbor a Large Inducible Reservoir of Latent, Replication-competent Human Immunodeficiency Virus Type 1. Clin. Infect. Dis. 2019, 69, 1919–1925. [Google Scholar] [CrossRef]
- Rullo, E.V.; Pinzone, M.R.; Cannon, L.; Weissman, S.; Ceccarelli, M.; Zurakowski, R.; Nunnari, G.; O’Doherty, U. Persistence of an intact HIV reservoir in phenotypically naive T cells. JCI Insight 2020, 5, 20. [Google Scholar] [CrossRef]
- Golumbeanu, M.; Cristinelli, S.; Rato, S.; Munoz, M.; Cavassini, M.; Beerenwinkel, N.; Ciuffi, A. Single-Cell RNA-Seq Reveals Transcriptional Heterogeneity in Latent and Reactivated HIV-Infected Cells. Cell Rep. 2018, 23, 942–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahu, G.K.; Lee, K.; Ji, J.; Braciale, V.; Baron, S.; Cloyd, M.W. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology 2006, 355, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Cohn, L.B.; Da Silva, I.T.; Valieris, R.; Huang, A.; Lorenzi, J.C.C.; Cohen, Y.; Pai, J.A.; Butler, A.L.; Caskey, M.; Jankovic, M.; et al. Clonal CD4+ T cells in the HIV-1 latent reservoir display a distinct gene profile upon reactivation. Nat. Med. 2018, 24, 604–609. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Liu, R.; Yeh, Y.-H.J.; Varabyou, A.; Collora, J.A.; Sherrill-Mix, S.; Mehta, S.; Albrecht, K.; Hao, H.; Zhang, H.; Pollack, R.; et al. Single-cell transcriptional landscapes reveal HIV-1–driven aberrant host gene transcription as a potential therapeutic target. Sci. Transl. Med. 2020, 12, eaaz0802. [Google Scholar] [CrossRef]
- Stoeckius, M.; Zheng, S.; Houck-Loomis, B.; Hao, S.; Yeung, B.Z.; Mauck, W.M., 3rd; Smibert, P.; Satija, R. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol. 2018, 19, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Stegle, O.; Teichmann, S.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Hicks, S.C.; Townes, F.W.; Teng, M.; Irizarry, R. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics 2018, 19, 562–578. [Google Scholar] [CrossRef]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Estes, J.D.; Kityo, C.; Ssali, F.; Swainson, L.; Makamdop, K.N.; Del Prete, G.Q.; Deeks, S.; Luciw, P.A.; Chipman, J.G.; Beilman, G.J.; et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat. Med. 2017, 23, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Exposito, J.G.; Luque-Ballesteros, L.; Navarro, J.; Curran, A.; Burgos, J.; Ribera, E.; Torrella, A.; Planas, B.; Badía, R.; Martin-Castillo, M.; et al. Latency reversal agents affect differently the latent reservoir present in distinct CD4+ T subpopulations. PLoS Pathog. 2019, 15, e1007991. [Google Scholar] [CrossRef] [Green Version]
- Casuso, J.C.V.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardons, M.; Baxter, A.E.; Massanella, M.; Pagliuzza, A.; Fromentin, R.; Dufour, C.; Leyre, L.; Routy, J.-P.; Kaufmann, D.E.; Chomont, N. Single-cell characterization and quantification of translation-competent viral reservoirs in treated and untreated HIV infection. PLoS Pathog. 2019, 15, e1007619. [Google Scholar] [CrossRef]
- Navin, N.; Kendall, J.; Troge, J.; Andrews, P.; Rodgers, L.; McIndoo, J.; Cook, K.; Stepansky, A.; Levy, D.; Esposito, D.; et al. Tumour evolution inferred by single-cell sequencing. Nat. Cell Biol. 2011, 472, 90–94. [Google Scholar] [CrossRef] [Green Version]
- Adey, A.; Vitak, S.; Torkenczy, K.; Rosenkrantz, J.; Fields, A.; Christiansen, L.; Wong, M.; Carbone, L.; Steemers, F. Sequencing thousands of single-cell genomes with combinatorial indexing. Protoc. Exch. 2017, 14, 302–308. [Google Scholar] [CrossRef]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643.e4. [Google Scholar] [CrossRef] [Green Version]
- Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 2017, 6, e23203. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Buenrostro, J.D.; Wu, B.; Greenside, P.G.; Chan, S.M.; Koenig, J.L.; Snyder, M.P.; Pritchard, J.K.; Kundaje, A.; Greenleaf, J.; et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 2016, 48, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nat. Cell Biol. 2015, 523, 486–490. [Google Scholar] [CrossRef]
- Cusanovich, D.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, B.B.; Cheng, S.; Sos, B.C.; Fan, J.; Yung, Y.; Kaeser, G.E.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.; et al. Integrative single-cell analysis by transcriptional and epigenetic states in human adult brain. bioRxiv 2017, 36, 128520. [Google Scholar] [CrossRef]
- Stoeckius, M.; Hafemeister, C.; Stephenson, W.; Houck-Loomis, B.; Chattopadhyay, P.K.; Swerdlow, H.; Satija, R.; Smibert, P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods 2017, 14, 865–868. [Google Scholar] [CrossRef] [Green Version]
- Peterson, V.M.; Zhang, K.X.; Kumar, N.; Wong, J.; Li, L.; Wilson, D.C.; Moore, R.; McClanahan, T.K.; Sadekova, S.; Klappenbach, J.A. Multiplexed quantification of proteins and transcripts in single cells. Nat. Biotechnol. 2017, 35, 936–939. [Google Scholar] [CrossRef]
- Ramani, V.; Deng, X.; Qiu, R.; Gunderson, K.L.; Steemers, K.L.G.F.J.; Disteche, X.D.C.M.; Noble, W.S.; Duan, Z.; Shendure, J. Massively multiplex single-cell Hi-C. Nat. Methods 2017, 14, 263–266. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Lubling, Y.; Stevens, T.; Schoenfelder, S.; Yaffe, E.; Dean, W.; Laue, E.D.; Tanay, A.; Fraser, P. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nat. Cell Biol. 2013, 502, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef]
- Shema, E.; Bernstein, B.E.; Buenrostro, J.D. Single-cell and single-molecule epigenomics to uncover genome regulation at unprecedented resolution. Nat. Genet. 2019, 51, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Miragaia, R.J.; Natarajan, K.N.; Teichmann, S.A. A rapid and robust method for single cell chromatin accessibility profiling. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lareau, C.A.; Ludwig, L.S.; Muus, C.; Gohil, S.H.; Zhao, T.; Chiang, Z.; Pelka, K.; Verboon, J.M.; Luo, W.; Christian, E.; et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat. Biotechnol. 2021, 39, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.J.; Argelaguet, R.; Kapourani, C.A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Zhang, B.; LaFave, L.M.; Earl, A.S.; Chiang, Z.; Hu, Y.; Ding, J.; Brack, A.; Kartha, V.K.; Tay, T.; et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell 2020, 183, 1103–1116.e20. [Google Scholar] [CrossRef]
- Yan, F.; Powell, D.R.; Curtis, D.J.; Wong, N.C. From reads to insight: A hitchhiker’s guide to ATAC-seq data analysis. Genome Biol. 2020, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M.; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021. [Google Scholar] [CrossRef]
- Schep, A.N.; Wu, B.; Buenrostro, J.D.; Greenleaf, W.J. chromVAR: Inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 2017, 14, 975–978. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Sun, D.; Huang, X.; Wan, C.; Li, Z.; Han, Y.; Qin, Q.; Fan, J.; Qiu, X.; Xie, Y.; et al. Integrative analyses of single-cell transcriptome and regulome using MAESTRO. Genome Biol. 2020, 21, 198. [Google Scholar] [CrossRef]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef] [Green Version]
- Lareau, C.A.; Ma, S.; Duarte, F.M.; Buenrostro, J.D. Inference and effects of barcode multiplets in droplet-based single-cell assays. Nat. Commun. 2020, 11, 866. [Google Scholar] [CrossRef] [Green Version]
- Satpathy, A.T.; Saligrama, N.; Buenrostro, J.D.; Wei, Y.; Wu, B.; Rubin, A.J.; Granja, J.M.; Lareau, C.A.; Li, R.; Qi, Y.; et al. Transcript-indexed ATAC-seq for precision immune profiling. Nat. Med. 2018, 24, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Levitin, H.M.; Miron, M.; Snyder, M.E.; Senda, T.; Yuan, J.; Cheng, Y.L.; Bush, E.C.; Dogra, P.; Thapa, P.; et al. Single-cell transcriptomics of human T cells reveals tissue and activation signatures in health and disease. Nat. Commun. 2019, 10, 4706. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Corces, M.R.; Lareau, C.A.; Wu, B.; Schep, A.N.; Aryee, M.J.; Majeti, R.; Chang, H.Y.; Greenleaf, W.J. Integrated Single-Cell Analysis Maps the Continuous Regulatory Landscape of Human Hematopoietic Differentiation. Cell 2018, 173, 1535–1548.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gate, R.E.; Cheng, C.S.; Aiden, A.P.; Siba, A.; Tabaka, M.; Lituiev, D.; Machol, I.; Gordon, M.G.; Subramaniam, M.; Shamim, M.; et al. Genetic determinants of co-accessible chromatin regions in activated T cells across humans. Nat. Genet. 2018, 50, 1140–1150. [Google Scholar] [CrossRef]
- Stubbington, M.J.; Lönnberg, T.; Proserpio, V.; Clare, S.; Speak, A.O.; Dougan, G.; Teichmann, S.A. T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods 2016, 13, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Mimitou, E.P.; Cheng, A.; Montalbano, A.; Hao, S.; Stoeckius, M.; Legut, M.; Roush, T.; Herrera, A.; Papalexi, E.; Ouyang, Z.; et al. Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nat. Methods 2019, 16, 409–412. [Google Scholar] [CrossRef]
- Papalexi, E.; Mimitou, E.P.; Butler, A.W.; Foster, S.; Bracken, B.; Mauck, W.M.; Wessels, H.H.; Hao, Y.; Yeung, B.Z.; Smibert, P.; et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat. Genet. 2021, 53, 322–331. [Google Scholar] [CrossRef]
- Collora, J.A.; Pinto-Santini, D.; Pasalar, S.; Ravindra, N.; Ganoza, C.; Lama, J.; Alfaro, R.; Chiarella, J.; Spudich, S.; van Dijk, D. Single-cell immune profiling reveals the impact of antiretroviral therapy on HIV-1-induced immune dysfunction, T cell clonal expansion, and HIV-1 persistence in vivo. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chung, H.; Parkhurst, C.; Magee, E.M.; Phillips, D.; Habibi, E.; Chen, F.; Yeung, B.; Waldman, J.A.; Artis, D.; Regev, A. Simultaneous single cell measurements of intranuclear proteins and gene expression. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rosenberg, A.B.; Roco, C.M.; Muscat, R.A.; Kuchina, A.; Sample, P.; Yao, Z.; Graybuck, L.T.; Peeler, D.J.; Mukherjee, S.; Chen, W.; et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Hu, P.; Fabyanic, E.; Kwon, D.Y.; Tang, S.; Zhou, Z.; Wu, H. Dissecting Cell-Type Composition and Activity-Dependent Transcriptional State in Mammalian Brains by Massively Parallel Single-Nucleus RNA-Seq. Mol. Cell 2017, 68, 1006–1015.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, N.; Avraham-Davidi, I.; Basu, A.; Burks, T.; Shekhar, K.; Hofree, M.; Choudhury, S.R.; Aguet, F.; Gelfand, E.; Ardlie, K.; et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 2017, 14, 955–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grindberg, R.V.; Yee-Greenbaum, J.L.; McConnell, M.J.; Novotny, M.; O’Shaughnessy, A.L.; Lambert, G.M.; Araúzo-Bravo, M.J.; Lee, J.; Fishman, M.; Robbins, G.E.; et al. RNA-sequencing from single nuclei. Proc. Natl. Acad. Sci. USA 2013, 110, 19802–19807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Kirita, Y.; Donnelly, E.L.; Humphreys, B.D. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J. Am. Soc. Nephrol. 2018, 30, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; CHI Consortium; Cheung, F.; Shi, R.; Zhou, H.; Lu, W. PBMC fixation and processing for Chromium single-cell RNA sequencing. J. Transl. Med. 2018, 16, 198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Ramskold, D.; Luo, S.; Wang, Y.C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively Parallel Single-Cell RNA-Seq for Marker-Free Decomposition of Tissues into Cell Types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [Green Version]
- Klein, A.M.; Mazutis, L.; Akartuna, I.; Tallapragada, N.; Veres, A.; Li, V.; Peshkin, L.; Weitz, D.A.; Kirschner, M.W. Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 2015, 161, 1187–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacar, B.; Linker, S.B.; Jaeger, B.N.; Krishnaswami, S.R.; Barron, J.J.; Kelder, M.J.E.; Parylak, S.L.; Paquola, A.C.M.; Venepally, P.; Novotny, M.; et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat. Commun. 2016, 7, 11022. [Google Scholar] [CrossRef]
- Cao, J.; Packer, J.S.; Ramani, V.; Cusanovich, D.A.; Huynh, C.; Daza, R.; Qiu, X.; Lee, C.; Furlan, S.N.; Steemers, F.J.; et al. Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357, 661–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, J.R.; Roberts, N.A.; McGowan, S.J.; Hay, D.; Giannoulatou, E.; Lynch, M.; De Gobbi, M.; Taylor, S.S.; Gibbons, R.; Higgs, D.R. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 2014, 46, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Rotem, A.; Ram, O.; Shoresh, N.; Sperling, R.A.; Goren, A.; Weitz, D.A.; Bernstein, B.E. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat. Biotechnol. 2015, 33, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Sos, B.C.; Fung, H.-L.; Gao, D.R.; Osothprarop, T.F.; Kia, A.; He, M.M.; Zhang, K. Characterization of chromatin accessibility with a transposome hypersensitive sites sequencing (THS-seq) assay. Genome Biol. 2016, 17, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mumbach, M.R.; Rubin, A.J.; Flynn, R.A.; Dai, C.; Khavari, P.A.; Greenleaf, W.J.; Chang, H.Y. HiChIP: Efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 2016, 13, 919–922. [Google Scholar] [CrossRef] [Green Version]
- Cusanovich, D.; Hill, A.J.; Aghamirzaie, D.; Daza, R.M.; Pliner, H.A.; Berletch, J.B.; Filippova, G.N.; Huang, X.; Christiansen, L.; DeWitt, W.S.; et al. A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility. Cell 2018, 174, 1309–1324.e18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wan, C.; Wang, P.; Chang, W.; Huo, Y.; Chen, J.; Ma, Q.; Cao, S.; Zhang, C. M3S: A comprehensive model selection for multi-modal single-cell RNA sequencing data. BMC Bioinform. 2019, 20, 672. [Google Scholar] [CrossRef]
- Satija, R.; Farrell, J.A.; Gennert, D.; Schier, A.F.; Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015, 33, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M., 3rd; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef]
- Welch, J.D.; Kozareva, V.; Ferreira, A.; Vanderburg, C.; Martin, C.; Macosko, E.Z. Single-Cell Multi-omic Integration Compares and Contrasts Features of Brain Cell Identity. Cell 2019, 177, 1873–1887.e17. [Google Scholar] [CrossRef]
- Yang, Z.; Michailidis, G. A non-negative matrix factorization method for detecting modules in heterogeneous omics multi-modal data. Bioinform. 2015, 32, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.-R.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Liang, S.; Mohanty, V.; Cheng, X.; Kim, S.; Choi, J.; Li, Y.; Rezvani, K.; Chen, R.; Chen, K. Unbiased integration of single cell multi-omics data. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jin, S.; Zhang, L.; Nie, Q. scAI: An unsupervised approach for the integrative analysis of parallel single-cell transcriptomic and epigenomic profiles. Genome Biol. 2020, 21, 25. [Google Scholar] [CrossRef] [PubMed]
| Study | CD4+ T-Cell Source | HIV-1 Source | Studied Cells | Latency Model | Studied LRA | Single-Cell Approach |
|---|---|---|---|---|---|---|
| Golumbeanu et al. |
|
|
|
|
| SMART-Seq |
| Bradley et al. | HIV donors | CXCR4-using pNL43-Δ6-dreGFP | Cocultured CD4+ T cells | H80 feeder model, 8-week culture |
| 3′ 10× Genomics |
| Cohn et al. | Three treated, suppressed participants with HIV |
| Env+Gag+ cells obtained after 36 hr PHA activation + pancaspase inhibitor and enrichment with 3BNC117/10-1074/PG16 bnAbs | N/A |
| SMART-Seq |
| Liu et al. | Fourteen treated, suppressed participants with HIV |
|
| N/A | PMA/ionomycin, 16 h incubation | SMART-Seq |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, S.; Tsibris, A. Leveraging Novel Integrated Single-Cell Analyses to Define HIV-1 Latency Reversal. Viruses 2021, 13, 1197. https://doi.org/10.3390/v13071197
Zhao S, Tsibris A. Leveraging Novel Integrated Single-Cell Analyses to Define HIV-1 Latency Reversal. Viruses. 2021; 13(7):1197. https://doi.org/10.3390/v13071197
Chicago/Turabian StyleZhao, Suhui, and Athe Tsibris. 2021. "Leveraging Novel Integrated Single-Cell Analyses to Define HIV-1 Latency Reversal" Viruses 13, no. 7: 1197. https://doi.org/10.3390/v13071197
APA StyleZhao, S., & Tsibris, A. (2021). Leveraging Novel Integrated Single-Cell Analyses to Define HIV-1 Latency Reversal. Viruses, 13(7), 1197. https://doi.org/10.3390/v13071197