
Improving Phage-Biofilm In Vitro Experimentation

 ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
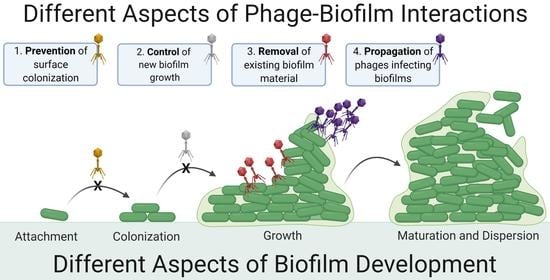
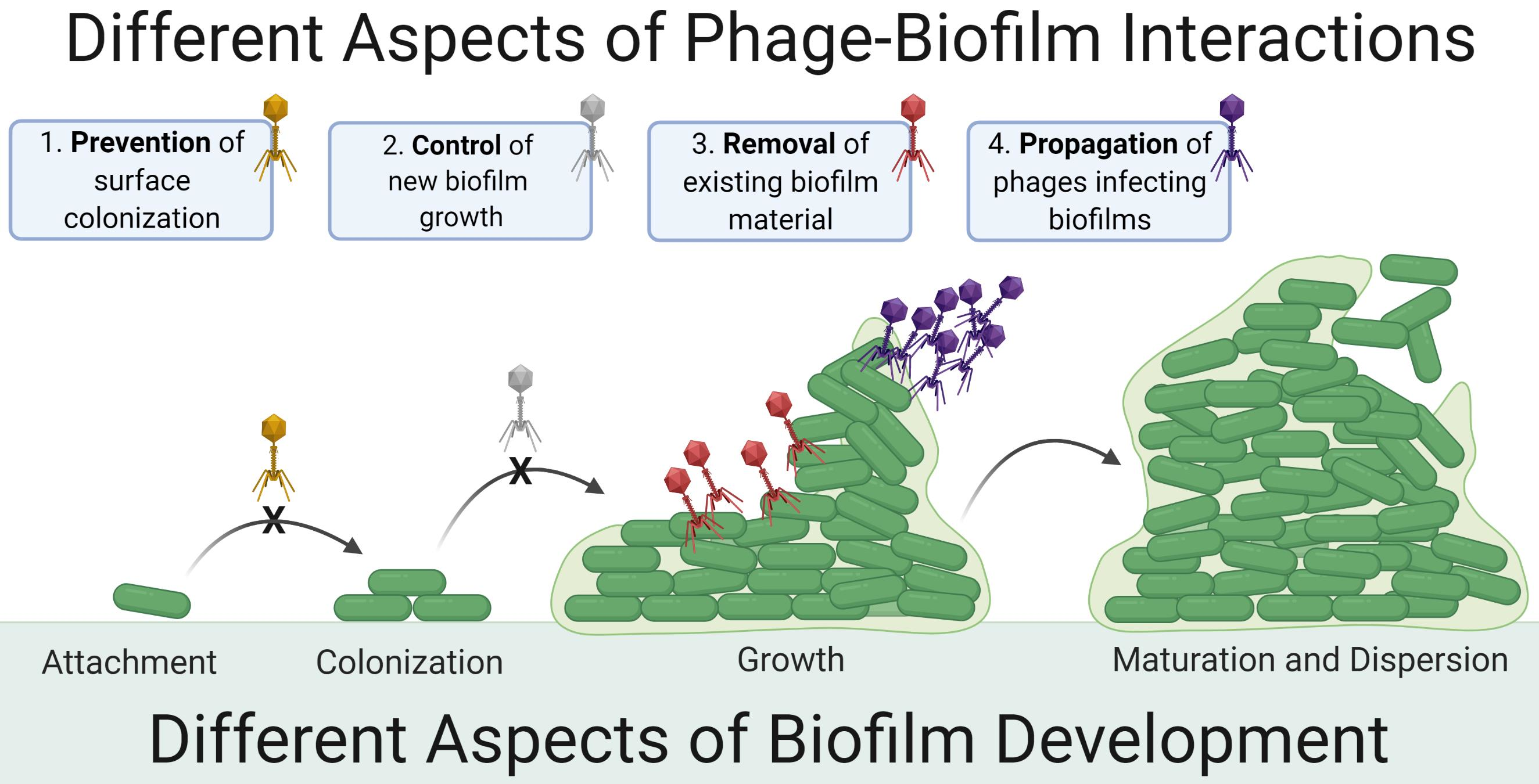
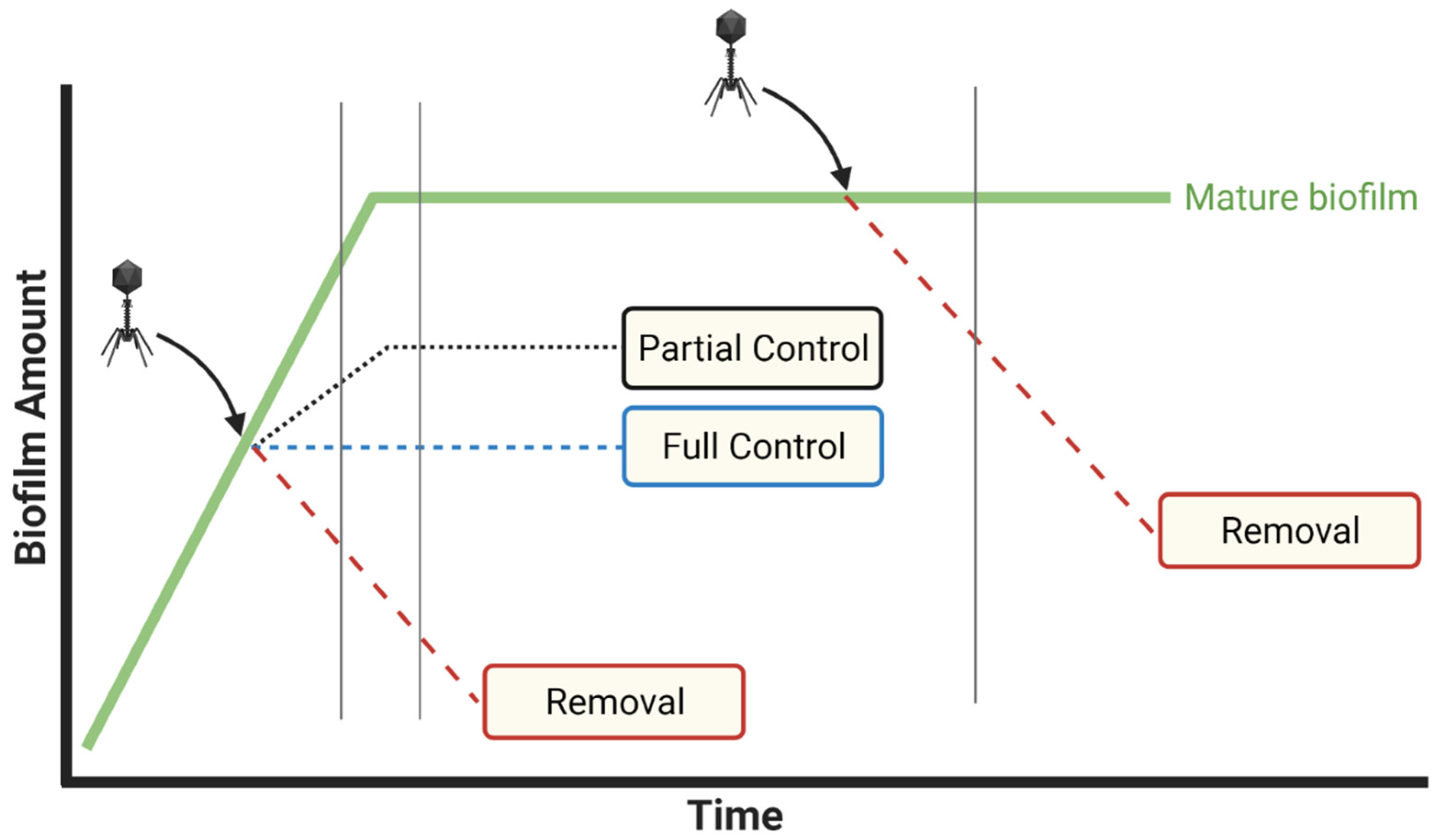
- Distinguishing biofilm control from biofilm removal: importance of zero-time-point determinations
- Measure biofilm properties (CFUs, thickness, etc.) just prior to phage treatment, i.e., at time zero
- Following treatments, compare biofilm properties to both zero-point and mock-treatment controls
- Knowledge of phage titers is needed for the interpretation and reproducibility of experiments
- Explicitly report titers of each phage applied, including, if possible, the expected resulting in situ titers
- MOI-based dosing, if used, should unambiguously report CFU concentrations as measured at time zero
- There are conceptual problems with per-area rather than titer-based dosing
- Report dosing as phage titers, i.e., PFUs/mL of phage-containing volumes applied to surfaces
- Report per-area dosing also as volumes applied, e.g., 100 μL/cm2
- Overlying fluid small-volume effects
- If dosing with lower titers, e.g., <<108 PFUs/mL, then measure planktonic CFUs and PFUs over time
- Or use sufficient titers that substantial in situ phage propagation is not necessary, e.g., ≥108 PFUs/mL
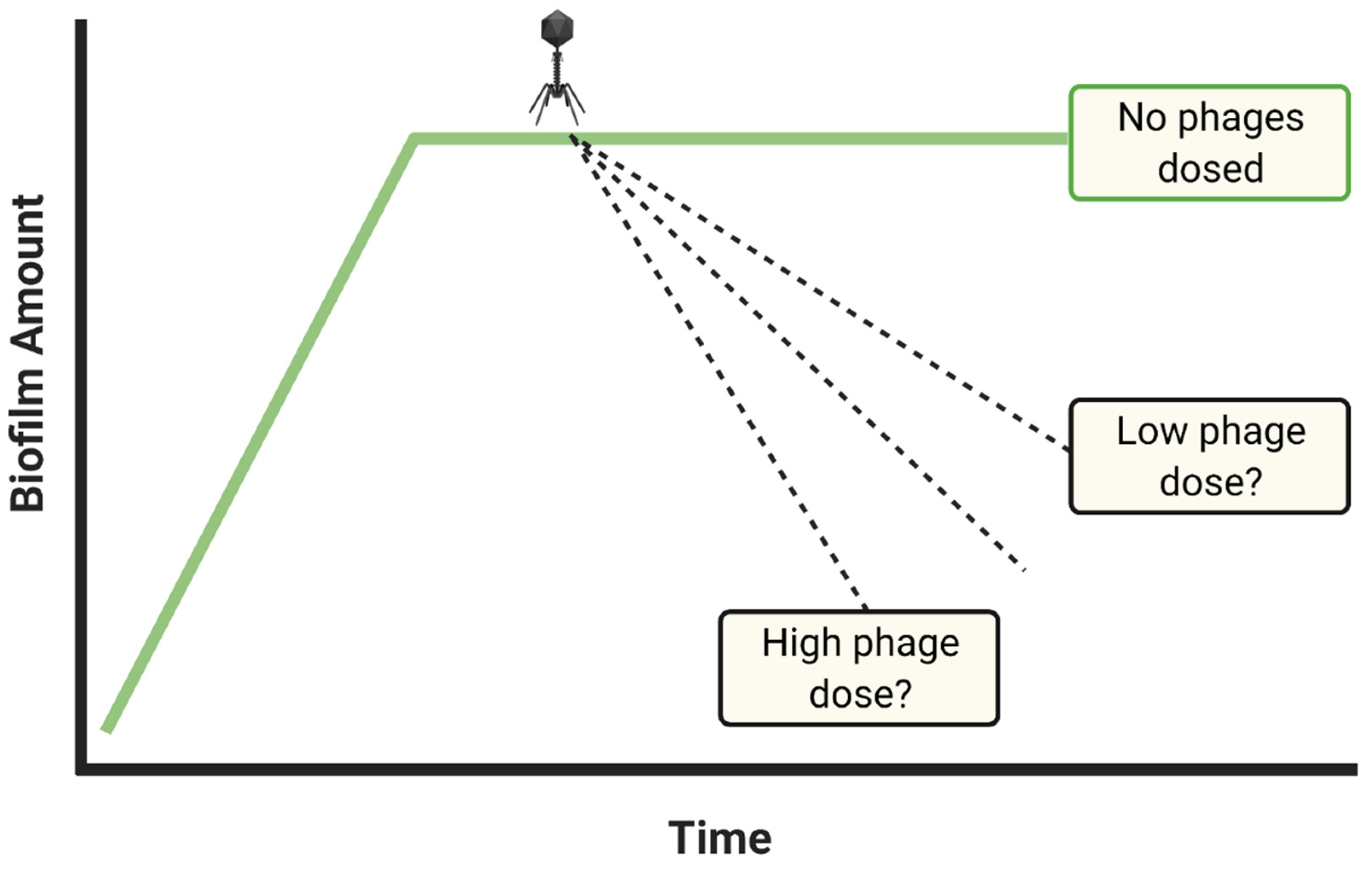
- Dosing with insufficient phage titers?
- If biofilm reductions are insufficient, repeat experiments with higher phage titers and/or multiple dosing
- Consider dosing with maximum achievable titers if biofilm reductions remain inadequate
- Insufficient numbers of time points?
- “Good laboratory practices include… determination of time courses…” [59]
- If possible, repeat experiments using alternative treatment durations, e.g., both 12 and 24 h
- Enzymatic biofilm matrix disruption
- Biofilm matrix degradation by EPS depolymerases can impact biofilms even without active phage infection
- Measure potential phage EPS depolymerase activity against all experimentally targeted bacterial strains
- Limitations of biofilm biomass determinations
- Quantifying biofilm presence using solely biomass determinations can be both inaccurate and imprecise
- If possible, characterize biofilm presence using additional methods, such as CFU counts
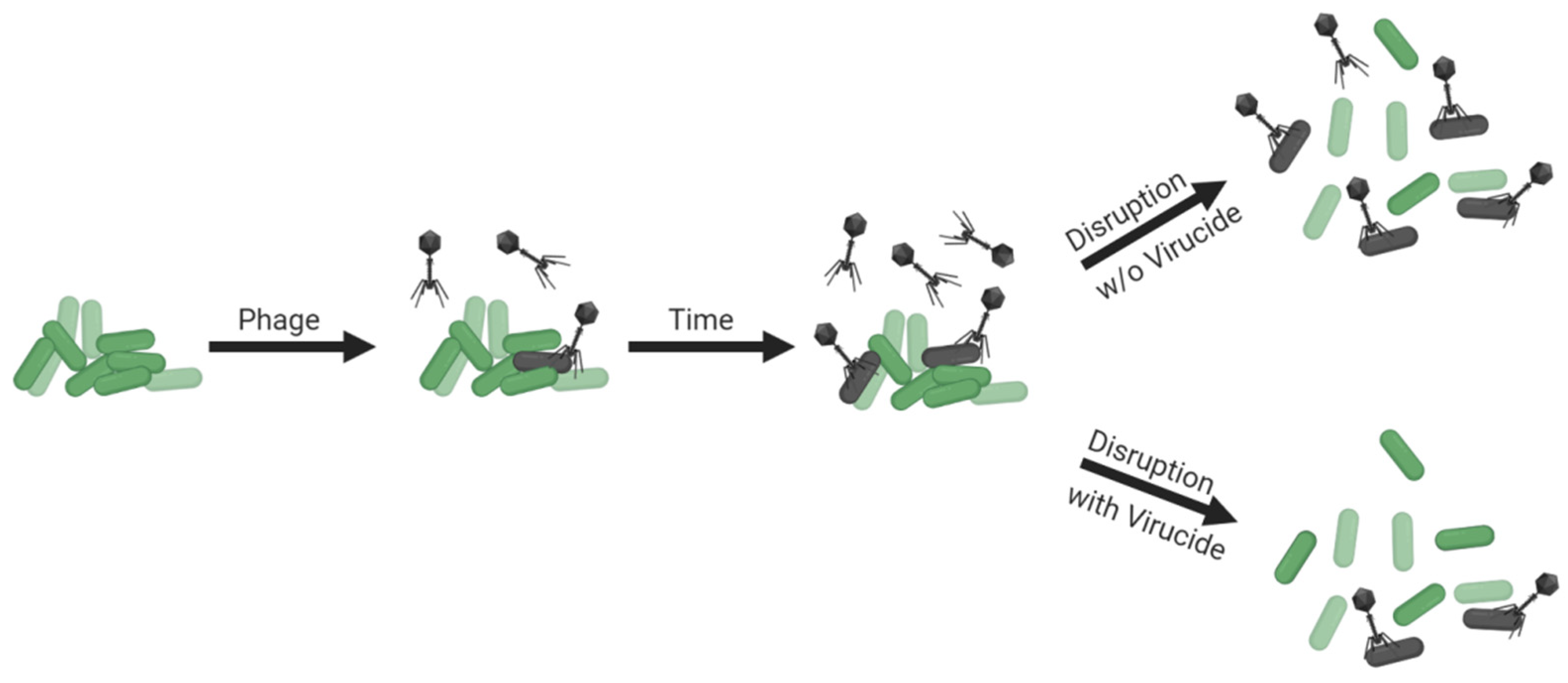
- Colony count complications (exposure to free phages during CFU enumeration)
- Disrupt biofilms within the largest volumes that are easily achieved and worked with
- Disrupt within a bacterium-tolerant virucide especially if sufficient disruption volumes are not achievable
- Avoid changing conditions mid-experiment, unless that is the intention of an experiment
- Report what medium is used during phage treatments, even if it is the same used for biofilm growth
- Discuss why, and possible consequences, of any changes to media or conditions made during experiments
- Characterization under multiple conditions toward improving the robustness of conclusions
- Seek out alternative conditions for biofilm growth toward better representing in vivo conditions
- Discuss limitations of conditions tested and possible alternatives that might also be tested
- Keeping in vitro biofilms real
- Describe conditions for biofilm growth and treatment that are thought to be present in situ or in vivo
- Discuss how in vitro conditions used may differ from those thought to be found in situ or in vivo
2. Contrasting Biofilm Prevention, Control, and Removal
3. Improving Phage-Biofilm In Vitro Experimentation
3.1. Distinguishing Control from Removal: Importance of Zero-Time-Point Determinations
3.2. Knowledge of Phage Titers Is Needed for Interpretation and Reproducibility of Experiments
3.3. The Conceptual Problem of per-Area Rather Than Titer-Based Dosing
3.4. Overlying Fluid Small-Volume Effects
3.5. Dosing with Insufficient Phage Titers?
3.6. Insufficient Numbers of Time Points?
3.7. Enzymatic Biofilm Matrix Disruption
3.8. Limitations of Biofilm Biomass Determinations
3.9. Colony Count Complications
3.10. Avoid Changing Conditions Mid-Experiment, Unless That Is the Intention of an Experiment
3.11. Characterization under Multiple Conditions toward Improving Robustness of Conclusions
3.12. Keeping In Vitro Biofilms Real
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flemming, H.C.; Neu, T.R.; Wozniak, D.J. The EPS matrix: The “house of biofilm cells”. J. Bacteriol. 2007, 189, 7945–7947. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Wei, Q.; Ma, L.Z. Biofilm matrix and its regulation in Pseudomonas aeruginosa. Int. J. Mol. Sci. 2013, 14, 20983–21005. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.R.; McGillicuddy, E.; Kaplan, L.J. Biofilm: Basic principles, pathophysiology, and implications for clinicians. Surg. Infect. 2014, 15, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Donlan, R.M.; Costerton, J.W. Biofilms: Survival mechanisms of clinically relevant microorganisms. Clin. Microbiol. Rev. 2002, 15, 167–193. [Google Scholar] [CrossRef] [PubMed]
- Flemming, H.C.; Wuertz, S. Bacteria and archaea on Earth and their abundance in biofilms. Nat. Rev. Microbiol. 2019, 17, 247–260. [Google Scholar] [CrossRef]
- Yan, J.; Bassler, B.L. Surviving as a community: Antibiotic tolerance and persistence in bacterial biofilms. Cell Host Microbe 2019, 26, 15–21. [Google Scholar] [CrossRef]
- Ferriol-Gonzalez, C.; Domingo-Calap, P. Phages for biofilm removal. Antibiotics 2020, 9, 268. [Google Scholar] [CrossRef]
- Parsek, M.R.; Singh, P.K. Bacterial biofilms: An emerging link to disease pathogenesis. Ann. Rev. Microbiol. 2003, 57, 677–701. [Google Scholar] [CrossRef]
- Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS 2013, 121, 1–51. [Google Scholar] [CrossRef]
- Scali, C.; Kunimoto, B. An update on chronic wounds and the role of biofilms. J. Cutan. Med. Surg. 2013, 17, 371–376. [Google Scholar] [CrossRef]
- Cooper, R.A.; Bjarnsholt, T.; Alhede, M. Biofilms in wounds: A review of present knowledge. J. Wound. Care 2014, 23, 570–580. [Google Scholar] [CrossRef]
- Macia, M.D.; Rojo-Molinero, E.; Oliver, A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 2014, 20, 981–990. [Google Scholar] [CrossRef]
- Percival, S.L.; McCarty, S.M.; Lipsky, B. Biofilms and wounds: An overview of the evidence. Adv. Wound. Care 2015, 4, 373–381. [Google Scholar] [CrossRef]
- Vestby, L.K.; Gronseth, T.; Simm, R.; Nesse, L.L. Bacterial biofilm and its role in the pathogenesis of disease. Antibiotics 2020, 9, 59. [Google Scholar] [CrossRef]
- De la Fuente-Núnez, C.; Reffuveille, F.; Fernández, L.; Hancock, R.E.W. Bacterial biofilm development as a multicellular adaptation: Antibiotic resistance and new therapeutic strategies. Curr. Opin. Microbiol. 2013, 16, 580–589. [Google Scholar] [CrossRef]
- Balcazar, J.L.; Subirats, J.; Borrego, C.M. The role of biofilms as environmental reservoirs of antibiotic resistance. Front. Microbiol. 2015, 6, 1216. [Google Scholar] [CrossRef]
- Penesyan, A.; Gillings, M.; Paulsen, I.T. Antibiotic discovery: Combatting bacterial resistance in cells and in biofilm communities. Molecules 2015, 20, 5286–5298. [Google Scholar] [CrossRef] [PubMed]
- France, M.T.; Cornea, A.; Kehlet-Delgado, H.; Forney, L.J. Spatial structure facilitates the accumulation and persistence of antibiotic-resistant mutants in biofilms. Evol. Appl. 2019, 12, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Jorge, P.; Magalhaes, A.P.; Grainha, T.; Alves, D.; Sousa, A.M.; Lopes, S.P.; Pereira, M.O. Antimicrobial resistance three ways: Healthcare crisis, major concepts and the relevance of biofilms. FEMS Microbiol. Ecol. 2019, 95, 95. [Google Scholar] [CrossRef] [PubMed]
- Ceri, H.; Olson, M.E.; Stremick, C.; Read, R.R.; Morck, D.; Buret, A. The Calgary Biofilm Device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 1999, 37, 1771–1776. [Google Scholar] [CrossRef] [PubMed]
- Hoiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef]
- Ciofu, O.; Rojo-Molinero, E.; Macia, M.D.; Oliver, A. Antibiotic treatment of biofilm infections. APMIS 2017, 125, 304–319. [Google Scholar] [CrossRef]
- Hall, C.W.; Mah, T.F. Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol. Rev. 2017, 41, 276–301. [Google Scholar] [CrossRef]
- Kumaran, D.; Taha, M.; Yi, Q.; Ramirez-Arcos, S.; Diallo, J.S.; Carli, A.; Abdelbary, H. Does treatment order matter? Investigating the ability of bacteriophage to augment antibiotic activity against Staphylococcus aureus biofilms. Front. Microbiol. 2018, 9, 127. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophage clinical use as antibactertial “drugs”: Utility, precedent. Microbiol. Spectr. 2017, 5, BAD-0003-2016. [Google Scholar] [CrossRef]
- Abedon, S.T. Kinetics of phage-mediated biocontrol of bacteria. Foodborne Pathog. Dis. 2009, 6, 807–815. [Google Scholar] [CrossRef]
- Harper, D.R. Biological control by microorganisms. In eLS; John Wiley & Sons: Chichester, UK, 2013. [Google Scholar] [CrossRef]
- Alves, D.R.; Clark, J.; Abedon, S.T. Viruses as biocontrol agents of microorganisms. In Viruses of Microorganisms; Hyman, P., Abedon, S.T., Eds.; Caister Academic Press: Norwich, UK, 2018; pp. 313–330. [Google Scholar]
- Abedon, S.T. Use of phage therapy to treat long-standing, persistent, or chronic bacterial infections. Adv. Drug Deliv. Rev. 2019, 145, 18–39. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Singh, H.S.; Shukla, V.K.; Nath, G.; Bhartiya, S.K. Bacteriophage therapy of chronic nonhealing wound: Clinical study. Int. J. Low Extrem. Wounds 2019, 18, 171–175. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Wu, S.; Beck, J.P. Phage therapy of wounds and related purulent infections. In Bacteriophages in Health and Disease; Hyman, P., Abedon, S.T., Eds.; CABI Press: Wallingford, UK, 2012; pp. 185–202. [Google Scholar]
- Abedon, S.T. Bacteriophage-mediated biocontrol of wound infections, and ecological exploitation of biofilms by phages. In Biofilm, Pilonidal Cysts and Sinuses. Recent Clinical Techniques, Results, and Research in Wounds; Shiffman, M., Low, M., Eds.; Springer Nature: London, UK, 2020; Volume 1, pp. 121–158. [Google Scholar]
- Chang, R.Y.K.; Morales, S.; Okamoto, Y.; Chan, H.K. Topical application of bacteriophages for treatment of wound infections. Transl. Res. 2020, 220, 166. [Google Scholar] [CrossRef]
- Moghadam, M.T.; Khoshbayan, A.; Chegini, Z.; Farahani, I.; Shariati, A. Bacteriophages, a new therapeutic solution for inhibiting multidrug-resistant bacteria causing wound infection: Lesson from animal models and clinical trials. Drug Des. Devel. Ther. 2020, 14, 1867–1883. [Google Scholar] [CrossRef]
- Pinto, A.M.; Cerqueira, M.A.; Banobre-Lopes, M.; Pastrana, L.M.; Sillankorva, S. Bacteriophages for chronic wound treatment: From traditional to novel delivery systems. Viruses 2020, 12, 235. [Google Scholar] [CrossRef] [PubMed]
- Semler, D.D.; Lynch, K.H.; Dennis, J.J. The promise of bacteriophage therapy for Burkholderia cepacia complex respiratory infections. Front. Cell. Infect. Microbiol. 2011, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Hoe, S.; Semler, D.D.; Goudie, A.D.; Lynch, K.H.; Matinkhoo, S.; Finlay, W.H.; Dennis, J.J.; Vehring, R. Respirable bacteriophages for the treatment of bacterial lung infections. J. Aerosol Med. Pulm. Drug Deliv. 2013, 26, 317–335. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy of pulmonary infections. Bacteriophage 2015, 5, e1020260. [Google Scholar] [CrossRef]
- Waters, E.M.; Neill, D.R.; Kaman, B.; Sahota, J.S.; Clokie, M.R.; Winstanley, C.; Kadioglu, A. Phage therapy is highly effective against chronic lung infections with Pseudomonas aeruginosa. Thorax 2017, 72, 666–667. [Google Scholar] [CrossRef]
- Trend, S.; Fonceca, A.M.; Ditcham, W.G.; Kicic, A.; AREST CF. The potential of phage therapy in cystic fibrosis: Essential human-bacterial-phage interactions and delivery considerations for use in Pseudomonas aeruginosa-infected airways. J. Cyst. Fibros. 2017, 16, 663–670. [Google Scholar] [CrossRef]
- Chang, R.Y.K.; Wallin, M.; Lin, Y.; Leung, S.S.Y.; Wang, H.; Morales, S.; Chan, H.K. Phage therapy for respiratory infections. Adv. Drug Deliv. Rev. 2018, 133, 76–86. [Google Scholar] [CrossRef]
- Aslam, S.; Courtwright, A.M.; Koval, C.; Lehman, S.M.; Morales, S.; Furr, C.-L.L.; Rosas, F.; Brownstein, M.J.; Fackler, J.R.; Sisson, B.M.; et al. Early clinical experience of bacteriophage therapy in three lung transplant recipients. Am. J. Transplant. 2019, 19, 2631–2639. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophages and Biofilms: Ecology, Phage Therapy, Plaques; Nova Science Publishers: Hauppauge, NY, USA, 2011. [Google Scholar]
- Brüssow, H. Bacteriophage-host interaction: From splendid isolation into a messy reality. Curr. Opin. Microbiol. 2013, 16, 500–506. [Google Scholar] [CrossRef]
- Fan, X.; Li, W.; Zheng, F.; Xie, J. Bacteriophage inspired antibiotics discovery against infection involved biofilm. Crit Rev. Eukaryot. Gene Expr. 2013, 23, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.R.; Parracho, H.M.R.; Walker, J.; Sharp, R.; Hughes, G.; Werthrén, M.; Lehman, S.; Morales, S. Bacteriophages and biofilms. Antibiotics 2014, 3, 270–284. [Google Scholar] [CrossRef]
- Parasion, S.; Kwiatek, M.; Gryko, R.; Mizak, L.; Malm, A. Bacteriophages as an alternative strategy for fighting biofilm development. Pol. J. Microbiol. 2014, 63, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Sillankorva, S.; Azeredo, J. Bacteriophage attack as an anti-biofilm strategy. Meth. Mol. Biol. 2014, 1147, 277–285. [Google Scholar]
- Sillankorva, S.; Azeredo, J. The use of bacteriophages and bacteriophage-derived enzymes for clinically relevant biofilm control. In Phage Therapy: Current Research and Applications; Borysowski, J., Międzybrodzki, R., Górski, A., Eds.; Caister Academic Press: Norfolk, UK, 2014. [Google Scholar]
- Abedon, S.T. Ecology of anti-biofilm agents I. Antibiotics versus bacteriophages. Pharmaceuticals 2015, 8, 525–558. [Google Scholar] [CrossRef]
- Abedon, S.T. Ecology of anti-biofilm agents II. Bacteriophage exploitation and biocontrol of biofilm bacteria. Pharmaceuticals 2015, 8, 559–589. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T. Bacteriophages and their enzymes in biofilm control. Curr. Pharm. Des. 2015, 21, 85–99. [Google Scholar] [CrossRef]
- Hansen, M.F.; Svenningsen, S.L.; Roder, H.L.; Middelboe, M.; Burmolle, M. Big impact of the tiny: Bacteriophage-bacteria interactions in biofilms. Trends Microbiol. 2019, 27, 739–752. [Google Scholar] [CrossRef]
- Kifelew, G.L.; Mitchell, J.G.; Speck, P. Mini-review: Efficacy of lytic bacteriophages on multispecies biofilms. Biofouling 2019, 35, 472–481. [Google Scholar] [CrossRef]
- Chegini, Z.; Khoshbayan, A.; Taati, M.M.; Farahani, I.; Jazireian, P.; Shariati, A. Bacteriophage therapy against Pseudomonas aeruginosa biofilms: A review. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 45. [Google Scholar] [CrossRef]
- Doub, J.B. Bacteriophage therapy for clinical biofilm infections: Parameters that influence treatment protocols and current treatment approaches. Antibiotics 2020, 9, 799. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Górski, A. Bacteriophages and lysins in biofilm control. Virol. Sin. 2020, 35, 125–133. [Google Scholar] [CrossRef]
- Casadevall, A.; Fang, F.C. Rigorous science: A how-to guide. MBio 2016, 7, e01902-16. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Harjai, K.; Chhibber, S. Restricting ciprofloxacin-induced resistant variant formation in biofilm of Klebsiella pneumoniae B5055 by complementary bacteriophage treatment. J. Antimicrob. Chemother. 2009, 64, 1212–1218. [Google Scholar] [CrossRef]
- Dickey, J.; Perrot, V. Adjunct phage treatment enhances the effectiveness of low antibiotic concentration against Staphylococcus aureus biofilms in vitro. PLoS ONE 2019, 14, e0209390. [Google Scholar] [CrossRef] [PubMed]
- Holguin, A.V.; Rangel, G.; Clavijo, V.; Prada, C.; Mantilla, M.; Gomez, M.C.; Kutter, E.; Taylor, C.; Fineran, P.C.; Barrios, A.F.; et al. Phage ΦPan70, a putative temperate phage, controls Pseudomonas aeruginosa in planktonic, biofilm and burn mouse model assays. Viruses 2015, 7, 4602–4623. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Colwell, R.R. Virioplankton: Viruses in aquatic ecosystems. Microbiol. Mol. Biol. Rev. 2000, 64, 69–114. [Google Scholar] [CrossRef]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, S.; Zhong, X.; Peduzzi, P.; Thingstad, T.F.; Parikka, K.J.; Weinbauer, M.G. Methods and technologies to assess viral interactions in the aquatic world. In Viruses of Microorganisms; Hyman, P., Abedon, S.T., Eds.; Caister Academic Press: Norwich, UK, 2018; pp. 331–349. [Google Scholar]
- Trubl, G.; Hyman, P.; Roux, S.; Abedon, S.T. Coming-of-age characterization of soil viruses: A user’s guide to virus isolation, detection within metagenomes, and viromics. Soil Sys. 2020, 4, 23. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy dosing: The problem(s) with multiplicity of infection (MOI). Bacteriophage 2016, 6, e1220348. [Google Scholar] [CrossRef]
- Stent, G.S. Molecular Biology of Bacterial Viruses; WH Freeman and, Co.: San Francisco, CA, USA, 1963. [Google Scholar]
- Abedon, S.T. Phage therapy: Killing titers, multiplicity of infection, adsorption theory, and passive versus active treatments. In Advances on the Applications of Bacteriophages; Kurtboke, D.I., Aminov, R., Eds.; Nova Science Publishers: Hauppauge, NY, USA, 2021. [Google Scholar]
- Dennehy, J.J.; Abedon, S.T. Adsorption: Phage acquisition of bacteria. In Bacteriophages: Biology, Technology, Therapy; Harper, D., Abedon, S.T., Burrowes, B.H., McConville, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2020; pp. 93–117. [Google Scholar]
- Abedon, S.T. Active bacteriophage biocontrol and therapy on sub-millimeter scales towards removal of unwanted bacteria from foods and microbiomes. AIMS Microbiol. 2017, 3, 649–688. [Google Scholar] [CrossRef]
- Weber-Dąbrowska, B.; Mulczyk, M.; Górski, A. Bacteriophage therapy of bacterial infections: An update of our institute’s experience. Arch. Immunol. Ther. Exp. 2000, 48, 547–551. [Google Scholar]
- Międzybrodzki, R.; Borysowski, J.; Weber-Dąbrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawelczyk, Z.; Rogoz, P.; Klak, M.; Wojtasik, E.; et al. Clinical aspects of phage therapy. Adv. Virus Res. 2012, 83, 73–121. [Google Scholar] [PubMed]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Bacteriophage treatment of intransigent diabetic toe ulcers: A case series. J. Wound Care 2016, 25 (Suppl. 7), S27–S33. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Dabrowska, K.; Abedon, S.T. Phage therapy: The pharmacology of antibacterial viruses. In Exploitation of Bacteriophages for Biocontrol and Therapeutics; Coffey, A., Ed.; Caister Academic Press: Norwich, UK, 2020. [Google Scholar]
- Cano, E.J.; Caflisch, K.M.; Bollyky, P.L.; Van Belleghem, J.D.; Patel, R.; Fackler, J.; Brownstein, M.J.; Horne, B.; Biswas, B.; Henry, M.; et al. Phage therapy for limb-threatening prosthetic knee Klebsiella pneumoniae infection: Case report and in vitro characterization of anti-biofilm activity. Clin. Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Lebeaux, D.; Merabishvili, M.; Caudron, E.; Lannoy, D.; Van, S.L.; Duyvejonck, H.; Guillemain, R.; Thumerelle, C.; Podglajen, I.; Compain, F.; et al. A case of phage therapy against pandrug-resistant Achromobacter xylosoxidans in a 12-year-old lung-transplanted cystic fibrosis patient. Viruses 2021, 13, 60. [Google Scholar] [CrossRef]
- Leitner, L.; Ujmajuridze, A.; Chanishvili, N.; Goderdzishvili, M.; Chkonia, I.; Rigvava, S.; Chkhotua, A.; Changashvili, G.; McCallin, S.; Schneider, M.P.; et al. Intravesical bacteriophages for treating urinary tract infections in patients undergoing transurethral resection of the prostate: A randomised, placebo-controlled, double-blind clinical trial. Lancet Infect. Dis. 2021, 21, 427–436. [Google Scholar] [CrossRef]
- Tan, X.; Chen, H.; Zhang, M.; Zhao, Y.; Jiang, Y.; Liu, X.; Huang, W.; Ma, Y. Clinical experience of personalized phage therapy against carbapenem-resistant Acinetobacter baumannii lung infection in a patient with chronic obstructive pulmonary disease. Front. Cell. Infect. Microbiol. 2021, 11, 631585. [Google Scholar] [CrossRef]
- Abedon, S.T.; Duffy, S.; Turner, P.E. Bacteriophage ecology. In Encyclopedia of Microbiology; Schaecter, M., Ed.; Elsevier: Oxford, UK, 2009; pp. 42–57. [Google Scholar]
- Abedon, S.T. Information phage therapy research should report. Pharmaceuticals 2017, 10, 43. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; Wozniak, D.J. Psl produced by mucoid Pseudomonas aeruginosa contributes to the establishment of biofilms and immune evasion. MBio 2017, 8, e00864-17. [Google Scholar] [CrossRef] [PubMed]
- Thomen, P.; Robert, J.; Monmeyran, A.; Bitbol, A.F.; Douarche, C.; Henry, N. Bacterial biofilm under flow: First a physical struggle to stay, then a matter of breathing. PLoS ONE 2017, 12, e0175197. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, M.M.; Cooney, J.J.; Caldwell, D.E. Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J. Indust. Microbiol. 1996, 16, 331–341. [Google Scholar] [CrossRef]
- Nale, J.Y.; Chutia, M.; Carr, P.; Hickenbotham, P.T.; Clokie, M.R. ‘Get in early’; biofilm and wax moth (Galleria mellonella) models reveal new insights into the therapeutic potential of Clostridium difficile bacteriophages. Front. Microbiol. 2016, 7, 1383. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.R. Viral interference. Some considerations of basic mechanisms and their potential relationship to host resistance. Bacteriol. Rev. 1960, 24, 151–166. [Google Scholar] [CrossRef]
- Callaway, T.R.; Edrington, T.S.; Brabban, A.D.; Anderson, R.C.; Rossman, M.L.; Engler, M.J.; Carr, M.A.; Genovese, K.J.; Keen, J.E.; Looper, M.L.; et al. Bacteriophage isolated from feedlot cattle can reduce Escherichia coli O157:H7 populations in ruminant gastrointestinal tracts. Foodborne Pathog. Dis. 2008, 5, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Abedon, S.T. Phage therapy pharmacology: Phage cocktails. Adv. Appl. Microbiol. 2012, 78, 1–23. [Google Scholar]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a rationale for phage therapy using phage cocktails. Peer J. 2014, 2, e590. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophage secondary infection. Virol. Sin. 2015, 30, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Chen, P.; Lin, Z.; Wang, T. Characterization of two Pseudomonas aeruginosa viruses vB_PaeM_SCUT-S1 and vB_PaeM_SCUT-S2. Viruses 2019, 11, 318. [Google Scholar] [CrossRef]
- Yuan, Y.; Qu, K.; Tan, D.; Li, X.; Wang, L.; Cong, C.; Xiu, Z.; Xu, Y. Isolation and characterization of a bacteriophage and its potential to disrupt multi-drug resistant Pseudomonas aeruginosa biofilms. Microb. Pathog. 2019, 128, 329–336. [Google Scholar] [CrossRef]
- Maszewska, A. Phage associated polysaccharide depolymerases—Characteristics and application. Postepy Hig. Med. Dosw. 2015, 69, 690–702. [Google Scholar] [CrossRef]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and function of phage encoded depolymerases. Front. Microbiol. 2019, 10, 2949. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Oliveira, H.; Melo, L.D.; Sillankorva, S.; Azeredo, J. Bacteriophage-encoded depolymerases: Their diversity and biotechnological applications. Appl. Microbiol. Biotechnol. 2016, 100, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Sutherland, I.W.; Jones, M.V. Biofilm susceptibility to bacteriophage attack: The role of phage-borne polysaccharide depolymerase. Microbiology 1998, 144, 3039–3047. [Google Scholar] [CrossRef] [PubMed]
- Tait, K.; Skilman, L.C.; Sutherland, I.W. The efficacy of bacteriophage as a method of biofilm eradication. Biofouling 2002, 18, 305–311. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage-antibiotic combination treatments: Antagonistic impacts of antibiotics on the pharmacodynamics of phage therapy? Antibiotics 2019, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Sutherland, I.W.; Clark, J.; Jones, M.V. Bacteriophage and associated polysaccharide depolymerases-novel tools for study of bacterial biofilms. J. Appl. Microbiol. 1998, 85, 583–590. [Google Scholar] [CrossRef]
- Glonti, T.; Chanishvili, N.; Taylor, P.W. Bacteriophage-derived enzyme that depolymerizes the alginic acid capsule associated with cystic fibrosis isolates of Pseudomonas aeruginosa. J. Appl. Microbiol. 2010, 108, 695–702. [Google Scholar] [CrossRef]
- Cornelissen, A.; Ceyssens, P.J.; T’Syen, J.; Van, P.H.; Noben, J.P.; Shaburova, O.V.; Krylov, V.N.; Volckaert, G.; Lavigne, R. The T7-related Pseudomonas putida phage φ15 displays virion-associated biofilm degradation properties. PLoS ONE 2011, 6, e18597. [Google Scholar] [CrossRef]
- Guo, Z.; Huang, J.; Yan, G.; Lei, L.; Wang, S.; Yu, L.; Zhou, L.; Gao, A.; Feng, X.; Han, W.; et al. Identification and characterization of Dpo42, a novel depolymerase derived from the Escherichia coli phage vB_EcoM_ECOO78. Front. Microbiol. 2017, 8, 1460. [Google Scholar] [CrossRef]
- Olszak, T.; Shneider, M.M.; Łątka, A.; Maciejewska, B.; Browning, C.; Sycheva, L.V.; Cornelissen, A.; Danis-Wlodarczyk, K.; Senchenkova, S.N.; Shashkov, A.S.; et al. The O-specific polysaccharide lyase from the phage LKA1 tailspike reduces Pseudomonas virulence. Sci. Rep. 2017, 7, 16302. [Google Scholar] [CrossRef]
- Lin, H.; Paff, M.L.; Molineux, I.J.; Bull, J.J. Antibiotic therapy using phage depolymerases: Robustness across a range of conditions. Viruses. 2018, 10, 622. [Google Scholar] [CrossRef] [PubMed]
- Mi, L.; Liu, Y.; Wang, C.; He, T.; Gao, S.; Xing, S.; Huang, Y.; Fan, H.; Zhang, X.; Yu, W.; et al. Identification of a lytic Pseudomonas aeruginosa phage depolymerase and its anti-biofilm effect and bactericidal contribution to serum. Virus Genes 2019, 55, 394–405. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, R.; Xu, M.; Liu, Y.; Zhu, X.; Qiu, J.; Liu, Q.; He, P.; Li, Q. A novel polysaccharide depolymerase encoded by the phage SH-KP152226 confers specific activity against multidrug-resistant Klebsiella pneumoniae via biofilm degradation. Front. Microbiol. 2019, 10, 2768. [Google Scholar] [CrossRef] [PubMed]
- Łątka, A.; Drulis-Kawa, Z. Advantages and limitations of microtiter biofilm assays in the model of antibiofilm activity of Klebsiella phage KP34 and its depolymerase. Sci. Rep. 2020, 10, 20338. [Google Scholar] [CrossRef] [PubMed]
- Danis-Wlodarczyk, K.; Vandenheuvel, D.; Jang, H.B.; Briers, Y.; Olszak, T.; Arabski, M.; Wasik, S.; Drabik, M.; Higgins, G.; Tyrrell, J.; et al. A proposed integrated approach for the preclinical evaluation of phage therapy in Pseudomonas infections. Sci. Rep. 2016, 6, 28115. [Google Scholar] [CrossRef]
- Darch, S.E.; Kragh, K.N.; Abbott, E.A.; Bjarnsholt, T.; Bull, J.J.; Whiteley, M. Phage inhibit pathogen dissemination by targeting bacterial migrants in a chronic infection model. MBio 2017, 8, e00240-17. [Google Scholar] [CrossRef]
- Peeters, E.; Nelis, H.J.; Coenye, T. Comparison of multiple methods for quantification of microbial biofilms grown in microtiter plates. J. Microbiol. Methods 2008, 72, 157–165. [Google Scholar] [CrossRef]
- Knezevic, P.; Petrovic, O. A colorimetric microtiter plate method for assessment of phage effect on Pseudomonas aeruginosa biofilm. J. Microbiol. Meth. 2008, 74, 114–118. [Google Scholar] [CrossRef]
- Tote, K.; Berghe, D.V.; Maes, L.; Cos, P. A new colorimetric microtitre model for the detection of Staphylococcus aureus biofilms. Lett. Appl. Microbiol. 2008, 46, 249–254. [Google Scholar] [CrossRef]
- Skogman, M.E.; Vuorela, P.M.; Fallarero, A. Combining biofilm matrix measurements with biomass and viability assays in susceptibility assessments of antimicrobials against Staphylococcus aureus biofilms. J. Antibiot. 2012, 65, 453–459. [Google Scholar] [CrossRef]
- Schuch, R.; Khan, B.K.; Raz, A.; Rotolo, J.A.; Wittekind, M. Bacteriophage lysin CF-301, a potent antistaphylococcal biofilm agent. Antimicrob. Agents Chemother. 2017, 61, e02666-16. [Google Scholar] [CrossRef] [PubMed]
- Chhibber, S.; Nag, D.; Bansal, S. Inhibiting biofilm formation by Klebsiella pneumoniae B5055 using an iron antagonizing molecule and a bacteriophage. BMC Microbiol. 2013, 13, 174. [Google Scholar] [CrossRef] [PubMed]
- Dakheel, K.H.; Rahim, R.A.; Neela, V.K.; Al-Obaidi, J.R.; Hun, T.G.; Isa, M.N.M.; Yusoff, K. Genomic analyses of two novel biofilm-degrading methicillin-resistant Staphylococcus aureus phages. BMC Microbiol. 2019, 19, 114. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.Y.K.; Das, T.; Manos, J.; Kutter, E.; Morales, S.; Chan, H.K. Bacteriophage PEV20 and ciprofloxacin combination treatment enhances removal of Pseudomonas aeruginosa biofilm isolated from cystic fibrosis and wound patients. AAPS J. 2019, 21, 49. [Google Scholar] [CrossRef] [PubMed]
- Townsend, E.M.; Moat, J.; Jameson, E. CAUTI’s next top model—Model dependent Klebsiella biofilm inhibition by bacteriophages and antimicrobials. Biofilm 2020, 2, 100038. [Google Scholar] [CrossRef]
- Brown, H.L.; van Vliet, A.H.; Betts, R.P.; Reuter, M. Tetrazolium reduction allows assessment of biofilm formation by Campylobacter jejuni in a food matrix model. J. Appl. Microbiol. 2013, 115, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Sabaeifard, P.; Abdi-Ali, A.; Soudi, M.R.; Dinarvand, R. Optimization of tetrazolium salt assay for Pseudomonas aeruginosa biofilm using microtiter plate method. J. Microbiol. Methods 2014, 105, 134–140. [Google Scholar] [CrossRef]
- Lehman, S.M.; Donlan, R.M. Bacteriophage-mediated control of a two-species biofilm formed by microorganisms causing catheter-associated urinary tract infections in an in vitro urinary catheter model. Antimicrob. Agents Chemother. 2015, 59, 1127–1137. [Google Scholar] [CrossRef]
- Kelly, D.; McAuliffe, O.; Ross, R.P.; Coffey, A. Prevention of Staphylococcus aureus biofilm formation and reduction in established biofilm density using a combination of phage K and modified derivatives. Lett. Appl. Microbiol. 2012, 54, 286–291. [Google Scholar] [CrossRef]
- Fong, S.A.; Drilling, A.; Morales, S.; Cornet, M.E.; Woodworth, B.A.; Fokkens, W.J.; Psaltis, A.J.; Vreugde, S.; Wormald, P.J. Activity of bacteriophages in removing biofilms of Pseudomonas aeruginosa isolates from chronic rhinosinusitis patients. Front. Cell. Infect. Microbiol. 2017, 7, 418. [Google Scholar] [CrossRef]
- Pallavali, R.R.; Degati, V.L.; Durbaka, V.R.P. Bacteriophages inhibit biofilms formed by multi-drug resistant bacteria isolated from septic wounds. bioRxiv 2019. [Google Scholar] [CrossRef]
- Magana, M.; Sereti, C.; Ioannidis, A.; Mitchell, C.A.; Ball, A.R.; Magiorkinis, E.; Chatzipanagiotou, S.; Hamblin, M.R.; Hadjifrangiskou, M.; Tegos, G.P. Options and limitations in clinical investigation of bacterial biofilms. Clin. Microbiol. Rev. 2018, 31. [Google Scholar] [CrossRef]
- Floyd, K.A.; Moore, J.L.; Eberly, A.R.; Good, J.A.; Shaffer, C.L.; Zaver, H.; Almqvist, F.; Skaar, E.P.; Caprioli, R.M.; Hadjifrangiskou, M. Adhesive fiber stratification in uropathogenic Escherichia coli biofilms unveils oxygen-mediated control of type 1 pili. PLoS Pathog. 2015, 11, e1004697. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Trimble, M.J.; Cheng, J.T.; Valle, Q.; Hancock, R.E.W. Critical assessment of methods to quantify biofilm growth and evaluate antibiofilm activity of host defence peptides. Biomolecules 2018, 8, 29. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Olszak, T.; Arabski, M.; Wasik, S.; Majkowska-Skrobek, G.; Augustyniak, D.; Gula, G.; Briers, Y.; Jang, H.B.; Vandenheuvel, D.; et al. Characterization of the newly isolated lytic bacteriophages KTN6 and KT28 and their efficacy against Pseudomonas aeruginosa biofilm. PLoS ONE 2015, 10, e0127603. [Google Scholar]
- Alves, D.R.; Perez-Esteban, P.; Kot, W.; Bean, J.E.; Arnot, T.; Hansen, L.H.; Enright, M.C.; Jenkins, A.T. A novel bacteriophage cocktail reduces and disperses Pseudomonas aeruginosa biofilms under static and flow conditions. Microb. Biotechnol. 2015, 9, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Melo, L.D.R. In vitro activity of bacteriophages against planktonic and biofilm populations assessed by flow cytometry. Meth. Mol. Biol. 2018, 1693, 33–41. [Google Scholar]
- Rodríguez-Melcón, C.; Capita, R.; García-Fernández, C.; Alonso-Calleja, C. Effects of bacteriophage P100 at different concentrations on the structural parameters of Listeria monocytogenes biofilms. J. Food Prot. 2018, 81, 2040–2044. [Google Scholar] [CrossRef]
- Kabwe, M.; Brown, T.L.; Dashper, S.; Speirs, L.; Ku, H.; Petrovski, S.; Chan, H.T.; Lock, P.; Tucci, J. Genomic, morphological and functional characterisation of novel bacteriophage FNU1 capable of disrupting Fusobacterium nucleatum biofilms. Sci. Rep. 2019, 9, 9107. [Google Scholar] [CrossRef]
- Guła, G.; Szymanowska, P.; Piasecki, T.; Goras, S.; Gotszalk, T.; Drulis-Kawa, Z. The application of impedance spectroscopy for Pseudomonas biofilm monitoring during phage infection. Viruses 2020, 12, 407. [Google Scholar] [CrossRef]
- Tkhilaishvili, T.; Wang, L.; Perka, C.; Trampuz, A.; Gonzalez, M.M. Using bacteriophages as a Trojan Horse to the killing of dual-species biofilm formed by Pseudomonas aeruginosa and methicillin resistant Staphylococcus aureus. Front. Microbiol. 2020, 11, 695. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.W.; Hughes, K.A.; Skillman, L.C.; Tait, K. The interaction of phage and biofilms. FEMS Microbiol. Lett. 2004, 232, 1–6. [Google Scholar] [CrossRef]
- Filippini, M.; Buesing, N.; Bettarel, Y.; Sime-Ngando, T.; Gessner, M.O. Infection paradox: High abundance but low impact of freshwater benthic viruses. Appl. Environ. Microbiol. 2006, 72, 4893–4898. [Google Scholar] [CrossRef] [PubMed]
- Abedon, S.T. Spatial vulnerability: Bacterial arrangements, microcolonies, and biofilms as responses to low rather than high phage densities. Viruses 2012, 4, 663–687. [Google Scholar] [CrossRef]
- Dunsing, V.; Irmscher, T.; Barbirz, S.; Chiantia, S. Purely polysaccharide-based biofilm matrix provides size-selective diffusion barriers for nanoparticles and bacteriophages. Biomacromolecules 2019, 20, 3842–3854. [Google Scholar] [CrossRef]
- Melo, L.D.R.; Pinto, G.; Oliveira, F.; Vilas-Boas, D.; Almeida, C.; Sillankorva, S.; Cerca, N.; Azeredo, J. The protective effect of Staphylococcus epidermidis biofilm matrix against phage predation. Viruses 2020, 12, 1076. [Google Scholar] [CrossRef]
- Ganegama Arachchi, G.J.; Cridge, A.G.; Dias-Wanigasekera, B.M.; Cruz, C.D.; McIntyre, L.; Liu, R.; Flint, S.H.; Mutukumira, A.N. Effectiveness of phages in the decontamination of Listeria monocytogenes adhered to clean stainless steel, stainless steel coated with fish protein, and as a biofilm. J. Ind. Microbiol. Biotechnol. 2013, 40, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Brown-Jaque, M.; Muniesa, M.; Navarro, F. Bacteriophages in clinical samples can interfere with microbiological diagnostic tools. Sci. Rep. 2016, 6, 33000. [Google Scholar] [CrossRef]
- Boas, D.V.; Almeida, C.; Azevedo, N.; Sillankorva, S.; Azeredo, J. Techniques to assess phage-biofilm interaction. Meth. Mol. Biol. 2019, 1898, 137–146. [Google Scholar]
- de Siqueira, R.S.; Dodd, C.E.R.; Rees, C.E.D. Evaluation of the natural virucidal activity of teas for use in the phage amplification assay. Int. J. Food Microbiol. 2006, 111, 259–262. [Google Scholar] [CrossRef]
- Friedman, M. Overview of antibacterial, antitoxin, antiviral, and antifungal activities of tea flavonoids and teas. Mol. Nutr. Food Res. 2007, 51, 116–134. [Google Scholar] [CrossRef] [PubMed]
- Jassim, S.A.; Naji, M.A. In vitro evaluation of the antiviral activity of an extract of date palm (Phoenix dactylifera L.) pits on a Pseudomonas phage. Evid. Based Complement. Alternat. Med. 2007, 7, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.; Willner, D.; Oechel, W.C.; Lipson, D. Top-down control of microbial activity and biomass in an Arctic soil ecosystem. Environ. Microbiol. 2010, 12, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Chibeu, A.; Agius, L.; Gao, A.; Sabour, P.M.; Kropinski, A.M.; Balamurugan, S. Efficacy of bacteriophage LISTEX™ P100 combined with chemical antimicrobials in reducing Listeria monocytogenes in cooked turkey and roast beef. Int. J. Food Microbiol. 2013, 167, 208–214. [Google Scholar] [CrossRef]
- Helsley, K.R.; Brown, T.M.; Furlong, K.; Williamson, K.E. Applications and limitations of tea extract as a virucidal agent to assess the role of phage predation in soils. Biol. Fertil. Soils 2014, 50, 263–274. [Google Scholar] [CrossRef]
- Liu, H.; Meng, R.; Wang, J.; Niu, Y.D.; Li, J.; Stanford, K.; McAllister, T.A. Inactivation of Escherichia coli O157 bacteriophages by using a mixture of ferrous sulfate and tea extract. J. Food Prot. 2015, 78, 2220–2226. [Google Scholar] [CrossRef] [PubMed]
- Chibeu, A.; Balamurugan, S. Application of a virucidal agent to avoid overestimation of phage kill during phage decontamination assays on ready-to-eat meats. Meth. Mol. Biol. 2018, 1681, 97–105. [Google Scholar]
- Miller, R.V.; Day, M. Contribution of lysogeny, pseudolysogeny, and starvation to phage ecology. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 114–143. [Google Scholar]
- Hunt, S.M.; Werner, E.M.; Huang, B.; Hamilton, M.A.; Stewart, P.S. Hypothesis for the role of nutrient starvation in biofilm detachment. Appl. Environ. Microbiol. 2004, 70, 7418–7425. [Google Scholar] [CrossRef] [PubMed]
- Fung, D.K.; Chan, E.W.; Chin, M.L.; Chan, R.C. Delineation of a bacterial starvation stress response network which can mediate antibiotic tolerance development. Antimicrob. Agents Chemother. 2010, 54, 1082–1093. [Google Scholar] [CrossRef]
- Brown, D.R. Nitrogen starvation induces persister cell formation in Escherichia coli. J. Bacteriol. 2019, 201, e00622-18. [Google Scholar] [CrossRef] [PubMed]
- Gray, D.A.; Dugar, G.; Gamba, P.; Strahl, H.; Jonker, M.J.; Hamoen, L.W. Extreme slow growth as alternative strategy to survive deep starvation in bacteria. Nat. Commun. 2019, 10, 890. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.; Jiang, X. Application of bacteriophages to reduce biofilms formed by hydrogen sulfide producing bacteria on surfaces in a rendering plant. Can. J. Microbiol. 2015, 61, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, C.; Colak, M.; Yilmaz, B.C.; Ersoz, G.; Kutateladze, M.; Gozlugol, M. Bacteriophage therapy in implant-related infections: An experimental study. J. Bone Joint Surg. Am. 2013, 95, 117–125. [Google Scholar] [CrossRef]
- Khalifa, L.; Shlezinger, M.; Beyth, S.; Houri-Haddad, Y.; Coppenhagen-Glazer, S.; Beyth, N.; Hazan, R. Phage therapy against Enterococcus faecalis in dental root canals. J. Oral Microbiol. 2016, 8, 32157. [Google Scholar] [CrossRef]
- Alves, D.R.; Booth, S.P.; Scavone, P.; Schellenberger, P.; Salvage, J.; Dedi, C.; Thet, N.T.; Jenkins, A.T.A.; Waters, R.; Ng, K.W.; et al. Development of a high-throughput ex-vivo burn wound model using porcine skin, and its application to evaluate new approaches to control wound infection. Front. Cell. Infect. Microbiol. 2018, 8, 196. [Google Scholar] [CrossRef]
- Milho, C.; Andrade, M.; Vilas, B.D.; Alves, D.; Sillankorva, S. Antimicrobial assessment of phage therapy using a porcine model of biofilm infection. Int. J. Pharm. 2019, 557, 112–123. [Google Scholar] [CrossRef]
- Grygorcewicz, B.; Wojciuk, B.; Roszak, M.; Lubowska, N.; Blazejczak, P.; Jursa-Kulesza, J.; Rakoczy, R.; Masiuk, H.; Dolegowska, B. Environmental phage-based cocktail and antibiotic combination effects on Acinetobacter baumannii biofilm in a human urine model. Microb. Drug Resist. 2021, 27, 25–35. [Google Scholar] [CrossRef]
- Alemayehu, D.; Casey, P.G.; McAuliffe, O.; Guinane, C.M.; Martin, J.G.; Shanahan, F.; Coffey, A.; Ross, R.P.; Hill, C. Bacteriophages ϕMR299-2 and ϕNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. MBio 2012, 3, e00029-12. [Google Scholar] [CrossRef]
- Schmerer, M.; Molineux, I.J.; Ally, D.; Tyerman, J.; Cecchini, N.; Bull, J.J. Challenges in predicting the evolutionary maintenance of a phage transgene. J. Biol. Eng. 2014, 8, 21. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abedon, S.T.; Danis-Wlodarczyk, K.M.; Wozniak, D.J.; Sullivan, M.B. Improving Phage-Biofilm In Vitro Experimentation. Viruses 2021, 13, 1175. https://doi.org/10.3390/v13061175
Abedon ST, Danis-Wlodarczyk KM, Wozniak DJ, Sullivan MB. Improving Phage-Biofilm In Vitro Experimentation. Viruses. 2021; 13(6):1175. https://doi.org/10.3390/v13061175
Chicago/Turabian StyleAbedon, Stephen T., Katarzyna M. Danis-Wlodarczyk, Daniel J. Wozniak, and Matthew B. Sullivan. 2021. "Improving Phage-Biofilm In Vitro Experimentation" Viruses 13, no. 6: 1175. https://doi.org/10.3390/v13061175
APA StyleAbedon, S. T., Danis-Wlodarczyk, K. M., Wozniak, D. J., & Sullivan, M. B. (2021). Improving Phage-Biofilm In Vitro Experimentation. Viruses, 13(6), 1175. https://doi.org/10.3390/v13061175