
Characterization of a Novel SARS-CoV-2 Genetic Variant with Distinct Spike Protein Mutations

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Extraction and Reverse Transcription qPCR
2.3. Primer Design for Near-Complete Genome Sequencing
2.4. Library Preparation and Near-Complete Genome Sequencing
2.5. In Silico Analysis
2.5.1. Genome Assembly
2.5.2. Phylogenetic Reconstructions
2.5.3. Protein Analysis
3. Results
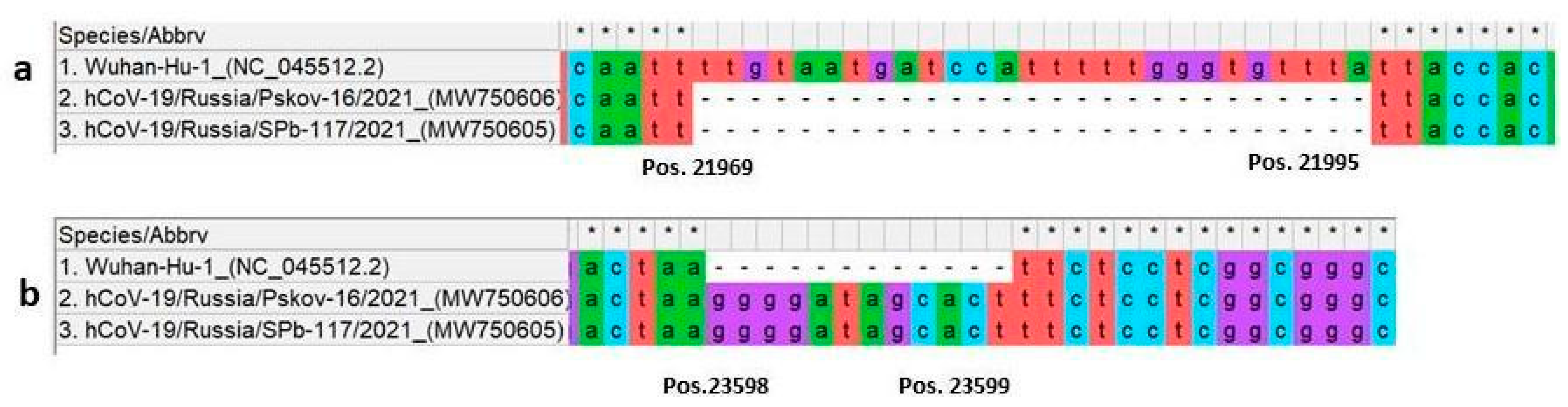
3.1. Sequencing
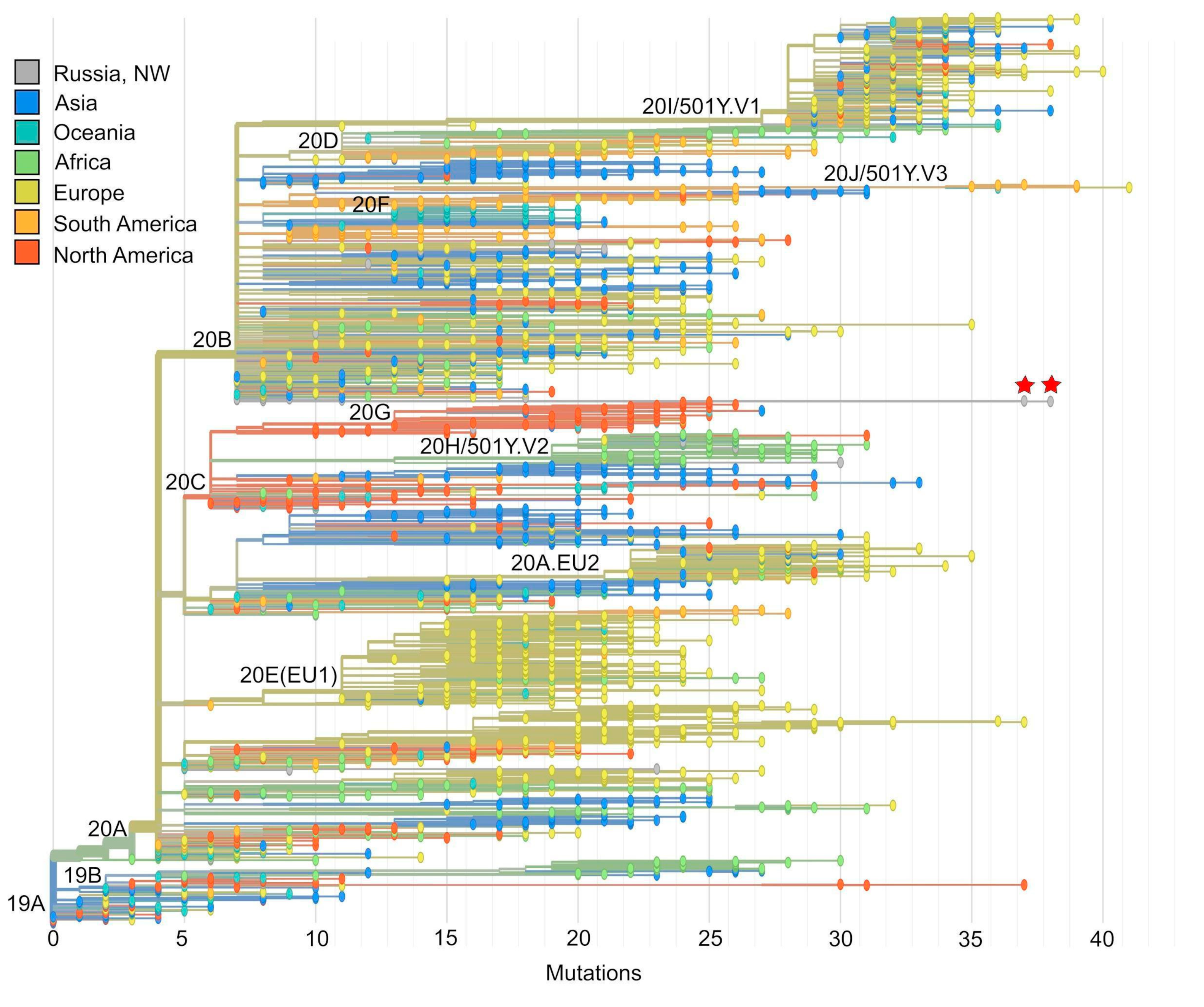
3.2. Phylogenetic Analysis
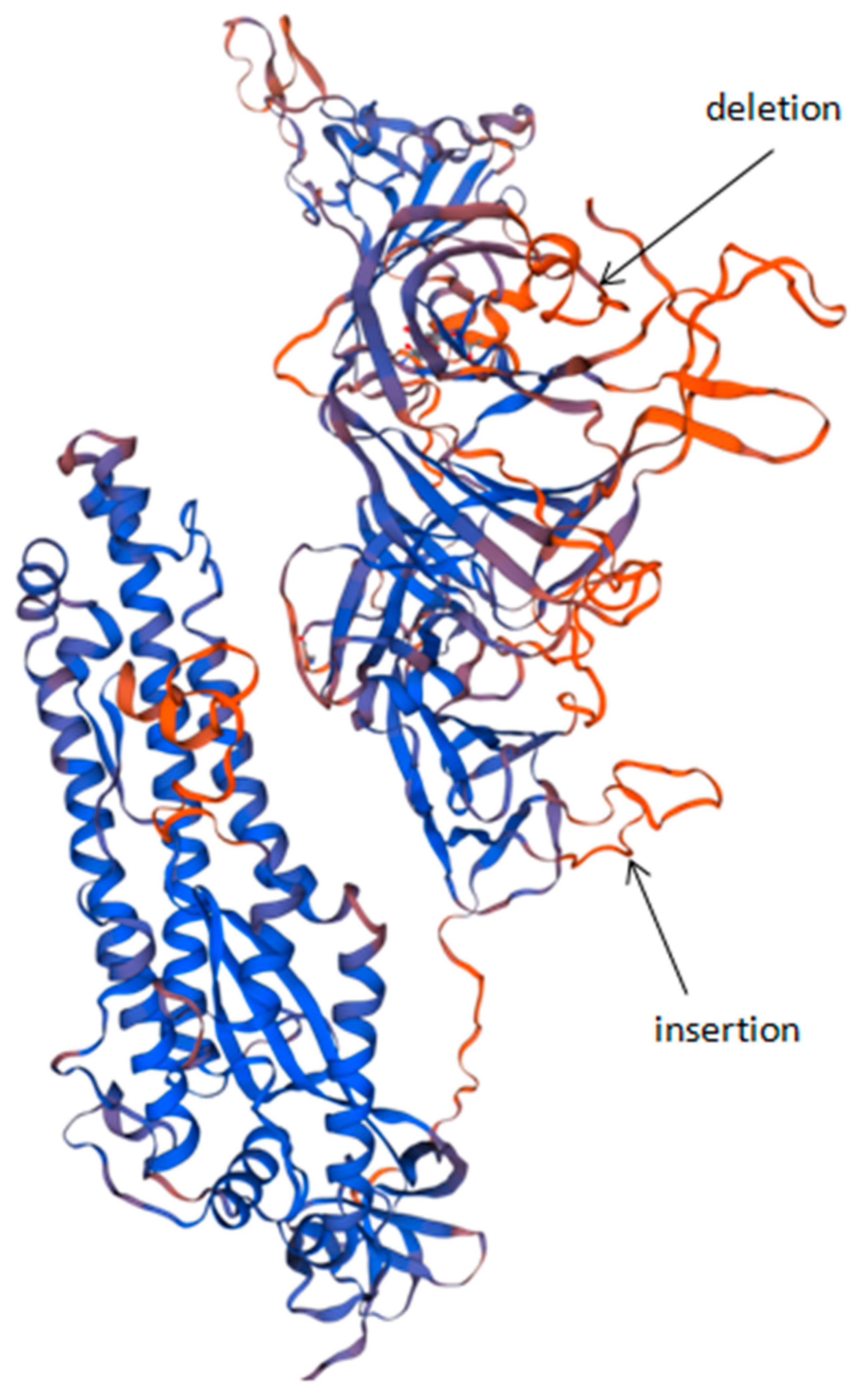
3.3. Protein Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Komissarov, A.B.; Safina, K.R.; Garushyants, S.K.; Fadeev, A.V.; Sergeeva, M.V.; Ivanova, A.A.; Danilenko, D.M.; Lioznov, D.; Shneider, O.V.; Shvyrev, N.; et al. Genomic epidemiology of the early stages of the SARS-CoV-2 outbreak in Russia. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coro-navirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [PubMed]
- Goncharova, E.A.; Dedkov, V.G.; Dolgova, A.S.; Kassirov, I.S.; Safonova, M.V.; Voytsekhovskaya, Y.; Totolian, A.A. One-step quantitative RT-PCR assay with armored RNA controls for detection of SARS-CoV-2. J. Med. Virol. 2021, 93, 1694–1701. [Google Scholar] [CrossRef] [PubMed]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 28 May 2021).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fou-rier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Lu, G. Vector NTI, a balanced all-in-one sequence analysis suite. Brief. Bioinform. 2004, 5, 378–388. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. Addendum: A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2021, 6, 415. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784.e5. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Čalyševa, J.; et al. ELM—the eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2019, 48, D296–D306. [Google Scholar] [CrossRef]
- Owen, D.; Vallis, Y.; Pearse, B.; McMahon, H.; Evans, P. The structure and function of the beta2-adaptin appendage domain. EMBO J. 2000, 19, 4216–4227. [Google Scholar] [CrossRef]
- Mardones, G.A.; Burgos, P.V.; Lin, Y.; Kloer, D.P.; Magadán, J.G.; Hurley, J.H.; Bonifacino, J.S. Structural Basis for the Recognition of Tyrosine-based Sorting Signals by the μ3A Subunit of the AP-3 Adaptor Complex. J. Biol. Chem. 2013, 288, 9563–9571. [Google Scholar] [CrossRef]
- Owen, D. Linking endocytic cargo to clathrin: Structural and functional insights into coated vesicle formation. Biochem. Soc. Trans. 2004, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular Structure, Function, and Dynamics of Clathrin-Mediated Membrane Traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar] [CrossRef]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus into Target Cells Expressing ACE2 with the Cytoplasmic Tail Deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef]
- Wang, N.; Sun, Y.; Feng, R.; Wang, Y.; Guo, Y.; Zhang, L.; Deng, Y.-Q.; Wang, L.; Cui, Z.; Cao, L.; et al. Structure-based development of human antibody cocktails against SARS-CoV-2. Cell Res. 2021, 31, 101–103. [Google Scholar] [CrossRef]
- Bayati, A.; Kumar, R.; Francis, V.; McPherson, P.S. SARS-CoV-2 infects cells after viral entry via clathrin-mediated endocytosis. J. Biol. Chem. 2021, 296, 100306. [Google Scholar] [CrossRef] [PubMed]
- Dell’Angelica, E.C. Clathrin-binding proteins: Got a motif? Join the network! Trends Cell Biol. 2001, 11, 315–318. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | NW Variant Strain of SARS-CoV-2 | |||||
|---|---|---|---|---|---|---|
| hCoV-19/Russia/SPb-117/2021(MW750605/EPI_ISL_1259282) | hCoV-19/Russia/Pskov-16/2021(MW750606/EPI_ISL_1259283) | |||||
| Synonymous Substitution, nt | Nonsynonymous Substitution/Indel, nt | Substitution/Indel, aa | Synonymous Substitution/Indel, nt | Nonsynonymous Substitution/Indel, nt | Substitution/Indel, aa | |
| 5’ UTR | 241C>T | |||||
| ORF 1a | 3037C>T 5176A>G 9070T>C 9778C>T | 1392C>T 3281G>T 3542A>G 7005C>A 10029C>T 11451A>G 12620T>A | S376L V1006F T1093A T2247N T3255I Q3729R S4119T | 3037C>T 5176A>G 9070T>C 9778C>T | 1392C>T 3281G>T 3542A>G 7005C>A 10029C>T 11451A>G 12620T>A | S376L V1006F T1093A T2247N T3255I Q3729R S4119T |
| ORF 1b | 17562G>T | 14408C>T 16934T>C 16985C>A 17470C>A 19180G>T 20759C>T | P314L M1156T T1173N L1335I V1905L A2431V | 17562G>T | 14408C>T 16985C>A 19180G>T 20759C>T | P314L T1173N V1905L A2431V |
| S gene | 22882T>C | 21588C>T | P9L | 23449T>G | 21588C>T | P9L |
| S1 domain | Deletion 21967_21993del 22206A>G 22296A>C 23012G>A 23403A>G | C136_Y144del D215G H245P E484K D614G | Deletion 21967_21993del 22206A>G 22296A>C 23012G>A 23403A>G | C136_Y144del D215G H245P E484K D614G | ||
| insertion 23598_23599ins | N679delins KGIAL | insertion 23598_23599ins | N679delins KGIAL | |||
| S2 domain | 25000C>T | 23900G>A 24697G>T | 780E>K 1045K>N | 24370C>T 24721T>G 25000C>T | 23900G>A | 780E>K |
| ORF 3a | 25603C>T 26211G>T | 25675T>A | L95M | 25603C>T 26211G>T | 25675T>A | L95M |
| M gene | 26568C>A | L16I | 26568C>A 27102G>A | L16I A194T | ||
| ORF 7a | 27674A>G | Q94R | ||||
| ORF 8 | 28079G>T 28271A>G | 28079G>T 28271A>G | ||||
| N gene | 28881G>A 28882G>A 28883G>C | R203K G204R | 28881G>A 28882G>A 28883G>C | R203K G204R | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gladkikh, A.; Dolgova, A.; Dedkov, V.; Sbarzaglia, V.; Kanaeva, O.; Popova, A.; Totolian, A. Characterization of a Novel SARS-CoV-2 Genetic Variant with Distinct Spike Protein Mutations. Viruses 2021, 13, 1029. https://doi.org/10.3390/v13061029
Gladkikh A, Dolgova A, Dedkov V, Sbarzaglia V, Kanaeva O, Popova A, Totolian A. Characterization of a Novel SARS-CoV-2 Genetic Variant with Distinct Spike Protein Mutations. Viruses. 2021; 13(6):1029. https://doi.org/10.3390/v13061029
Chicago/Turabian StyleGladkikh, Anna, Anna Dolgova, Vladimir Dedkov, Valeriya Sbarzaglia, Olga Kanaeva, Anna Popova, and Areg Totolian. 2021. "Characterization of a Novel SARS-CoV-2 Genetic Variant with Distinct Spike Protein Mutations" Viruses 13, no. 6: 1029. https://doi.org/10.3390/v13061029
APA StyleGladkikh, A., Dolgova, A., Dedkov, V., Sbarzaglia, V., Kanaeva, O., Popova, A., & Totolian, A. (2021). Characterization of a Novel SARS-CoV-2 Genetic Variant with Distinct Spike Protein Mutations. Viruses, 13(6), 1029. https://doi.org/10.3390/v13061029