
Age, Disease Severity and Ethnicity Influence Humoral Responses in a Multi-Ethnic COVID-19 Cohort

 , , , , , and add
Show full author list
, , , , , and add
Show full author list
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design
2.1.1. Cohort 1
2.1.2. Cohort 2
2.1.3. Cohort 3
2.2. Selection, Cloning, and Expression of SARS-CoV-2 Antigens
2.2.1. Antigen Selection for Immunoassay Platform
2.2.2. Gene Synthesis and Cloning
2.2.3. Expression of Nucleocapsid Proteins as Fusions to a BCCP Tag
2.3. Fabrication of Prototype and Final Protein Microarray
2.4. Serological Assays
Optimization of Serum Concentration and Determination of Linear Range
2.5. Bioinformatic Analysis
2.5.1. Image Analysis: Raw Data Extraction
2.5.2. Data Pre-Processing
2.5.3. Statistical Tests
3. Results
3.1. Developing a High-Sensitivity, High-Specificity SARS-CoV-2 Antigen Microarray
3.1.1. Selecting N-Protein Constructs for the Final Microarray Design
3.1.2. Selecting Peptides from the N Protein for Microarray Fabrication
3.2. Technical Performance of the SARS-CoV-2 Antigen Microarray Platform in an Independent Validation Cohort
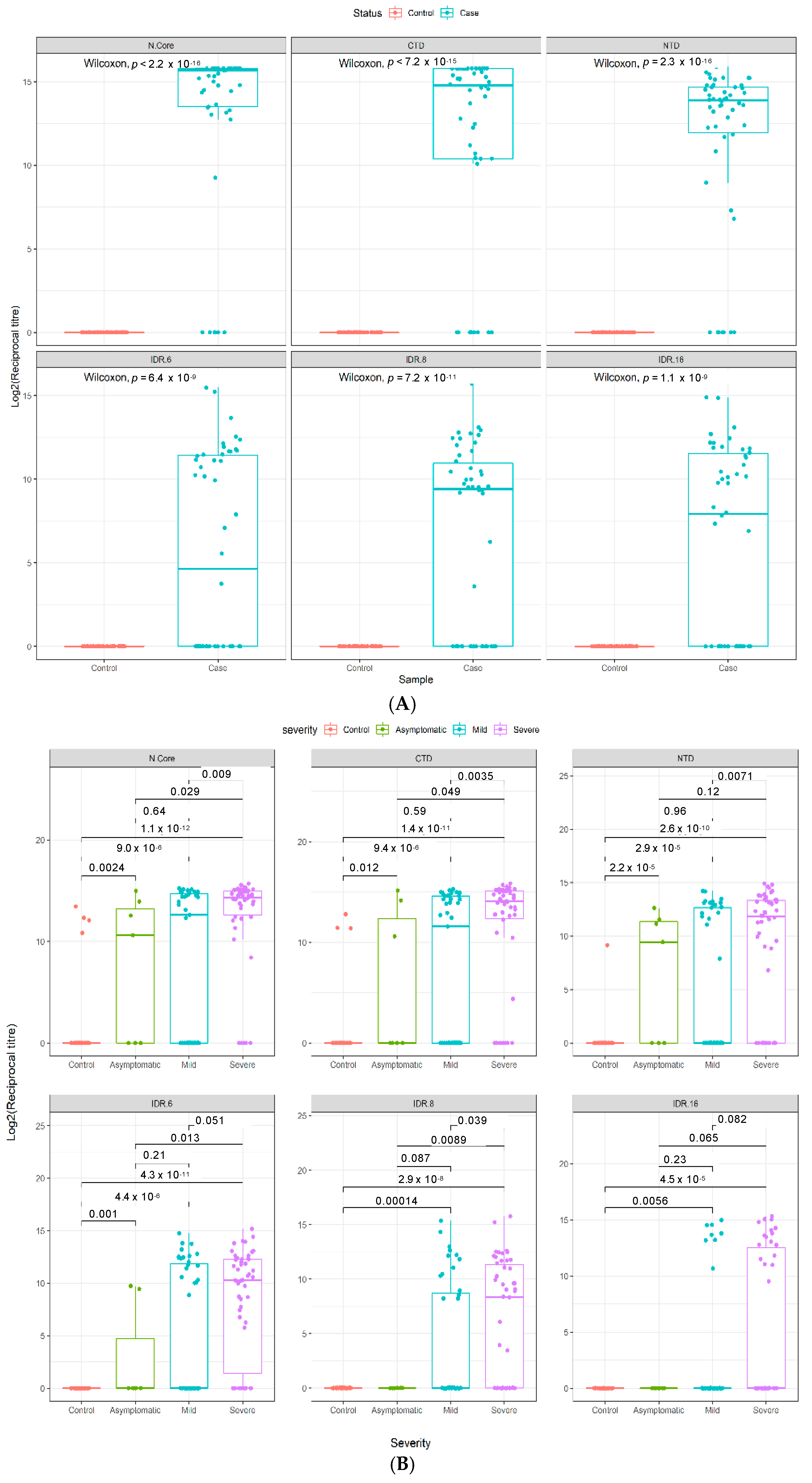
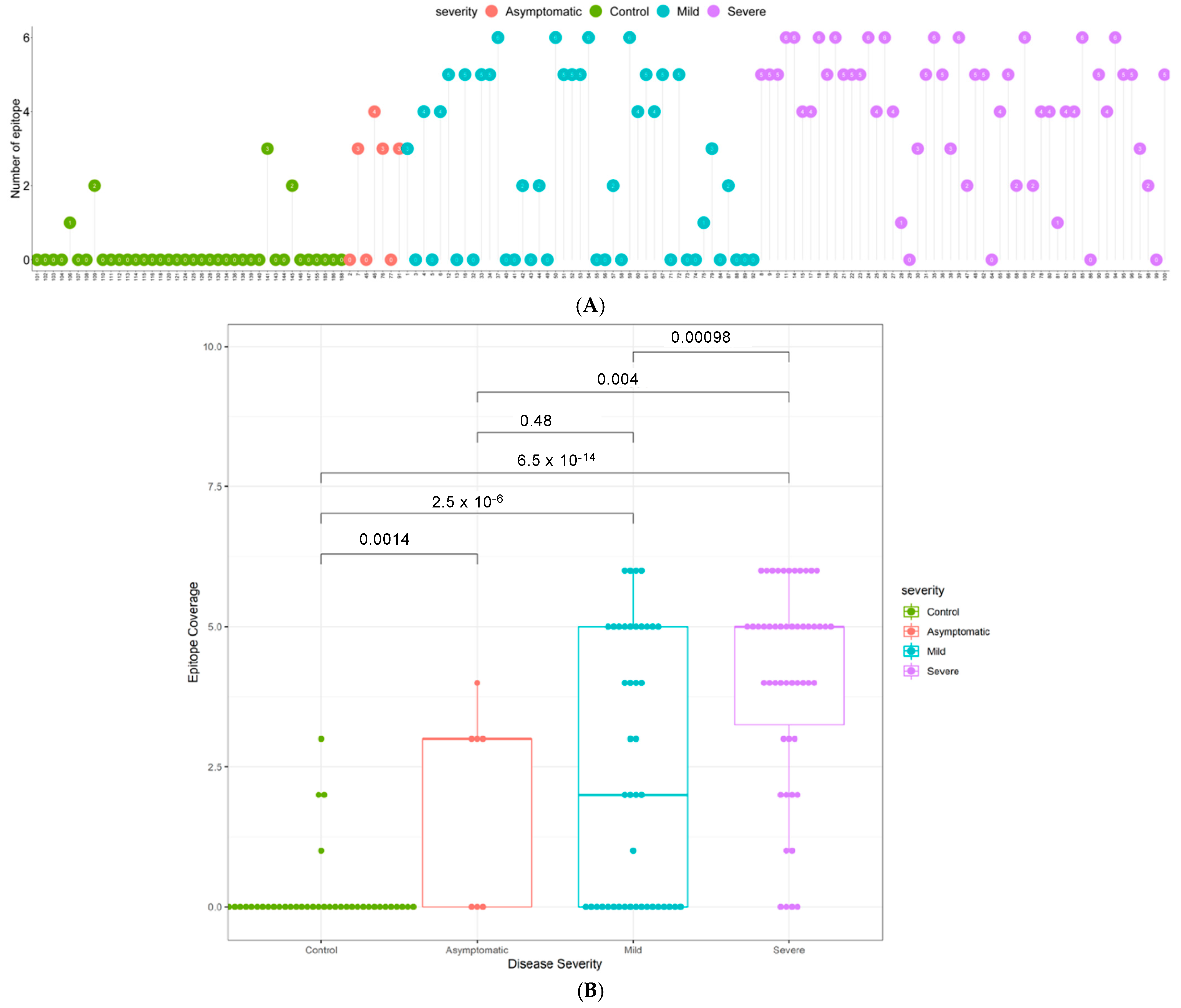
3.3. Quantitative Analysis of an Independent, Multi-Ethnic Cohort Reveals Differences in Antibody Titers and Epitope Coverage Scores Associated with Age, Disease Severity, and Ethnicity
3.4. Elevated N-Specific Antibody Titers and Broader Epitope Coverage Observed in Patients with Severe Disease
3.4.1. Increasing Antibody Titers and Epitope Coverage with Increasing Age
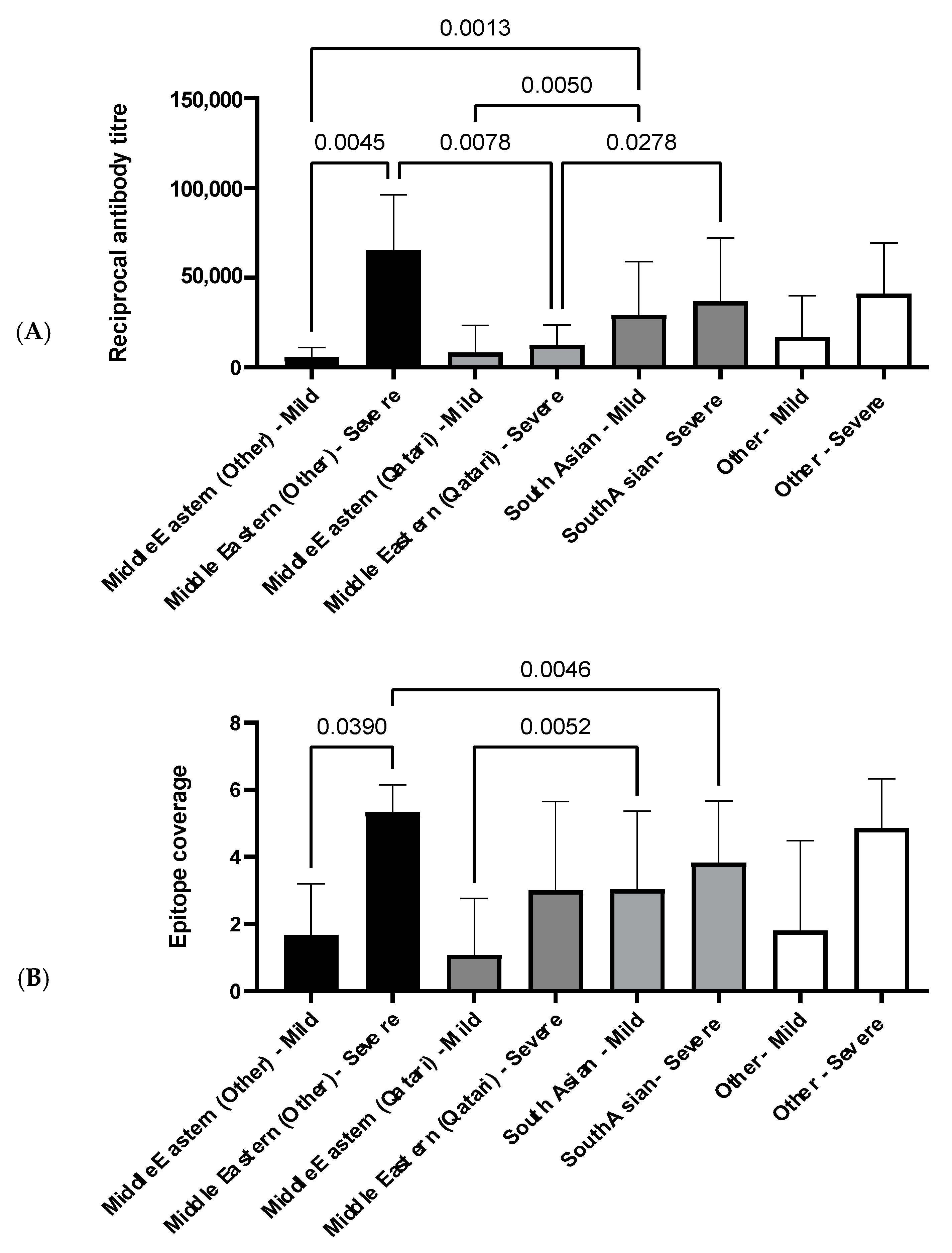
3.4.2. The Influence of Ethnicity on N-Specific Antibody Titers and the Breadth of Epitope Coverage
4. Discussion
Limitations and Further Work
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organisation. Rolling Updates on Coronavirus Disease (COVID-19). 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen (accessed on 27 April 2021).
- Khurshid, Z.; Asiri, F.Y.I.; Al Wadaani, H. Human Saliva: Non-Invasive Fluid for Detecting Novel Coronavirus (2019-nCoV). Int. J. Environ. Res. Public Health 2020, 17, 2225. [Google Scholar] [CrossRef]
- Sohrabia, C.; Alsafib, Z.; O’Neilla, N.; Khanb, M.; Kerwanc, A.; Al-Jabirc, A.; Iosifidisa, C.; Aghad, R. World Health Organization declares global emergency: A review of the 2019 novel coronavirus (COVID-19). Int. J. Surg. 2020, 76, 71–76. [Google Scholar] [CrossRef]
- de Walque, D.B.C.M.; Friedman, J.; Gatti, R.V.; Mattoo, A. How Two Tests Can Help Contain COVID-19 and Revive the Economy; Research & Policy Briefs; World Bank Group: Washington, DC, USA, 2020; Volume 29, Available online: http://documents.worldbank.org/curated/en/766471586360658318/How-Two-Tests-Can-Help-Contain-COVID-19-and-Revive-the-Economy (accessed on 27 April 2021).
- World Health Organisation. Coronavirus Disease (COVID-19) Outbreak Situation. 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 27 April 2021).
- Escobedo-de la Peña, J.; Rascón Pacheco, R.A.; de Jesús Ascencio-Montiel, I.; González-Figueroa, E.; Fernández-Gárate, J.E.; Medina-Gómez, O.S.; Borja-Bustamante, P.; Santillán-Oropeza, J.A.; Borja-Aburto, V.H. Hypertension, diabetes and obesity, major risk factors for death in patients with COVID-19 in Mexico. Arch. Med Res. 2020. [Google Scholar] [CrossRef]
- Ye, C.; Zhang, S.; Zhang, X.; Cai, H.; Gu, J.; Lian, J.; Lu, Y.; Jia, H.; Hu, J.; Jin, C.; et al. Impact of comorbidities on patients with COVID-19: A large retrospective study in Zhejiang, China. J. Med. Virol. 2020, 92, 2821–2829. [Google Scholar] [CrossRef]
- Kopel, J.; Perisetti, A.; Roghani, A.; Aziz, M.; Gajendran, M.; Goyal, H. Racial and Gender-Based Differences in COVID-19. Front. Public Health 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.J.; Swadling, L.; Gibbons, J.M.; Pade, C.; Jensen, M.P.; Diniz, M.O.; Schmidt, N.M.; Butler, D.K.; Amin, O.E.; Bailey, S.N.L.; et al. Discordant neutralizing antibody and T cell responses in asymptomatic and mild SARS-CoV-2 infection. Sci. Immunol. 2020, 5, eabf3698. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, J. Immunologic testing for SARS-CoV-2 infection from the antigen perspective. J. Clin. Microbiol. 2020, 59. [Google Scholar] [CrossRef]
- van Tol, S.; Mögling, R.; Li, W.; Godeke, G.-J.; Swart, A.; Bergmans, B.; Brandenburg, A.; Kremer, K.; Murk, J.-L.; van Beek, J.; et al. Accurate serology for SARS-CoV-2 and common human coronaviruses using a multiplex approach. Emerg. Microbes Infect. 2020, 9, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- de Assis, R.R.; Jain, A.; Nakajima, R.; Jasinskas, A.; Felgner, J.; Obiero, J.M.; Norris, P.J.; Stone, M.; Simmons, G.; Bagri, A.; et al. Analysis of SARS-CoV-2 antibodies in COVID-19 convalescent blood using a coronavirus antigen microarray. Nat. Commun. 2021, 12, 6. [Google Scholar] [CrossRef]
- Zhao, J.; Yuan, Q.; Wang, H.; Liu, W.; Liao, X.; Su, Y.; Wang, X.; Yuan, J.; L, T.; Li, J.; et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 2027–2034. [Google Scholar] [CrossRef]
- Tang, F.; Quan, Y.; Xin, Z.; Wrammert, J.; Ma, M.-J.; Lv, H.; Wang, T.-B.; Yang, H.; Richardus, J.H.; Liu, W.; et al. Lack of Peripheral Memory B Cell Responses in Recovered Patients with Severe Acute Respiratory Syndrome: A Six-Year Follow-Up Study. J. Immunol. 2011, 186, 7264–7268. [Google Scholar] [CrossRef]
- Summary of Probable SARS Cases with Onset of Illness from 1 November 2002 to 31 July 2003. 2003. Available online: https://www.who.int/csr/sars/country/table2004_04_21/en/ (accessed on 27 April 2021).
- Zhou, W.; Wang, W.; Wang, H.; Lu, R.; Tan, W. First infection by all four non-severe acute respiratory syndrome human coronaviruses takes place during childhood. BMC Infect. Dis. 2013, 13, 1–8. [Google Scholar] [CrossRef]
- Blackburn, J.M.; Shoko, A.; Beeton-Kempen, N. Miniaturized, Microarray-Based Assays for Chemical Proteomic Studies of Protein Function. In Chemical Genomics and Proteomics: Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 800, pp. 133–162. [Google Scholar]
- Adeola, H.A.; Blackburn, J.M.; Rebbeck, T.R.; Zerbini, L.F. Novel potential serological prostate cancer biomarkers using CT100+ cancer antigen microarray platform in a multi-cultural South African cohort. Oncotarget 2016, 7, 13949–13960. [Google Scholar] [CrossRef]
- López-Ratón, M.; Rodríguez-Álvarez, M.X.; Cadarso-Suárez, C.; Gude-Sampedro, F. OptimalCutpoints: An R Package for Selecting Optimal Cutpoints in Diagnostic Tests. J. Stat. Softw. 2014, 61, 36. [Google Scholar] [CrossRef]
- Beeton-Kempen, N.; Duarte, J.; Shoko, A.; Serufuri, J.-M.; John, T.; Cebon, J.; Blackburn, J. Development of a novel, quantitative protein microarray platform for the multiplexed serological analysis of autoantibodies to cancer-testis antigens. Int. J. Cancer 2014, 135, 1842–1851. [Google Scholar] [CrossRef]
- Larkin, J.G. Family History of rheumatoid arthritis—a non-predictor of inflammatory disease? Rheumatology 2009, 49, 608–609. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Barlow, D.J.; Edwards, M.S.; Thornton, J.M. Continuous and discontinuous protein antigenic determinants. Nature 1986, 322, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Van Regenmortel, M.H.V. Mapping Epitope Structure and Activity: From One-Dimensional Prediction to Four-Dimensional Description of Antigenic Specificity. Methods 1996, 9, 465–472. [Google Scholar] [CrossRef]
- Chapman-Smith, A.; Cronan, J.E., Jr. Molecular Biology of Biotin Attachment to Proteins. J. Nutr. 1999, 129, 477S–484S. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses 2020, 12, 254. [Google Scholar] [CrossRef]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e1415. [Google Scholar] [CrossRef]
- Peng, Y.; Mentzer, A.J.; Liu, G.; Yao, X.; Yin, Z.; Dong, D.; Dejnirattisai, W.; Rostron, T.; Supasa, P.; Liu, C.; et al. Broad and strong memory CD4+ and CD8+ T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat. Immunol. 2020, 21, 1336–1345. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e114. [Google Scholar] [CrossRef]
- Cele, S.; Gazy, I.; Jackson, L.; Hwa, S.-H.; Tegally, H.; Lustig, G.; Giandhari, J.; Pillay, S.; Wilkinson, E.; Naidoo, Y.; et al. Escape of SARS-CoV-2 501Y.V2 from neutralization by convalescent plasma. Nature 2021. [Google Scholar] [CrossRef]
- Wibmer, C.K.; Ayres, F.; Hermanus, T.; Madzivhandila, M.; Kgagudi, P.; Oosthuysen, B.; Lambson, B.E.; de Oliveira, T.; Vermeulen, M.; van der Berg, K.; et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021, 27, 622–625. [Google Scholar] [CrossRef]
- Manisty, C.; Treibel, T.A.; Jensen, M.; Semper, A.; Joy, G.; Gupta, R.K.; Cutino-Moguel, T.; Andiapen, M.; Jones, J.; Taylor, S.; et al. Time series analysis and mechanistic modelling of heterogeneity and sero-reversion in antibody responses to mild SARS-CoV-2 infection. EBioMedicine 2021, 65, 103259. [Google Scholar] [CrossRef] [PubMed]
- Castelo-Branco, C.; Soveral, I. The immune system and aging: A review. Gynecol. Endocrinol. 2014, 30, 16–22. [Google Scholar] [CrossRef]
- Gibson, K.L.; Wu, Y.-C.; Barnett, Y.; Duggan, O.; Vaughan, R.; Kondeatis, E.; Nilsson, B.-O.; Wikby, A.; Kipling, D.; Dunn-Walters, D.K. B-cell diversity decreases in old age and is correlated with poor health status. Aging Cell 2009, 8, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Wu, Y.-C.; Kipling, D.; Dunn-Walters, D. Ageing of the B-cell repertoire. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140237. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, K.; Viboud, C.; Simonsen, L. Antibody response to influenza vaccination in the elderly: A quantitative review. Vaccine 2006, 24, 1159–1169. [Google Scholar] [CrossRef]
- Davydov, A.N.; Obraztsova, A.S.; Lebedin, M.Y.; Turchaninova, M.A.; Staroverov, D.B.; Merzlyak, E.M.; Sharonov, G.V.; Kladova, O.; Shugay, M.; Britanova, O.V.; et al. Comparative Analysis of B-Cell Receptor Repertoires Induced by Live Yellow Fever Vaccine in Young and Middle-Age Donors. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; DeVies, J.; Caruso, E.; Radhakrishnan, L.; Sheppard, M.; Stein, Z.; Calanan, R.M.; Hartnett, K.P.; Kite-Powell, A.; Rodgers, L.; et al. Emergency Department Visits for COVID-19 by Race and Ethnicity—13 States, October–December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Bener, A.; Hussain, R.; Teebi, A.S. Consanguineous marriages and their effects on common adult diseases: Studies from an endogamous population. Med. Princ. Pract. 2007, 16, 262–267. [Google Scholar] [CrossRef] [PubMed]
- De Sanctis, V. Type 1 and Type 2 diabetes in children and adolescents: A public health problem in Qatar. The experience of Pediatric Diabetes Center at Hamad General Hospital (HGH) of Doha. Acta Biomed. 2018, 89, 5–6. [Google Scholar] [CrossRef]
- Al Naqbi, H.; Mawart, A.; Alshamsi, J.; Al Safar, H.; Tay, G.K. Major histocompatibility complex (MHC) associations with diseases in ethnic groups of the Arabian Peninsula. Immunogenetics 2021, 73, 131–152. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Cohort 1 | Cohort 2 | Cohort 3 | |
|---|---|---|---|---|
| Total number of patients | 174 | 100 | 138 | |
| Disease status | Pre-pandemic disease controls | 68 | 50 | 0 |
| COVID-19 PCR − ve | 23 | |||
| COVID-19 PCR + ve | 76 | 50 | 100 | |
| No COVID-19 PCR test data | 7 | 38 | ||
| Disease Severity | Asymptomatic (PCR − ve) | 4 | 0 | 0 |
| Symptomatic (PCR − ve) | 19 | 0 | 0 | |
| Asymptomatic (PCR + ve) | 14 | 0 | 7 | |
| Mild (PCR + ve) | 24 | 0 | 43 | |
| Severe (PCR + ve) | 34 | 50 | 50 | |
| Asymptomatic (no PCR test data) | 7 | 0 | 38 | |
| Not declared (PCR + ve) | 4 | 0 | 0 | |
| Gender | Female | 55 * | 30 * | 12 |
| Male | 49 * | 13 * | 126 | |
| Not declared | 2 * | 7 * | 0 | |
| Age distribution | 18–40 | 60 * | 10 * | 67 |
| 41–60 | 38 * | 24 * | 65 | |
| 61–73 | 6 * | 9 * | 6 | |
| Not declared | 2 * | 7 * | 0 | |
| Ethnicity | African | 9 * | 0 | |
| Caucasian | 72 * | 0 | 0 | |
| Colored | 1 * | 0 | 0 | |
| Half-Japanese, half-Caucasian | 1 * | 0 | 0 | |
| South Asian | 9 * | 100 | 94 | |
| Middle East (Other) | 0 * | 0 | 10 | |
| Middle East (Qatari) | 0 * | 0 | 18 | |
| Other | 0 * | 0 | 15 | |
| Not declared | 14 * | 0 | 1 | |
| Immunoassay Result | COVID-19 Status | |
|---|---|---|
| Positive | Negative | |
| Positive | 50 | 0 |
| Negative | 0 | 50 |
| Sensitivity = 100% | Specificity = 100% | |
| Disease Severity | Immunoassay Result | RT-PCR Status | Sensitivity | Specificity | |
|---|---|---|---|---|---|
| Positive | Unknown | ||||
| All samples (Case, n = 100; Control, n = 38) | Positive | 75/100 | 4/38 | 0.75 | 0.90 |
| Negative | 25/100 | 34/38 | |||
| Asymptomatic (Case, n = 7; Control, n = 38) | Positive | 4/7 | 4/38 | 0.57 | 0.90 |
| Negative | 3/7 | 34/38 | |||
| Mild (Case, n = 43; Control, n = 38) | Positive | 25/43 | 4/38 | 0.58 | 0.90 |
| Negative | 18/43 | 34/38 | |||
| Severe (Case, n = 50, Control, n = 38) | Positive | 46/50 | 4/38 | 0.92 | 0.90 |
| Negative | 4/50 | 34/38 | |||
| Characteristic | Number of Individuals in Cohort | Percentage of Cohort (%) | Percentage of Qatari Population (%) |
|---|---|---|---|
| Ethnic Group | |||
| Middle Eastern (Other) | 10 | 10 | 18.35 |
| Middle Eastern (Qatari) | 15 | 15 | 10.50 |
| South Asian | 70 | 70 | 64.32 |
| Gender | |||
| Male | 91 | 91 | 72.90 |
| Female | 9 | 9 | 27.10 |
| Age Group | |||
| 18–40 | 43 | 43 | 69.44 |
| 41–50 | 24 | 24 | 19.82 |
| 51–60 | 27 | 27 | 7.76 |
| >60 | 6 | 6 | 2.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smith, M.; Abdesselem, H.B.; Mullins, M.; Tan, T.-M.; Nel, A.J.M.; Al-Nesf, M.A.Y.; Bensmail, I.; Majbour, N.K.; Vaikath, N.N.; Naik, A.; et al. Age, Disease Severity and Ethnicity Influence Humoral Responses in a Multi-Ethnic COVID-19 Cohort. Viruses 2021, 13, 786. https://doi.org/10.3390/v13050786
Smith M, Abdesselem HB, Mullins M, Tan T-M, Nel AJM, Al-Nesf MAY, Bensmail I, Majbour NK, Vaikath NN, Naik A, et al. Age, Disease Severity and Ethnicity Influence Humoral Responses in a Multi-Ethnic COVID-19 Cohort. Viruses. 2021; 13(5):786. https://doi.org/10.3390/v13050786
Chicago/Turabian StyleSmith, Muneerah, Houari B. Abdesselem, Michelle Mullins, Ti-Myen Tan, Andrew J. M. Nel, Maryam A. Y. Al-Nesf, Ilham Bensmail, Nour K. Majbour, Nishant N. Vaikath, Adviti Naik, and et al. 2021. "Age, Disease Severity and Ethnicity Influence Humoral Responses in a Multi-Ethnic COVID-19 Cohort" Viruses 13, no. 5: 786. https://doi.org/10.3390/v13050786
APA StyleSmith, M., Abdesselem, H. B., Mullins, M., Tan, T.-M., Nel, A. J. M., Al-Nesf, M. A. Y., Bensmail, I., Majbour, N. K., Vaikath, N. N., Naik, A., Ouararhni, K., Mohamed-Ali, V., Al-Maadheed, M., Schell, D. T., Baros-Steyl, S. S., Anuar, N. D., Ismail, N. H., Morris, P. E., Mamat, R. N. R., ... Blackburn, J. M. (2021). Age, Disease Severity and Ethnicity Influence Humoral Responses in a Multi-Ethnic COVID-19 Cohort. Viruses, 13(5), 786. https://doi.org/10.3390/v13050786