
ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics

 , , , , , , , , , , , , , , , , and
, , , , , , , , , , , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
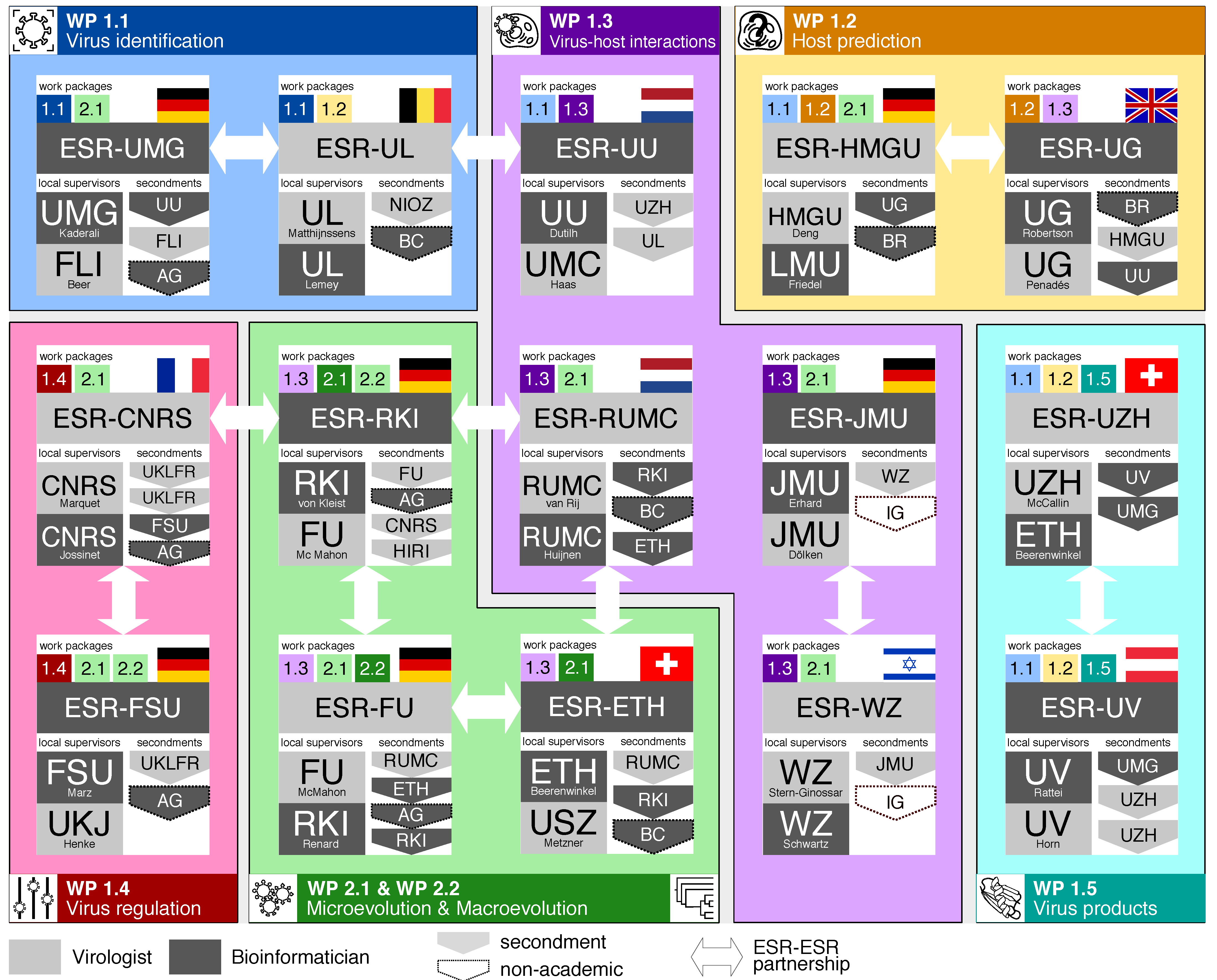
2. The Research Programme
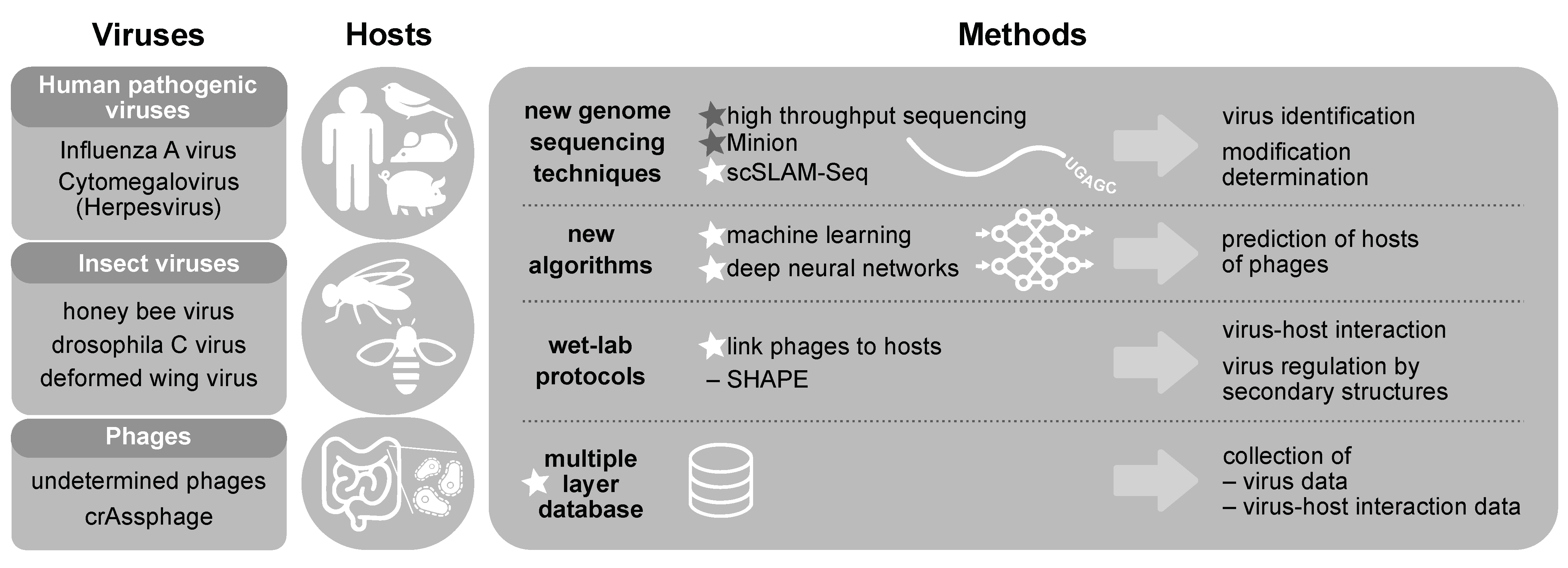
3. Modelling Virus-Host Interactions
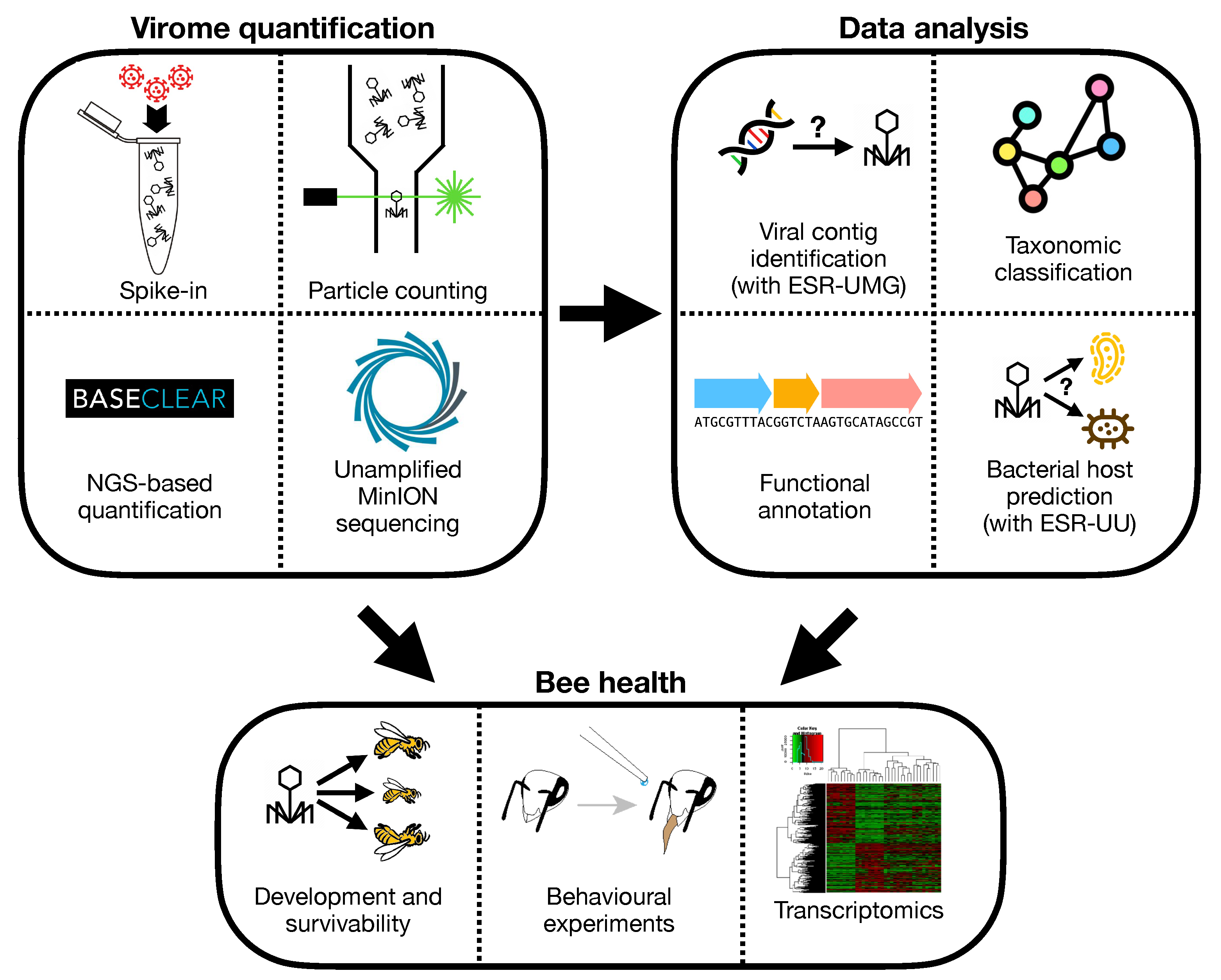
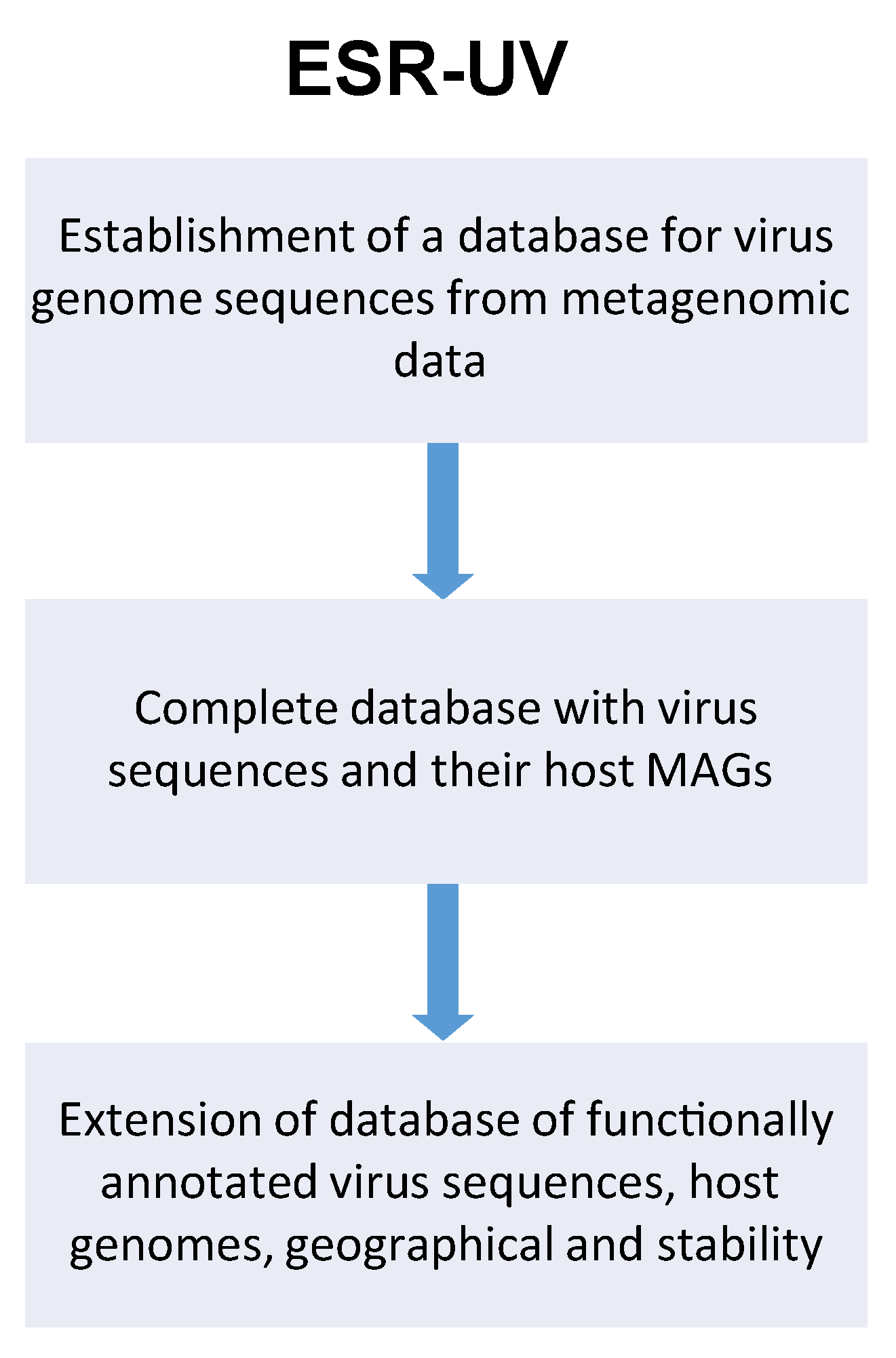
3.1. Virus Identification
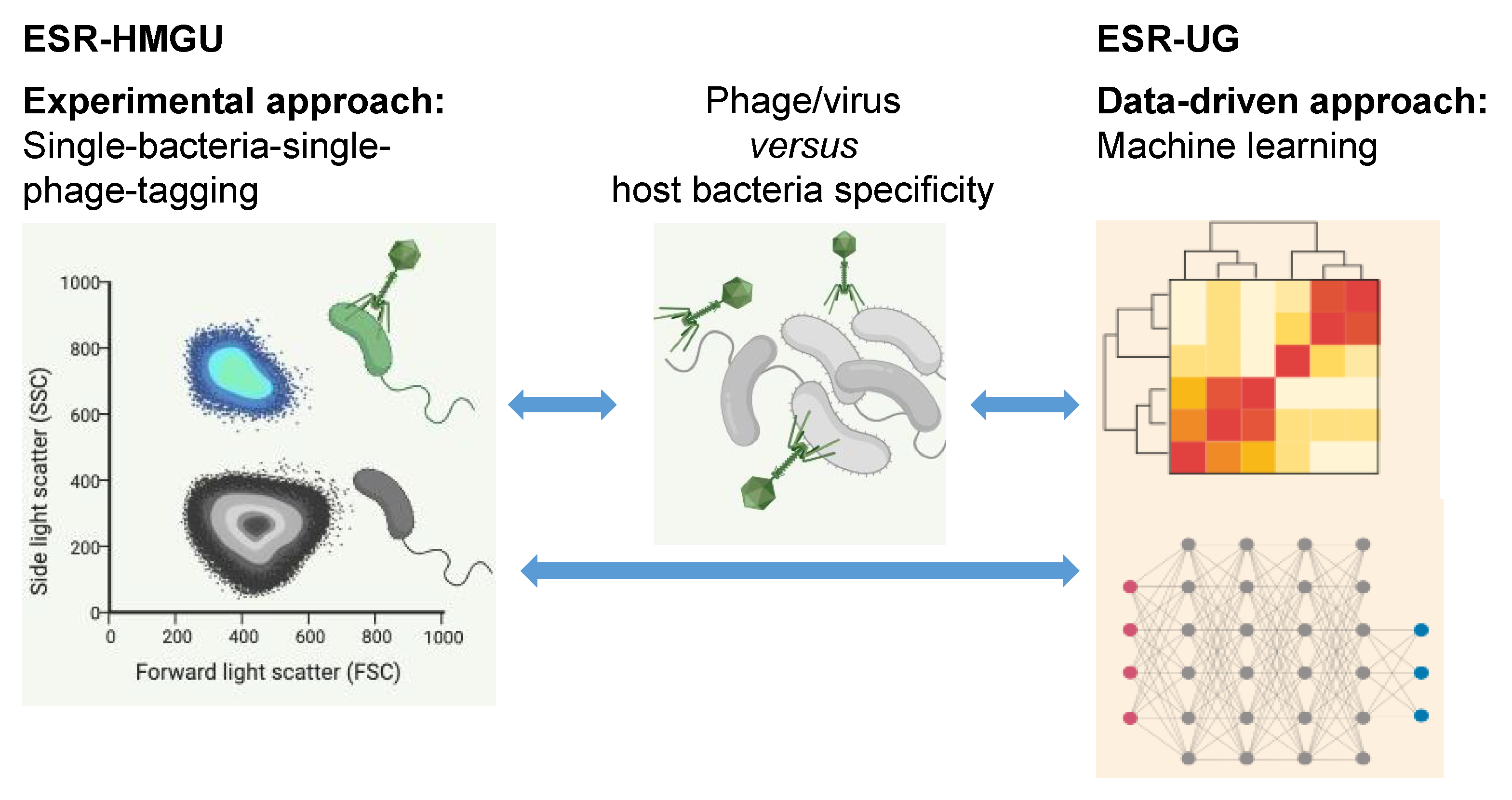
3.2. Host Prediction
3.3. Virus-Host Interactions
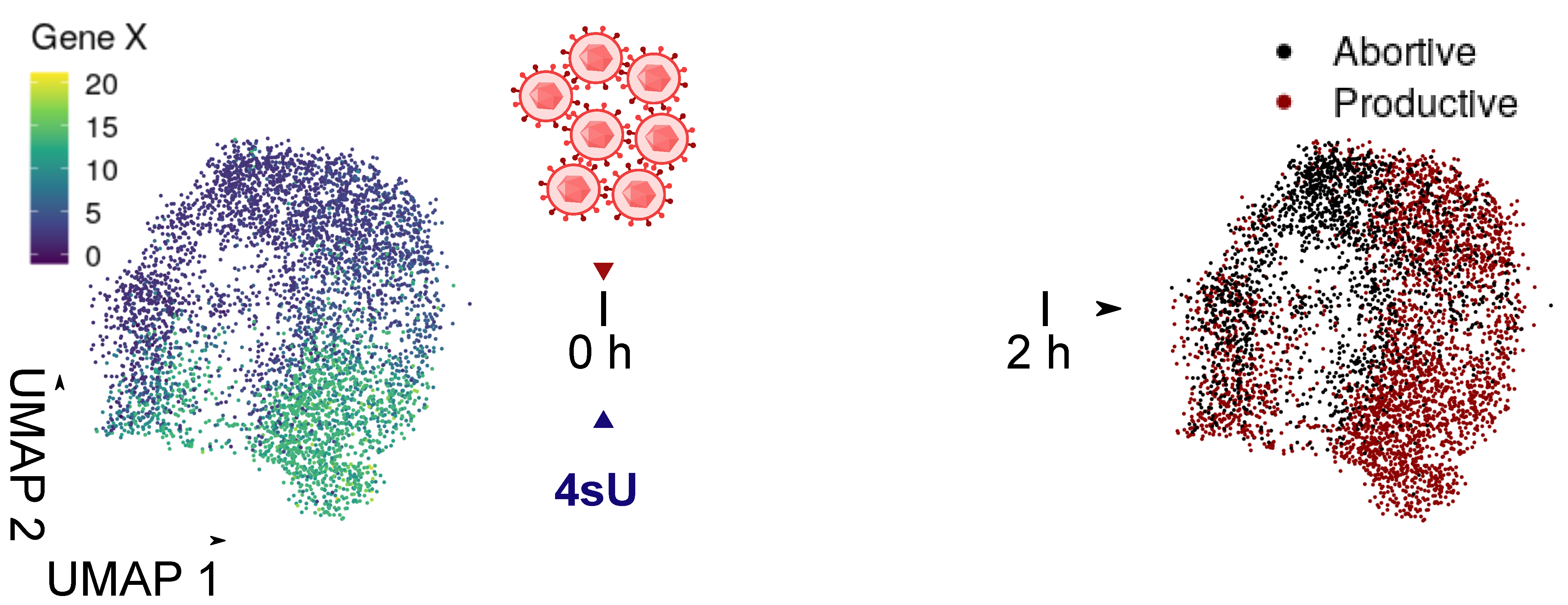
3.4. Virus Regulation
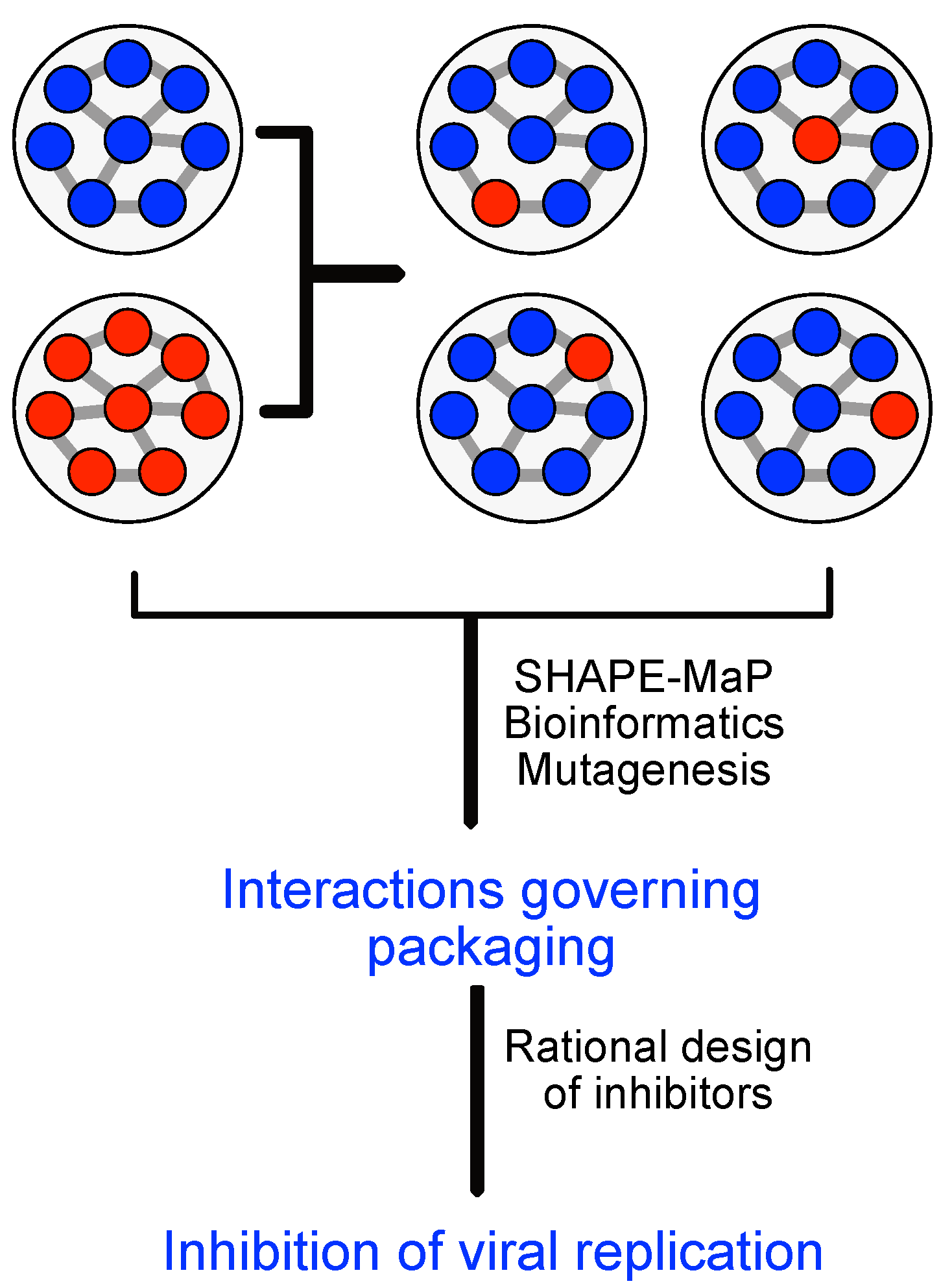
3.5. Virus Products
4. Modelling Virus Evolution in Hosts
4.1. Microevolution: Virus Quasispecies
4.2. Natural Selection of Viruses
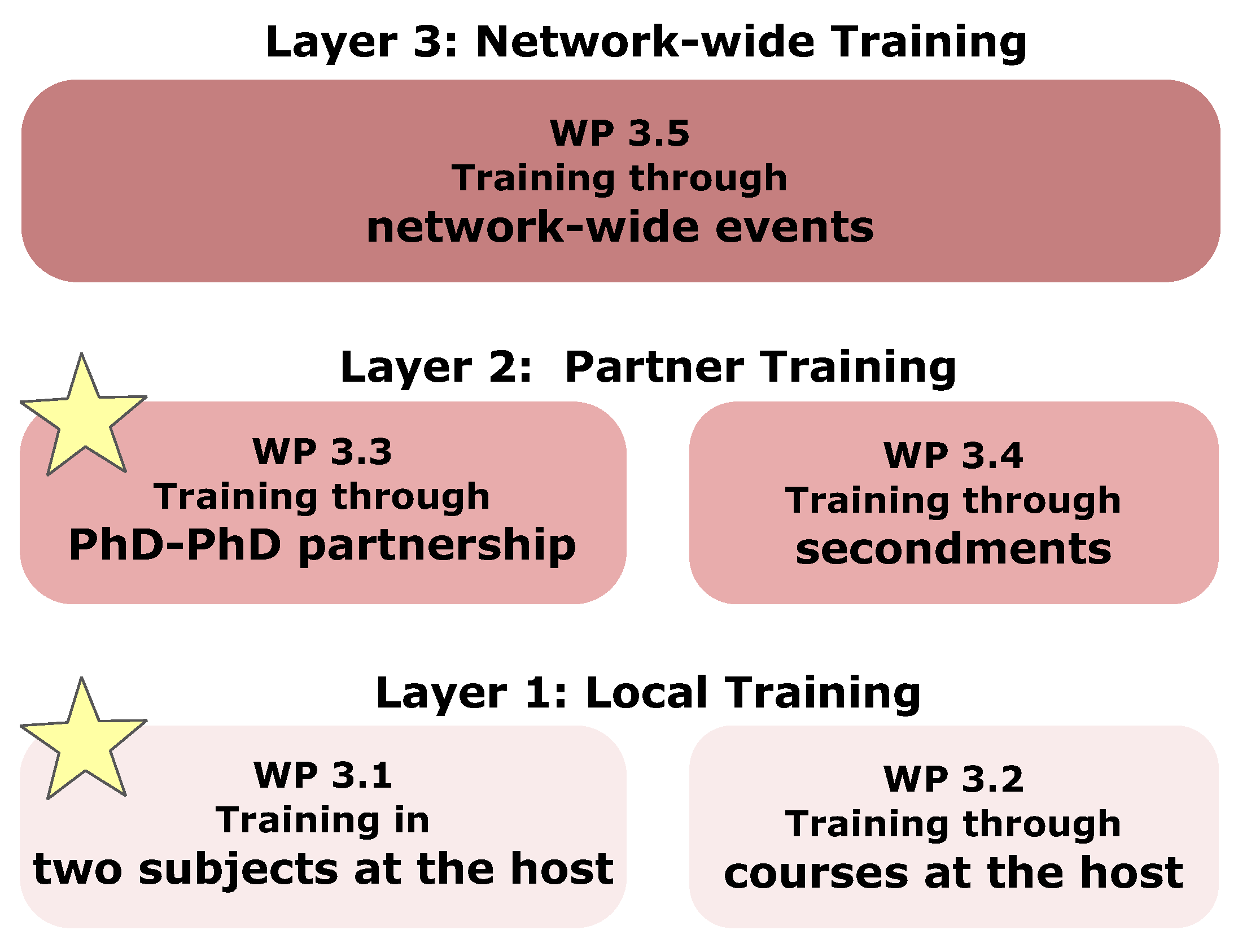
5. Training
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hufsky, F.; Ibrahim, B.; Modha, S.; Clokie, M.R.J.; Deinhardt-Emmer, S.; Dutilh, B.E.; Lycett, S.; Simmonds, P.; Thiel, V.; Abroi, A.; et al. The Third Annual Meeting of the European Virus Bioinformatics Center. Viruses 2019, 11, 420. [Google Scholar] [CrossRef]
- Ibrahim, B.; Arkhipova, K.; Andeweg, A.C.; Posada-Cespedes, S.; Enault, F.; Gruber, A.; Koonin, E.V.; Kupczok, A.; Lemey, P.; McHardy, A.C.; et al. Bioinformatics Meets Virology: The European Virus Bioinformatics Center’s Second Annual Meeting. Viruses 2018, 10, 256. [Google Scholar] [CrossRef]
- Marz, M.; Beerenwinkel, N.; Drosten, C.; Fricke, M.; Frishman, D.; Hofacker, I.L.; Hoffmann, D.; Middendorf, M.; Rattei, T.; Stadler, P.F.; et al. Challenges in RNA virus bioinformatics. Bioinformatics 2014. [Google Scholar] [CrossRef] [PubMed]
- Conceicao-Neto, N.; Yinda, K.C.; Van Ranst, M.; Matthijnssens, J. NetoVIR: Modular Approach to Customize Sample Preparation Procedures for Viral Metagenomics. Methods Mol. Biol. 2018, 1838, 85–95. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Börold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Prangishvili, D.; Hendrix, R.W.; Bamford, D.H. Genomics of bacterial and archaeal viruses: Dynamics within the prokaryotic virosphere. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 610–635. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ignacio-Espinoza, J.C.; Gregory, A.C.; Poulos, B.T.; Weitz, J.S.; Hugenholtz, P.; Sullivan, M.B. Viral tagging reveals discrete populations in Synechococcus viral genome sequence space. Nature 2014, 513, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef]
- Khan Mirzaei, M.; Xue, J.; Costa, R.; Ru, J.; Schulz, S.; Taranu, Z.E.; Deng, L. Challenges of Studying the Human Virome—Relevant Emerging Technologies. Trends Microbiol. 2020. [Google Scholar] [CrossRef]
- Deng, L.; Gregory, A.; Yilmaz, S.; Poulos, B.T.; Hugenholtz, P.; Sullivan, M.B. Contrasting Life Strategies of Viruses that Infect Photo- and Heterotrophic Bacteria, as Revealed by Viral Tagging. mBio 2012, 3. [Google Scholar] [CrossRef]
- Dzunkova, M.; Low, S.J.; Daly, J.N.; Deng, L.; Rinke, C.; Hugenholtz, P. Defining the human gut host-phage network through single-cell viral tagging. Nat. Microbiol. 2019, 4, 2192–2203. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.Z.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Fitzgerald, C.B.; Stockdale, S.R.; Draper, L.A.; Ross, R.P.; Hill, C. ΦCrAss001 represents the most abundant bacteriophage family in the human gut and infects Bacteroides intestinalis. Nat. Commun. 2018, 9, 4781. [Google Scholar] [CrossRef] [PubMed]
- Shkoporov, A.N.; Stockdale, S.R.; Adriaenssens, E.M.; Yutin, N.; Koonin, E.; Dutilh, B.E.; Krupovic, M.; Edwards, R.A.; Tolstoy, I.; Hill, C. Create One New Order (Crassvirales) Including Six New Families, Ten New Subfamilies, 78 New Genera and 279 New Species. ICTV Taxonomy Proposal 2020.039B. 2020. Available online: https://talk.ictvonline.org/files/proposals/taxonomy_proposals_prokaryote1 (accessed on 15 April 2021).
- Yutin, N.; Benler, S.; Shmakov, S.A.; Wolf, Y.I.; Tolstoy, I.; Rayko, M.; Antipov, D.; Pevzner, P.A.; Koonin, E.V. Unique genomic features of crAss-like phages, the dominant component of the human gut virome. bioRxiv 2020. [Google Scholar] [CrossRef]
- Erhard, F.; Baptista, M.A.P.; Krammer, T.; Hennig, T.; Lange, M.; Arampatzi, P.; Jurges, C.S.; Theis, F.J.; Saliba, A.E.; Dolken, L. scSLAM-seq reveals core features of transcription dynamics in single cells. Nature 2019, 571, 419–423. [Google Scholar] [CrossRef]
- Jürges, C.; Dölken, L.; Erhard, F. Dissecting newly transcribed and old RNA using GRAND-SLAM. Bioinformatics 2018, 34, i218–i226. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.W.; van Rij, R.P. The long and short of antiviral defense: Small RNA-based immunity in insects. Curr. Opin. Virol. 2014, 7, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.H.; Hadfield, J.D.; Obbard, D.J. RNA-Interference Pathways Display High Rates of Adaptive Protein Evolution in Multiple Invertebrates. Genetics 2018, 208, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- Obbard, D.J.; Linton, Y.M.; Jiggins, F.M.; Yan, G.; Little, T.J. Population genetics of Plasmodium resistance genes in Anopheles gambiae: No evidence for strong selection. Mol. Ecol. 2007, 16, 3497–3510. [Google Scholar] [CrossRef]
- van Rij, R.P.; Saleh, M.C.; Berry, B.; Foo, C.; Houk, A.; Antoniewski, C.; Andino, R. The RNA silencing endonuclease Argonaute 2 mediates specific antiviral immunity in Drosophila melanogaster. Genes Dev. 2006, 20, 2985–2995. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, J.T.; Bronkhorst, A.W.; Overheul, G.J.; Sadanandan, S.A.; Ekstrom, J.O.; Heestermans, M.; Hultmark, D.; Antoniewski, C.; van Rij, R.P. Convergent evolution of argonaute-2 slicer antagonism in two distinct insect RNA viruses. PLoS Pathog. 2012, 8, e1002872. [Google Scholar] [CrossRef] [PubMed]
- Desirò, D.; Hölzer, M.; Ibrahim, B.; Marz, M. SilentMutations (SIM): A tool for analyzing long-range RNA-RNA interactions in viral genomes and structured RNAs. Virus Res. 2019, 260, 135–141. [Google Scholar] [CrossRef]
- Li, A.X.; Marz, M.; Qin, J.; Reidys, C.M. RNA-RNA interaction prediction based on multiple sequence alignments. Bioinformatics 2011, 27, 456–463. [Google Scholar] [CrossRef]
- Fancello, L.; Raoult, D.; Desnues, C. Computational tools for viral metagenomics and their application in clinical research. Virology 2012, 434, 162–174. [Google Scholar] [CrossRef]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.S.; Mohammed, M.H.; Komanduri, D.; Mande, S.S. ProViDE: A software tool for accurate estimation of viral diversity in metagenomic samples. Bioinformation 2011, 6, 91–94. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palu, G. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef]
- Smyth, R.P.; Despons, L.; Huili, G.; Bernacchi, S.; Hijnen, M.; Mak, J.; Jossinet, F.; Weixi, L.; Paillart, J.C.; von Kleist, M.; et al. Mutational interference mapping experiment (MIME) for studying RNA structure and function. Nat. Methods 2015, 12, 866–872. [Google Scholar] [CrossRef]
- Smyth, R.P.; Smith, M.R.; Jousset, A.C.; Despons, L.; Laumond, G.; Decoville, T.; Cattenoz, P.; Moog, C.; Jossinet, F.; Mougel, M.; et al. In cell mutational interference mapping experiment (in cell MIME) identifies the 5’ polyadenylation signal as a dual regulator of HIV-1 genomic RNA production and packaging. Nucleic Acids Res. 2018, 46, e57. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Smyth, R.P.; Marquet, R.; von Kleist, M. MIMEAnTo: Profiling functional RNA in mutational interference mapping experiments. Bioinformatics 2016, 32, 3369–3370. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.P.; Wilfert, L.; Paxton, R.J.; Brown, M.J.F. Emerging Viruses in Bees: From Molecules to Ecology. Adv. Virus Res. 2018, 101, 251–291. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goettsch, W.; Beerenwinkel, N.; Deng, L.; Dölken, L.; Dutilh, B.E.; Erhard, F.; Kaderali, L.; von Kleist, M.; Marquet, R.; Matthijnssens, J.; et al. ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics. Viruses 2021, 13, 766. https://doi.org/10.3390/v13050766
Goettsch W, Beerenwinkel N, Deng L, Dölken L, Dutilh BE, Erhard F, Kaderali L, von Kleist M, Marquet R, Matthijnssens J, et al. ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics. Viruses. 2021; 13(5):766. https://doi.org/10.3390/v13050766
Chicago/Turabian StyleGoettsch, Winfried, Niko Beerenwinkel, Li Deng, Lars Dölken, Bas E. Dutilh, Florian Erhard, Lars Kaderali, Max von Kleist, Roland Marquet, Jelle Matthijnssens, and et al. 2021. "ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics" Viruses 13, no. 5: 766. https://doi.org/10.3390/v13050766
APA StyleGoettsch, W., Beerenwinkel, N., Deng, L., Dölken, L., Dutilh, B. E., Erhard, F., Kaderali, L., von Kleist, M., Marquet, R., Matthijnssens, J., McCallin, S., McMahon, D., Rattei, T., Van Rij, R. P., Robertson, D. L., Schwemmle, M., Stern-Ginossar, N., & Marz, M. (2021). ITN—VIROINF: Understanding (Harmful) Virus-Host Interactions by Linking Virology and Bioinformatics. Viruses, 13(5), 766. https://doi.org/10.3390/v13050766