
A Potential SARS-CoV-2 Variant of Interest (VOI) Harboring Mutation E484K in the Spike Protein Was Identified within Lineage B.1.1.33 Circulating in Brazil

 ,
,  , , , , ,
, , , , ,  , ,
, ,  , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
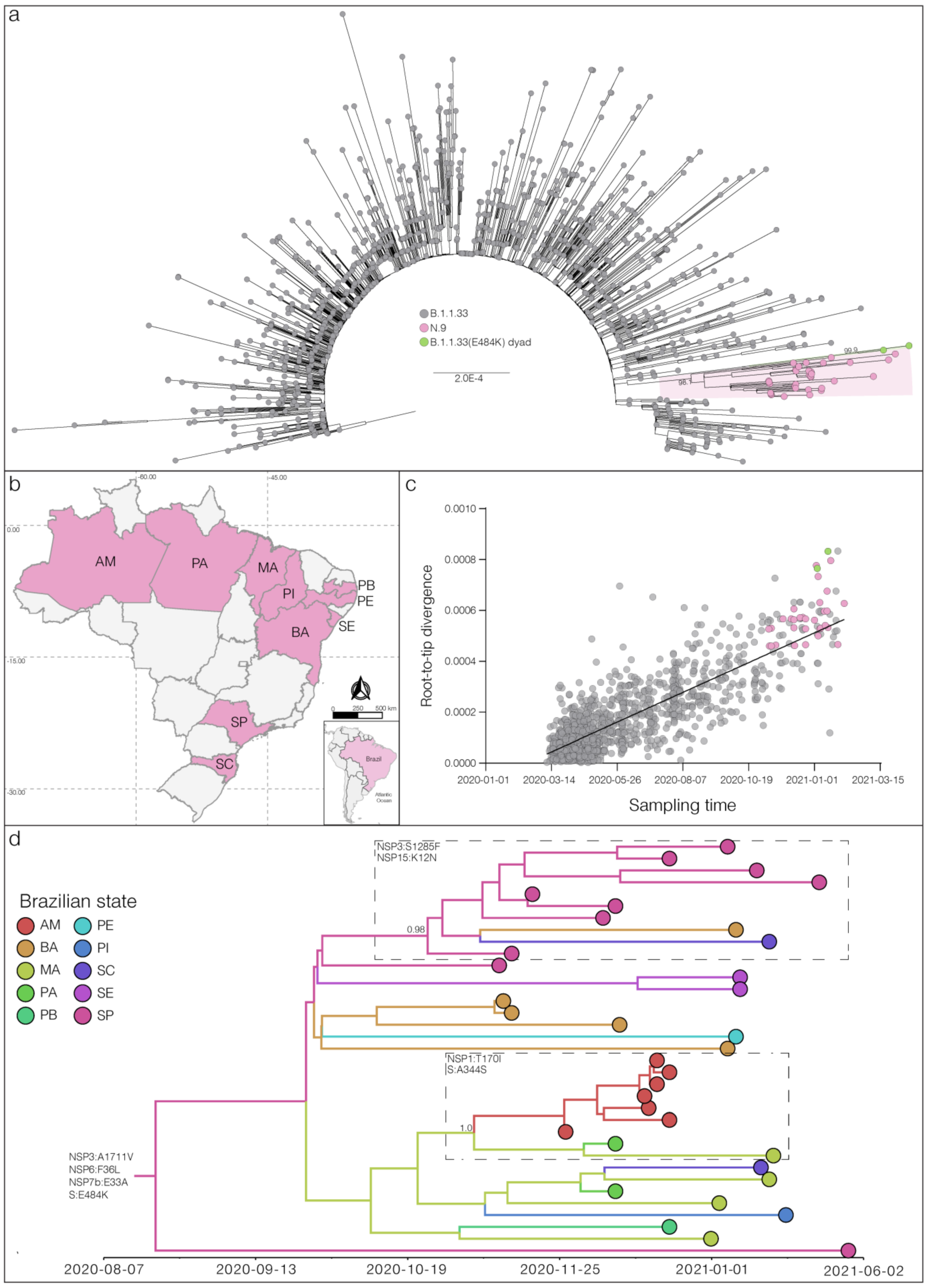
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimers
References
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.C.; Delatorre, E.; Graf, T.; Mir, D.; Motta, F.C.; Appolinario, L.R.; Dias da Paixao, A.C.; da Fonseca Mendonca, A.C.; Ogrzewalska, O.; Caetano, B.; et al. Evolutionary dynamics and dissemination pattern of the SARS-CoV-2 lineage B.1.1.33 during the early pandemic phase in Brazil. Front. Microbiol. 2021, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Nomoto, H.; Kutsuna, S.; Ujiie, M.; Suzuki, T.; Sato, R.; Fujimoto, T.; Kuroda, M.; Wakita, T.; Ohmagari, N. Novel SARS-CoV-2 variant identified in travelers from Brazil to Japan. Emerg. Infect. Dis. 2021, 27, 1243–1245. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimiento, F.; Silva, G.; Costa, A.; Duarte, D.; Pessoa, K.; Mejia, M.; et al. COVID-19 epidemic in the Brazilian state of Amazonas was driven by long-term persistence of endemic SARS-CoV-2 lineages and the recent emergence of the new Variant of Concern, P.1. Res. Sq 2021. preprint. Available online: https://www.researchsquare.com/article/rs-275494/v1 (accessed on 1 March 2021).
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; da Silva Candido, D.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerder, A.L.; Guimaraes, A.P.C.; Mariani, D.; da Costa, R.M.; Ferreisa, O.C., Jr.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, V.A.; de Lima Guerra Corado, A.; do Nascimento, F.O.; da Costa, A.K.A.; Gomes Duarte, D.C.; Bessa Luz, S.L.; Goncalves, L.M.F.; de Jesus, M.S.; da Costa, C.F.; Delatorre, E.; et al. Genomic and phylogenetic characterization of an imported case of SARS-CoV-2 in Amazonas State, Brazil. Mem. Inst. Oswaldo Cruz 2020, 115, e200310. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.C.; Motta, F.C.; Roy, S.; Applinario, L.; Fabri, A.; Xavier, J.; Harris, K.; Matos, A.R.; Caetano, B.; Orgeswalska, M.; et al. SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. bioRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schretmpf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; Von Haetseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, L.E.; Russo, V.; Giordano, S.; Lanza, K.; Negrom, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, D.K.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, P463–P476.E6. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liu, L.; Iketani, S.; Nair, M.S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Increased resistance of SARS-CoV-2 variants, B.1.351 and B.1.1.7 to antibody neutralization. bioRxiv 2021. preprint. [Google Scholar] [CrossRef]
- Gangavarapu, K.; Alkuzweny, M.; Cano, M.; Andersen, K.; Haag, E.; Hughes, L.; Mullen, J.; Su, A.; Latif, A.A.; Tsueng, G.; et al. outbreak.info. 2020. Available online: https://outbreak.info (accessed on 1 March 2021).
- Martin, D.P.; Weaver, S.; Tegally, H.; San, E.L.; Shank, S.D.; Wilkinson, E.; Giandhari, J.; Naidoo, S.; Pullay, Y.; Singh, L.; et al. The emergence and ongoing convergent evolution of the N501Y lineages coincides with a major global shift in the SARS-CoV-2 selective landscape. medRxiv 2021. preprint. [Google Scholar] [CrossRef]


{kind=link}
| Genomic Region (Protein) | Nucleotide | Amino Acid |
|---|---|---|
| ORF1a | G1264T | - |
| ORF1a | C7600T | - |
| ORF1a (NSP3) | C7851T | A2529V (A1711V) |
| ORF1a (NSP6) | T11078C | F3605L (F36L) |
| Spike (S) | G23012A | E484K |
| ORF7b (NSP7b) | A27853C | E33A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Resende, P.C.; Gräf, T.; Paixão, A.C.D.; Appolinario, L.; Lopes, R.S.; Mendonça, A.C.d.F.; da Rocha, A.S.B.; Motta, F.C.; Neto, L.G.L.; Khouri, R.; et al. A Potential SARS-CoV-2 Variant of Interest (VOI) Harboring Mutation E484K in the Spike Protein Was Identified within Lineage B.1.1.33 Circulating in Brazil. Viruses 2021, 13, 724. https://doi.org/10.3390/v13050724
Resende PC, Gräf T, Paixão ACD, Appolinario L, Lopes RS, Mendonça ACdF, da Rocha ASB, Motta FC, Neto LGL, Khouri R, et al. A Potential SARS-CoV-2 Variant of Interest (VOI) Harboring Mutation E484K in the Spike Protein Was Identified within Lineage B.1.1.33 Circulating in Brazil. Viruses. 2021; 13(5):724. https://doi.org/10.3390/v13050724
Chicago/Turabian StyleResende, Paola Cristina, Tiago Gräf, Anna Carolina Dias Paixão, Luciana Appolinario, Renata Serrano Lopes, Ana Carolina da Fonseca Mendonça, Alice Sampaio Barreto da Rocha, Fernando Couto Motta, Lidio Gonçalves Lima Neto, Ricardo Khouri, and et al. 2021. "A Potential SARS-CoV-2 Variant of Interest (VOI) Harboring Mutation E484K in the Spike Protein Was Identified within Lineage B.1.1.33 Circulating in Brazil" Viruses 13, no. 5: 724. https://doi.org/10.3390/v13050724
APA StyleResende, P. C., Gräf, T., Paixão, A. C. D., Appolinario, L., Lopes, R. S., Mendonça, A. C. d. F., da Rocha, A. S. B., Motta, F. C., Neto, L. G. L., Khouri, R., de Oliveira, C. I., Santos-Muccillo, P., Bezerra, J. F., Teixeira, D. L. F., Riediger, I., Debur, M. d. C., Ribeiro-Rodrigues, R., Leite, A. B., do Santos, C. A., ... Siqueira, M. M. (2021). A Potential SARS-CoV-2 Variant of Interest (VOI) Harboring Mutation E484K in the Spike Protein Was Identified within Lineage B.1.1.33 Circulating in Brazil. Viruses, 13(5), 724. https://doi.org/10.3390/v13050724