
Genetic Variation in the Domain II, 3′ Untranslated Region of Human and Mosquito Derived Dengue Virus Strains in Sri Lanka



Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
2.2. Sample Collection
2.3. Laboratory Rearing of Ae. aegypti Mosquitoes
2.4. Mosquito Inoculation: Oral Infection of Laboratory-Infected Mosquitoes
2.5. Molecular Diagnosis of Human Serum and Mosquito Samples
2.6. Nucleotide Sequence Analysis
2.7. Phylogenetic Analysis
2.8. Secondary Structure Analysis
3. Results
3.1. Results of RT-PCR and Sequence-Based Serotype Analysis of the Human-Derived and Mosquito-Derived DENV Samples to Examine the Domain II, 3′ UTR
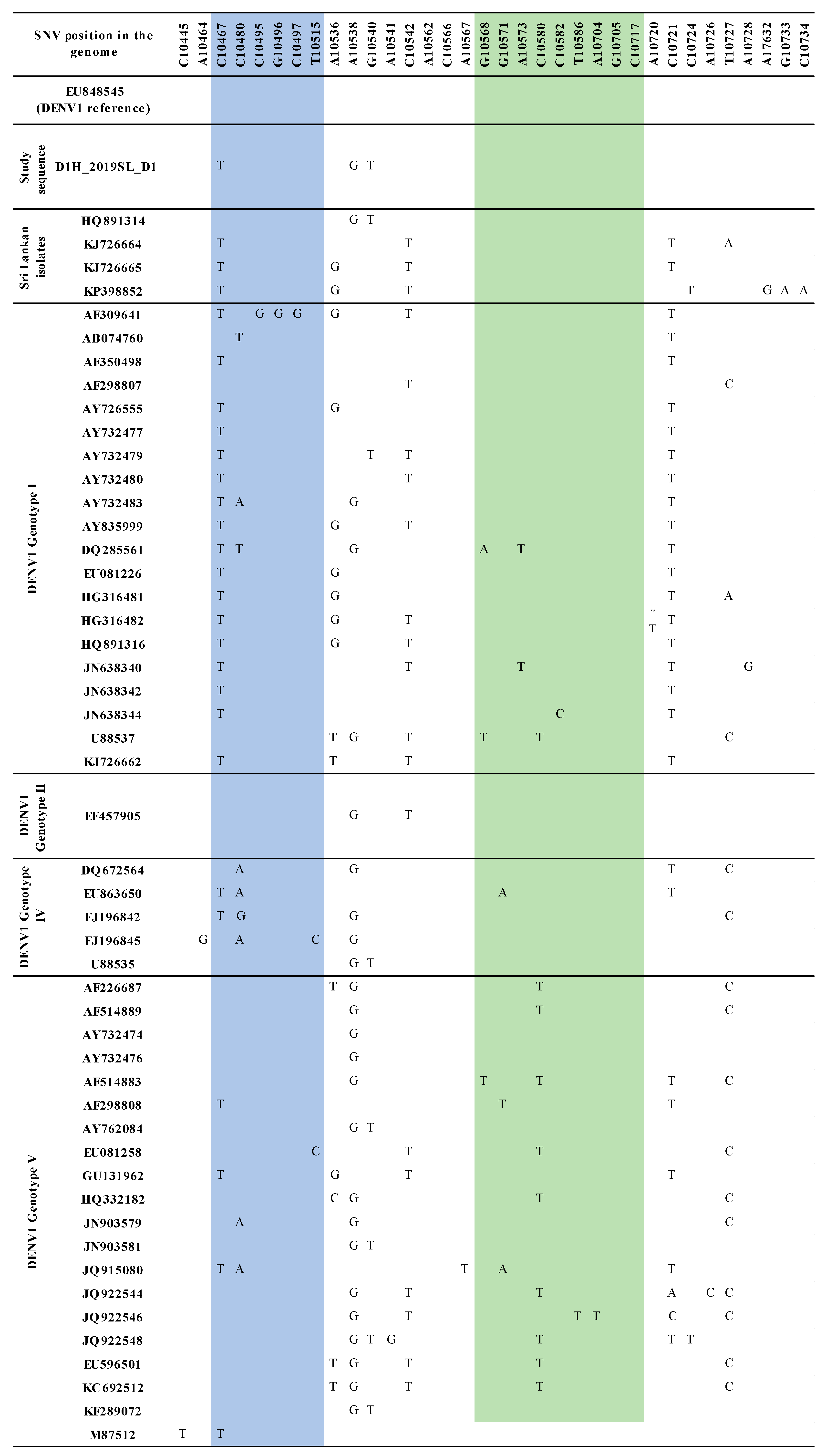
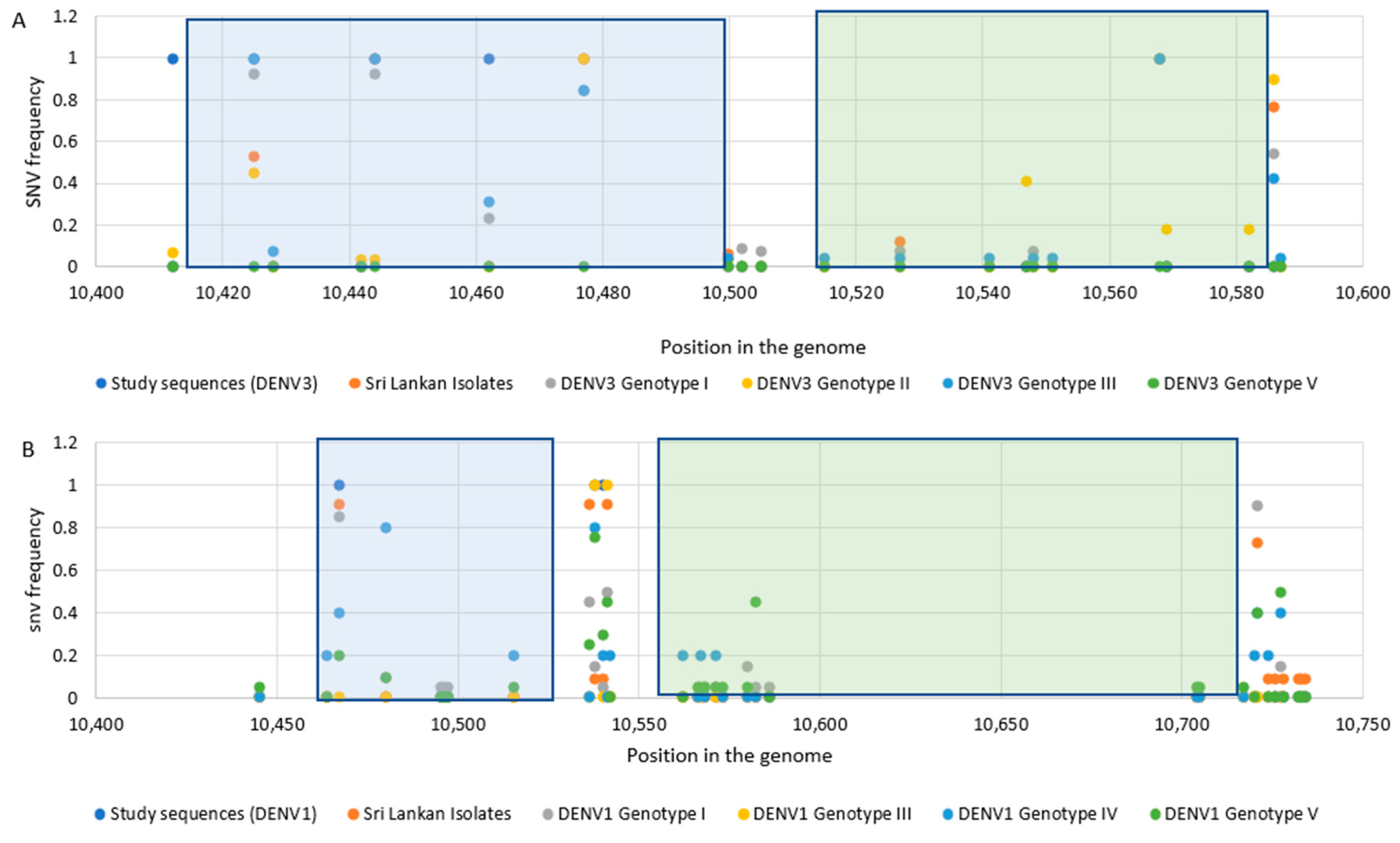
3.2. Sequence Analysis for Nucleotide Variation
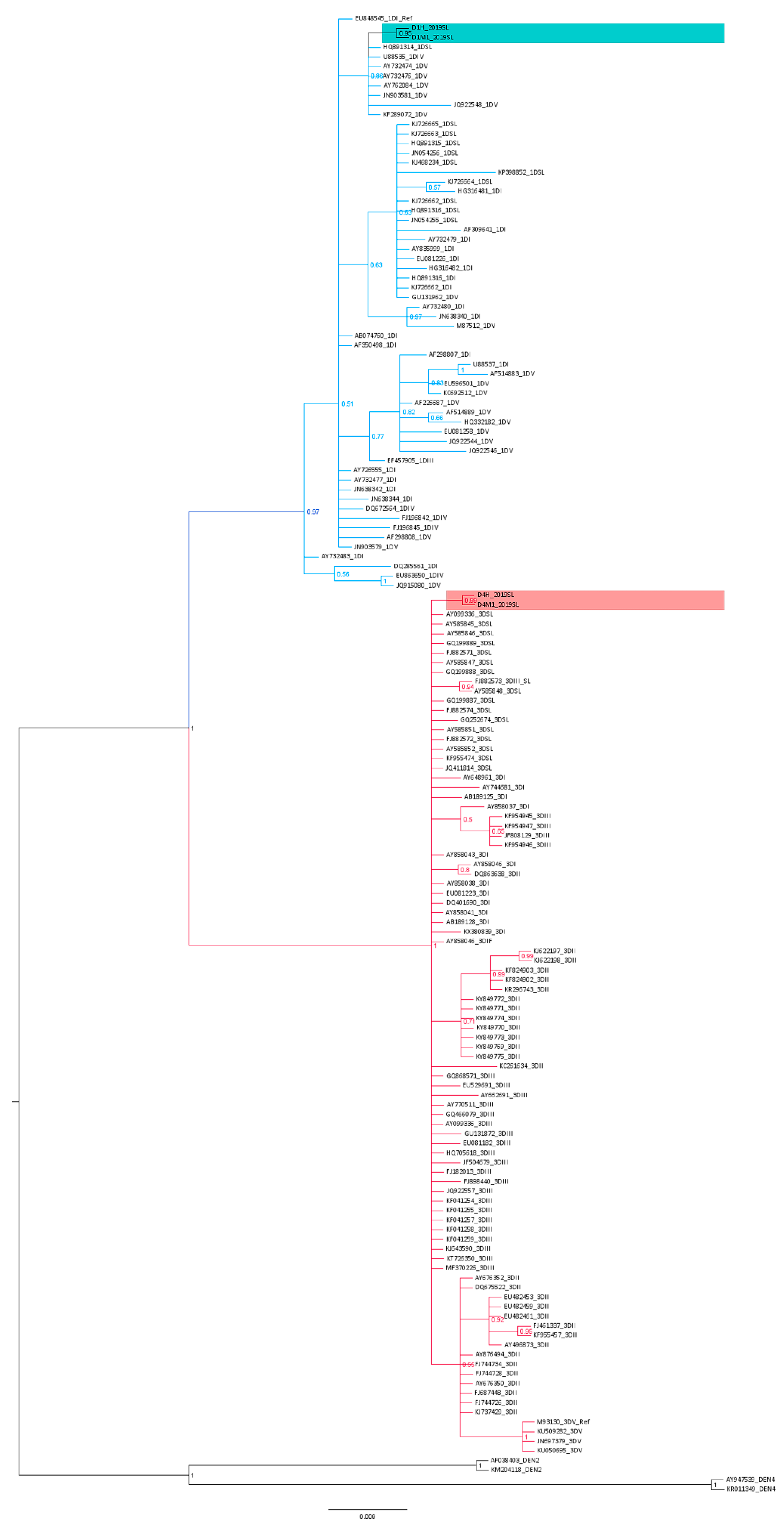
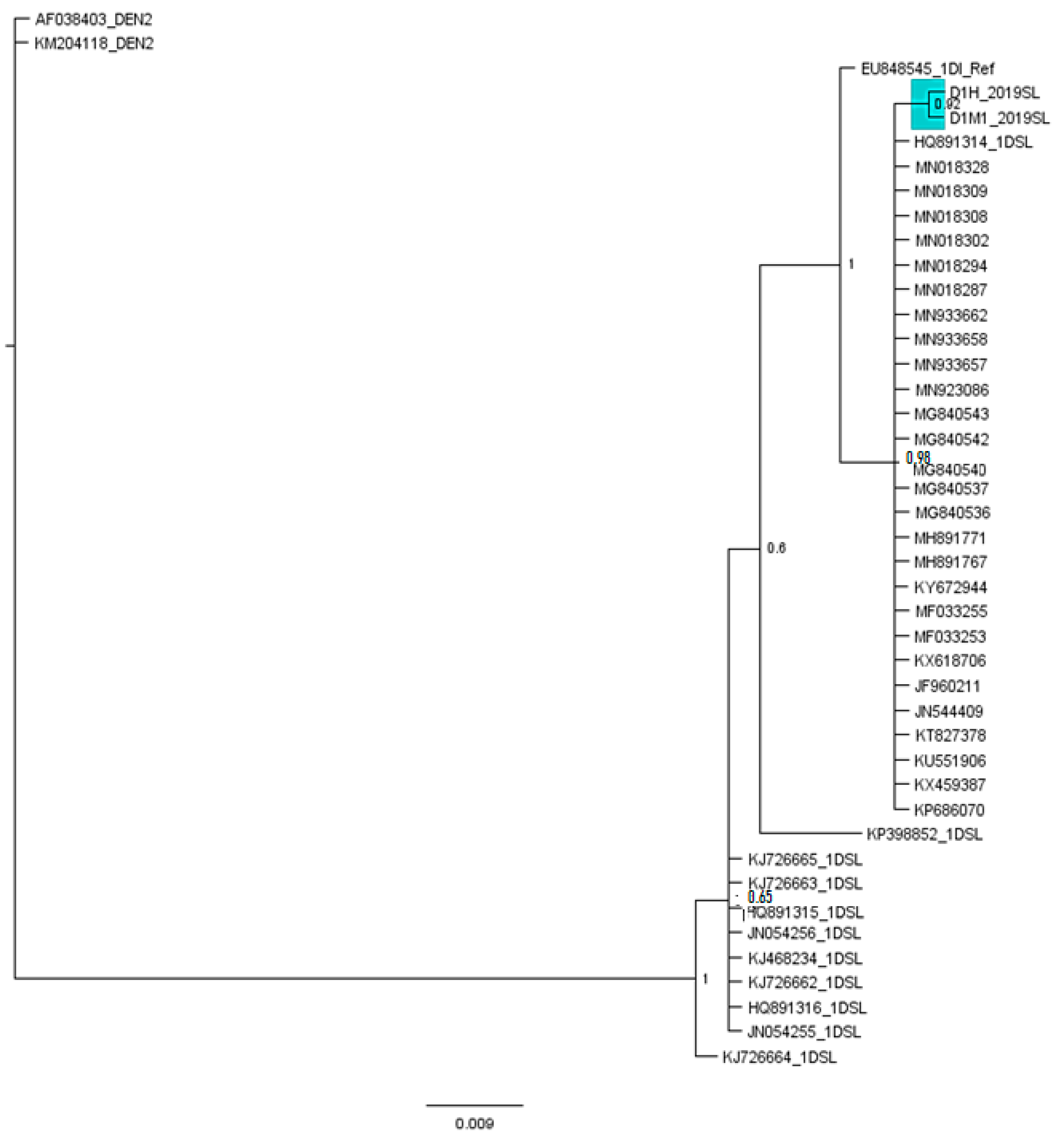
3.3. Phylogenetic Analysis
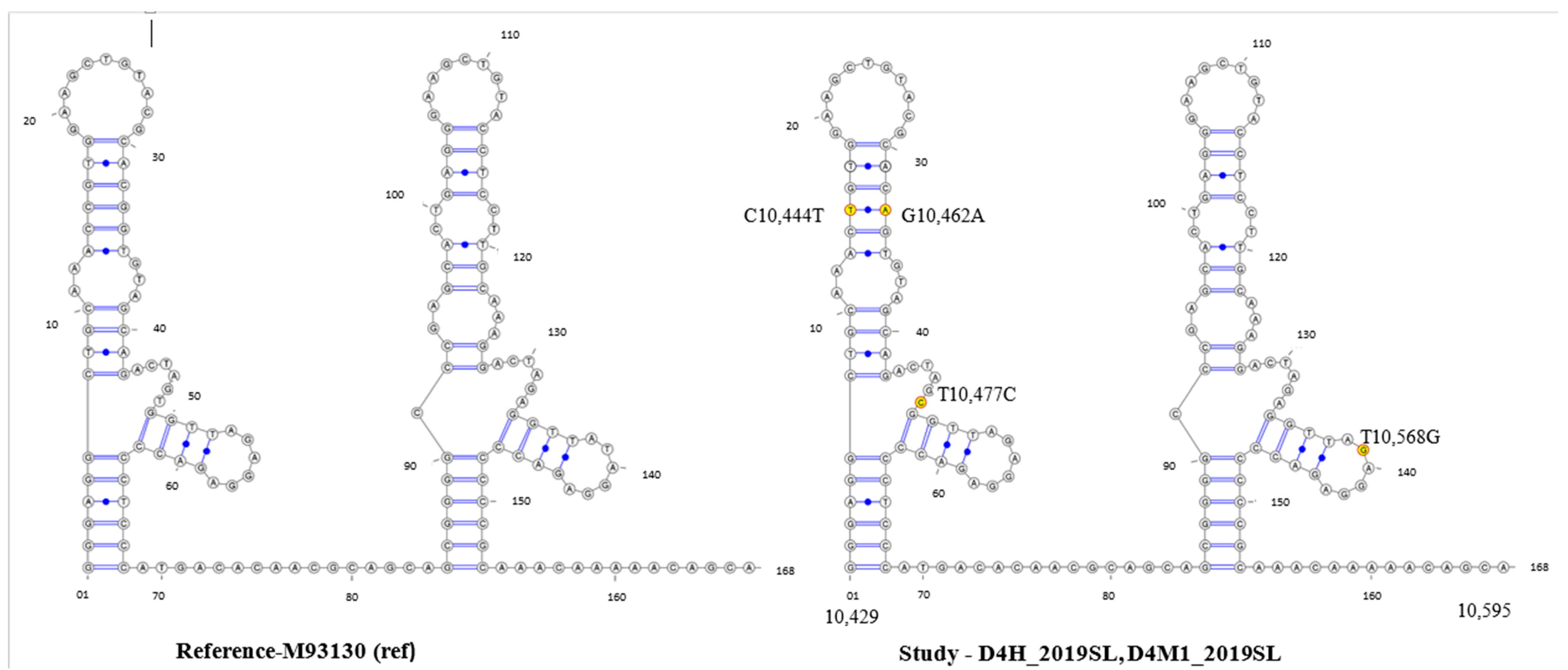
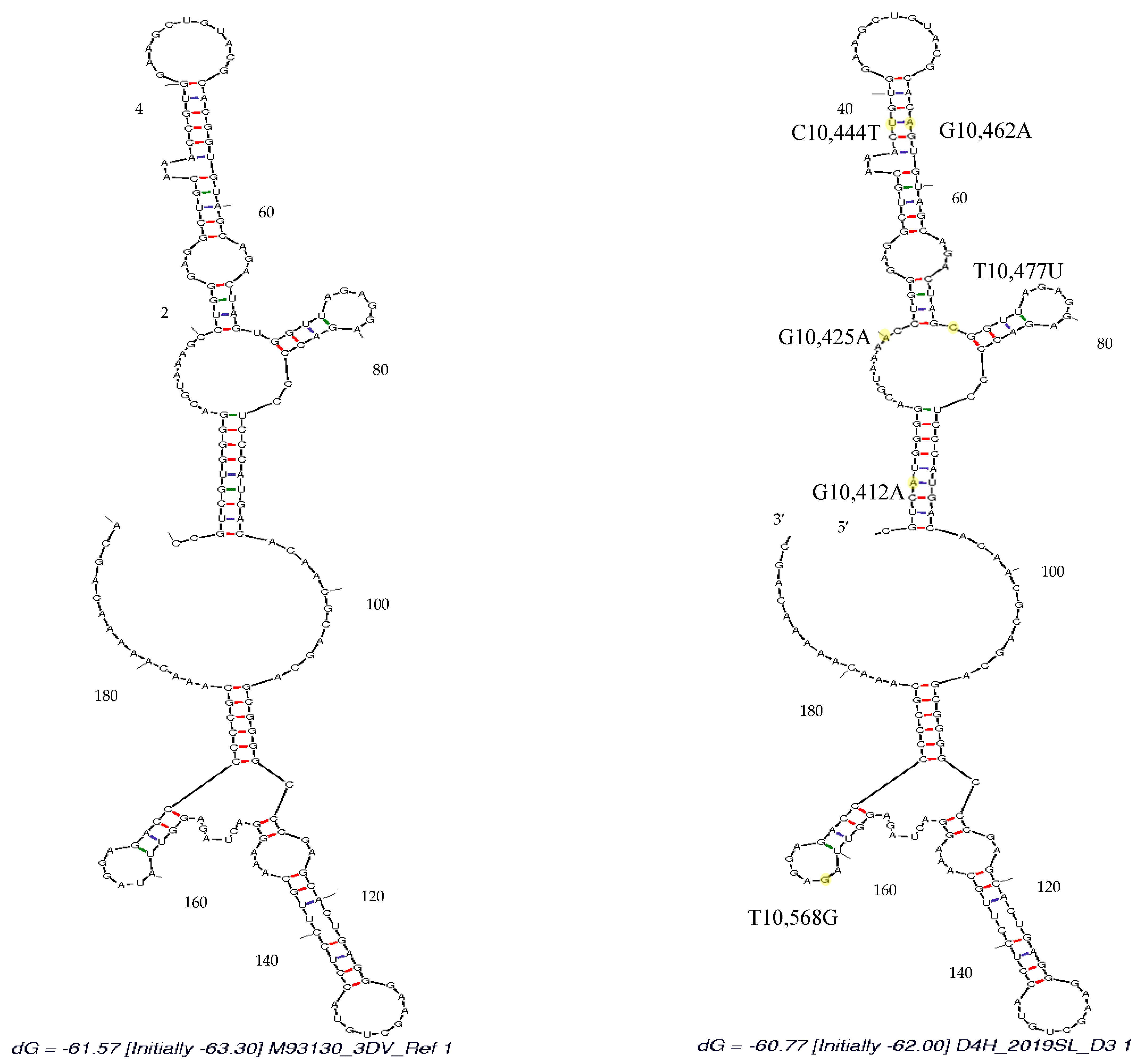
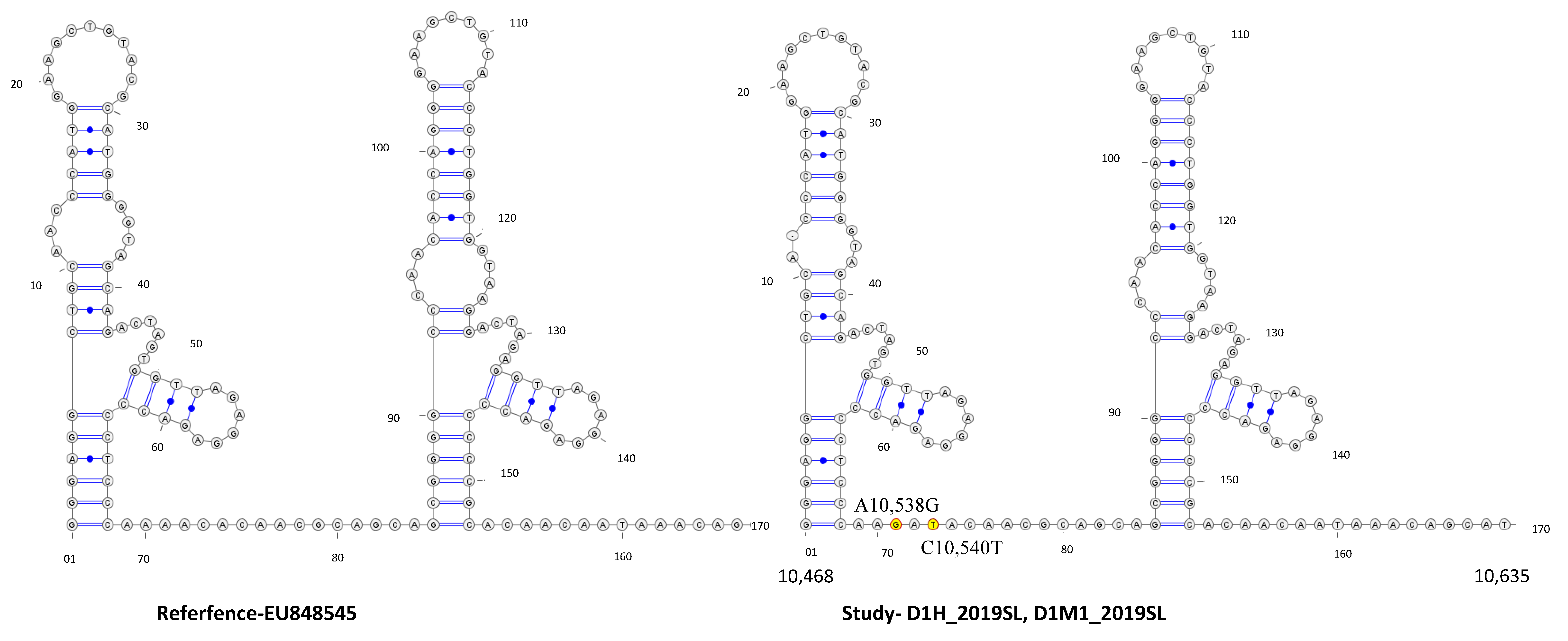
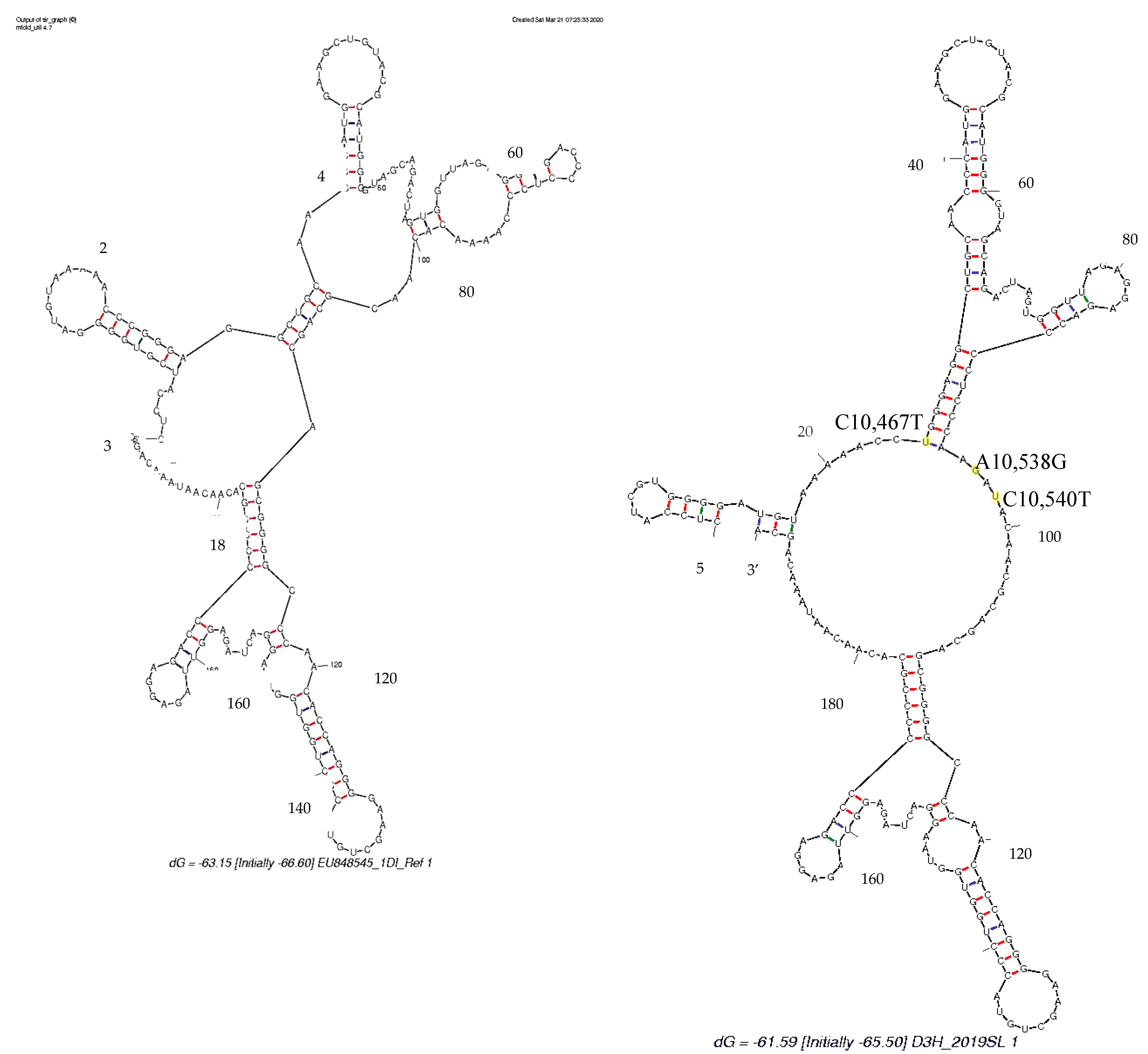
3.4. Secondary Structure Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holmes, E.C.; Twiddy, S.S. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 2003, 3, 19–28. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Shepard, D.S.; Undurraga, E.A.; Halasa, Y.A.; Coffeng, L.E.; Brady, O.J.; Hay, S.I.; Bedi, N.; Bensenor, I.M.; Castañeda-Orjuela, C.A.; et al. The global burden of dengue: An analysis from the Global Burden of Disease Study 2013. Lancet Infect. Dis. 2016, 16, 712–723. [Google Scholar] [CrossRef]
- James, S.; Abate, D.; Abate, K.; Abay, S.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 dis-eases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- OhAinle, M.; Balmaseda, A.; Macalalad, A.; Tellez, Y.; Zody, M.; Saborío, S.; Nuñez, A.; Lennon, N.; Birren, B.; Gordon, A.; et al. Dynamics of dengue disease severity determined by the interplay between viral genetics and sero-type-specific immunity. Sci. Transl. Med. 2011, 3, 114ra128. [Google Scholar] [CrossRef] [PubMed]
- Filomatori, C.V.; Carballeda, J.M.; Villordo, S.M.; Aguirre, S.; Pallarés, H.M.; Maestre, A.M.; Sánchez-Vargas, I.; Blair, C.D.; Fabri, C.; Morales, M.A.; et al. Dengue virus genomic variation associated with mosquito adaptation defines the pattern of viral non-coding RNAs and fitness in human cells. PLoS Pathog. 2017, 13, e1006265. [Google Scholar] [CrossRef]
- Gubler, D.; Clark, G. Dengue/dengue hemorrhagic fever: The emergence of a global health problem. Emerg. Infect. Dis. 1995, 1, 55. [Google Scholar] [CrossRef]
- Bennett, S.; Drummond, A.; Kapan, D.; Suchard, M.; Munoz-Jordan, J.; Pybus, O.; Holmes, E.; Gubler, D. Epidemic dynamics revealed in dengue evolution. Mol. Biol. Evol. 2010, 27, 811–818. [Google Scholar] [CrossRef]
- Yamanaka, A.; Mulyatno, K.C.; Susilowati, H.; Hendrianto, E.; Ginting, A.P.; Sary, D.D.; Rantam, F.A.; Soegijanto, S.; Konishi, E. Displacement of the Predominant Dengue Virus from Type 2 to Type 1 with a Subsequent Genotype Shift from IV to I in Surabaya, Indonesia 2008–2010. PLoS ONE 2011, 6, e27322. [Google Scholar] [CrossRef]
- Lambrechts, L.; Fansiri, T.; Pongsiri, A.; Thaisomboonsuk, B.; Klungthong, C.; Richardson, J.H.; Ponlawat, A.; Jarman, R.G.; Scott, T.W. Dengue-1 Virus Clade Replacement in Thailand Associated with Enhanced Mosquito Transmission. J. Virol. 2011, 86, 1853–1861. [Google Scholar] [CrossRef]
- Santiago, G.A.; McElroy-Horne, K.; Lennon, N.J.; Santiago, L.M.; Birren, B.W.; Henn, M.R.; Muñoz-Jordán, J.L. Reemergence and Decline of Dengue Virus Serotype 3 in Puerto Rico. J. Infect. Dis. 2012, 206, 893–901. [Google Scholar] [CrossRef]
- Drumond, B.P.; Mondini, A.; Schmidt, D.J.; Bronzoni, R.V.D.M.; Bosch, I.; Nogueira, M.L. Circulation of Different Lineages of Dengue Virus 2, Genotype American/Asian in Brazil: Dynamics and Molecular and Phylogenetic Characterization. PLoS ONE 2013, 8, e59422. [Google Scholar] [CrossRef] [PubMed]
- Messer, W.B.; Vitarana, U.T.; Elvtigala, J.; Sivananthan, K.; Preethimala, L.D.; Ramesh, R.; Gubler, D.J.; Withana, N.; De Silva, A.M. Epidemiology of dengue in Sri Lanka before and after the emergence of epidemic dengue hemorrhagic fever. Am. J. Trop. Med. Hyg. 2002, 66, 765–773. [Google Scholar] [CrossRef]
- Messer, W.; Gubler, D.; Harris, E.; Sivananthan, K.; De Silva, A. Emergence and global spread of a dengue sero-type 3, subtype III virus. Emerg. Infect. Dis. 2003, 9, 800. [Google Scholar] [CrossRef]
- Silva, R.; de Silva, A.; Harris, E.; MacDonald, G. Genetic analysis of Dengue 3 virus subtype III 5′ and 3′ non-coding regions. Virus Res. 2008, 135, 320–325. [Google Scholar] [CrossRef]
- Kanakaratne, N.; Wahala, W.M.; Messer, W.B.; Tissera, H.A.; Shahani, A.; Abeysinghe, N.; De Silva, A.M.; Gunasekera, M. Severe Dengue Epidemics in Sri Lanka, 2003–2006. Emerg. Infect. Dis. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Epidemiological Unit, Ministry of Health. Dengue Update. 2019. Available online: http://www.epid.gov.lk/web/index.php?option=com_content&view=article&id=171%3Adengue-update&catid=51%3Amessage-for-public&Itemid=487&lang=en (accessed on 30 January 2021).
- Rico-Hesse, R. Microevolution and virulence of dengue viruses. Adv. Appl. Microbiol. 2003, 59, 315–341. [Google Scholar] [CrossRef]
- Thu, H.M.; Lowry, K.; Jiang, L.; Hlaing, T.; Holmes, E.C.; Aaskov, J. Lineage extinction and replacement in den-gue type 1 virus populations are due to stochastic events rather than to natural selection. Virology 2005, 336, 163–172. [Google Scholar]
- Dash, P.K.; Sharma, S.; Soni, M.; Agarwal, A.; Sahni, A.K.; Parida, M. Complete genome sequencing and evolutionary phylogeography analysis of Indian isolates of Dengue virus type 1. Virus Res. 2015, 195, 124–134. [Google Scholar] [CrossRef]
- Finol, E.; Ooi, E. Evolution of subgenomic RNA shapes dengue virus adaptation and epidemiological fitness. iScience 2019, 16, 94–105. [Google Scholar] [CrossRef]
- Ng, W.; Soto-Acosta, R.; Bradrick, S.; Garcia-Blanco, M.; Ooi, E. The 5′ and 3′ untranslated regions of the flaviviral genome. Viruses 2017, 9, 137. [Google Scholar] [CrossRef]
- Alvarez, D.E.; Ezcurra, A.L.D.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef]
- Manzano, M.; Reichert, E.D.; Polo, S.; Falgout, B.; Kasprzak, W.; Shapiro, B.A.; Padmanabhan, R. Identification of Cis-Acting Elements in the 3′-Untranslated Region of the Dengue Virus Type 2 RNA That Modulate Translation and Replication. J. Biol. Chem. 2011, 286, 22521–22534. [Google Scholar] [CrossRef]
- De Borba, L.; Villordo, S.M.; Marsico, F.L.; Carballeda, J.M.; Filomatori, C.V.; Gebhard, L.G.; Pallarés, H.M.; Lequime, S.; Lambrechts, L.; Vargas, I.S.; et al. RNA Structure Duplication in the Dengue Virus 3′ UTR: Redundancy or Host Specificity? mBio 2019, 10, e02506-18. [Google Scholar] [CrossRef]
- Li, W.; Brinton, M. The 3′ stem loop of the West Nile virus genomic RNA can suppress translation of chimeric mRNAs. Virology 2001, 287, 49–61. [Google Scholar] [CrossRef]
- Villordo, S.M.; Alvarez, D.E.; Gamarnik, A.V. A balance between circular and linear forms of the dengue virus genome is crucial for viral replication. RNA 2010, 16, 2325–2335. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.N.; Holmes, E.C.; Chirivella, M.; Rodriguez, D.M.; Beltran, M.; Vorndam, V.; Gubler, D.J.; McMillan, W.O. Molecular evolution of dengue 2 virus in Puerto Rico: Positive selection in the viral envelope accompanies clade reintroduction. J. Gen. Virol. 2006, 87, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Pompon, J.; Manuel, M.; Ng, G.K.; Wong, B.; Shan, C.; Manokaran, G.; Soto-Acosta, R.; Bradrick, S.S.; Ooi, E.E.; Missé, D.; et al. Dengue subgenomic flaviviral RNA disrupts immunity in mosquito salivary glands to increase virus transmission. PLoS Pathog. 2017, 13, e1006535. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Filomatori, C.V.; Sánchez-Vargas, I.; Blair, C.D.; Gamarnik, A.V. Dengue Virus RNA Structure Specialization Facilitates Host Adaptation. PLoS Pathog. 2015, 11, e1004604. [Google Scholar] [CrossRef]
- Sessions, O.M.; Wilm, A.; Kamaraj, U.S.; Choy, M.M.; Chow, A.; Chong, Y.; Ong, X.M.; Nagarajan, N.; Cook, A.R.; Ooi, E.E. Analysis of Dengue Virus Genetic Diversity during Human and Mosquito Infection Reveals Genetic Constraints. PLoS Negl. Trop. Dis. 2015, 9, e0004044. [Google Scholar] [CrossRef] [PubMed]
- Villordo, S.M.; Carballeda, J.M.; Filomatori, C.V.; Gamarnik, A.V. RNA Structure Duplications and Flavivirus Host Adaptation. Trends Microbiol. 2016, 24, 270–283. [Google Scholar] [CrossRef]
- Clemons, A.; Mori, A.; Haugen, M.; Severson, D.; Duman-Scheel, M. Culturing and egg collection of Aedes aegypti. Cold Spring Harb. Protoc. 2010, 2010, pdb–rot5507. [Google Scholar] [CrossRef]
- Foggie, T.; Achee, N. Standard Operating Procedures: Rearing Aedes aegypti for the HITSS and Box Laboratory Assays. Vol 1 [pdf] Maryland: Uniformed Services University of the Health Sciences. 2009. Available online: https://www.usuhs.edu/sites/default/files/media/pmb/pdf/insectarysop.pdf (accessed on 14 April 2019).
- Sylvestre, G.; Gandini, M.; Maciel-De-Freitas, R. Age-Dependent Effects of Oral Infection with Dengue Virus on Aedes aegypti (Diptera: Culicidae) Feeding Behavior, Survival, Oviposition Success and Fecundity. PLoS ONE 2013, 8, e59933. [Google Scholar] [CrossRef]
- Tan, C.-H.; Wong, P.-S.J.; Li, M.-Z.I.; Yang, H.-T.; Chong, C.-S.; Lee, L.K.; Yuan, S.; Leo, Y.-S.; Ng, L.-C.; Lye, D.C. Membrane feeding of dengue patient’s blood as a substitute for direct skin feeding in studying Aedes-dengue virus interaction. Parasites Vectors 2016, 9, 211. [Google Scholar] [CrossRef]
- Rutledge, L.; Ward, R.; Gould, D. Studies on the feeding response of mosquitoes to nutritive solutions in a new membrane feeder. Mosq. News 1964, 24, 407–419. [Google Scholar]
- Maciel-De-Freitas, R.; Koella, J.; Lourenço-De-Oliveira, R. Lower survival rate, longevity and fecundity of Aedes aegypti (Diptera: Culicidae) females orally challenged with dengue virus serotype 2. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Sudiro, T.M.; Vaughn, D.W.; Kurane, I.; Ishiko, H.; Rothman, A.L.; Green, S.; Raengsakulrach, B.; Ennis, F.A.; Nisalak, A.; Kalayanarooj, S.; et al. Rapid Diagnosis of Dengue Viremia by Reverse Transcriptase-Polymerase Chain Reaction using 3′-Noncoding Region Universal Primers. Am. J. Trop. Med. Hyg. 1997, 56, 424–429. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Goncalvez, A.P.; Escalante, A.A.; Pujol, F.H.; Ludert, J.E.; Tovar, D.; Salas, R.A.; Liprandi, F. Diversity and Evolution of the Envelope Gene of Dengue Virus Type 1. Virology 2002, 303, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Pyke, A.; Moore, P.; Taylor, C.; Hall-Mendelin, S.; Cameron, J.; Hewitson, G.; Pukallus, D.; Huang, B.; Warrilow, D.; Van Den Hurk, A. Highly divergent dengue virus type 1 genotype sets a new distance record. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mo, L.; Shi, J.; Guo, X.; Zeng, Z.; Hu, N.; Sun, J.; Wu, M.; Zhou, H.; Hu, Y. Molecular characterization and phy-logenetic analysis of a dengue virus serotype 3 isolated from a Chinese traveler returned from Laos. Virol. J. 2018, 15, 113. [Google Scholar] [CrossRef]
- Tissera, H.A.; Ooi, E.E.; Gubler, D.J.; Tan, Y.; Logendra, B.; Wahala, W.M.; De Silva, A.M.; Abeysinghe, M.N.; Palihawadana, P.; Gunasena, S.; et al. New Dengue Virus Type 1 Genotype in Colombo, Sri Lanka. Emerg. Infect. Dis. 2011, 17, 2053–2055. [Google Scholar] [CrossRef]
- Ocwieja, K.E.; Sundararaman, S.A.; Fernando, A.N.; De Silva, A.D.; Krishnananthasivam, S.; Sherrill-Mix, S.; Tennekoon, R.N.; Premawansa, G.; Tippalagama, R.; Premawansa, S. Phylogeography and Molecular Epidemiology of an Epidemic Strain of Dengue Virus Type 1 in Sri Lanka. Am. J. Trop. Med. Hyg. 2014, 91, 225–234. [Google Scholar] [CrossRef]
- Añez, G.; Heisey, D.A.; Volkova, E.; Rios, M. Complete Genome Sequences of Dengue Virus Type 1 to 4 Strains Used for the Development of CBER/FDA RNA Reference Reagents and WHO International Standard Candidates for Nucleic Acid Testing. Genome Announc. 2016, 4, 01583-15. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.; Guindon, S. Partition Finder: Combined selection of partitioning schemes and sub-stitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.F.; Zu Siederdissen, C.H.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Waterhouse, A.; Procter, J.; Martin, D.; Clamp, M.; Barton, G. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L.; Fontana, W.; Stadler, P.F.; Bonhoeffer, L.S.; Tacker, M.; Schuster, P. Fast folding and comparison of RNA secondary structures. Mon. Chem. Chem. Mon. 1994, 125, 167–188. [Google Scholar] [CrossRef]
- Drake, J.W.; Holland, J.J. Mutation rates among RNA viruses. Proc. Natl. Acad. Sci. USA 1999, 96, 13910–13913. [Google Scholar] [CrossRef]
- de Castro, M.; de Nogueira, F.; Nogueira, R.; Lourenço-de-Oliveira, R.; dos Santos, F. Genetic variation in the 3′untranslated region of dengue virus serotype 3 strains isolated from mosquitoes and humans in Brazil. Virol. J. 2013, 10, 3. [Google Scholar] [CrossRef]
- Liu, Z.-Y.; Li, X.-F.; Jiang, T.; Deng, Y.-Q.; Zhao, H.; Wang, H.-J.; Ye, Q.; Zhu, S.-Y.; Qiu, Y.; Zhou, X.; et al. Novel cis-Acting Element within the Capsid-Coding Region Enhances Flavivirus Viral-RNA Replication by Regulating Genome Cyclization. J. Virol. 2013, 87, 6804–6818. [Google Scholar] [CrossRef]
- de Borba, L.; Villordo, S.; Iglesias, N.; Filomatori, C.; Gebhard, L.; Gamarnik, A. Overlapping local and long-range RNA-RNA interactions modulate dengue virus genome cyclization and replication. J. Virol. 2015, 89, 3430–3437. [Google Scholar] [CrossRef]
- Sim, S.; Aw, P.P.K.; Wilm, A.; Teoh, G.; Hue, K.D.T.; Nguyen, N.M.; Nagarajan, N.; Simmons, C.P.; Hibberd, M.L. Tracking Dengue Virus Intra-host Genetic Diversity during Human-to-Mosquito Transmission. PLoS Negl. Trop. Dis. 2015, 9, e0004052. [Google Scholar] [CrossRef]
- Lequime, S.; Fontaine, A.; Gouilh, M.A.; Moltini-Conclois, I.; Lambrechts, L. Genetic Drift, Purifying Selection and Vector Genotype Shape Dengue Virus Intra-host Genetic Diversity in Mosquitoes. PLoS Genet. 2016, 12, e1006111. [Google Scholar] [CrossRef]
- Ko, H.Y.; Salem, G.M.; Chang, G.J.J.; Chao, D.Y. Application of Next-Generation Sequencing to Reveal How Evolutionary Dynamics of Viral Population Shape Dengue Epidemiology. Front. Microbiol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Malavige, G.; Fernando, S.; Aaskov, J.; Sivayogan, S.; Dissanayaka, T.; Peelawattage, M.K.; Dabare, M. Seroprevalence of Anti-dengue Virus Antibodies in Children in Colombo District, Sri Lanka. Dengue Bull. 2006, 30, 68–71. [Google Scholar]
- Peyrefitte, C.N.; Couissinier-Paris, P.; Mercier-Perennec, V.; Bessaud, M.; Martial, J.; Kenane, N.; Durand, J.-P.A.; Tolou, H.J. Genetic Characterization of Newly Reintroduced Dengue Virus Type 3 in Martinique (French West Indies). J. Clin. Microbiol. 2003, 41, 5195–5198. [Google Scholar] [CrossRef]
- Yeh, S.-C.; Pompon, J. Flaviviruses Produce a Subgenomic Flaviviral RNA That Enhances Mosquito Transmission. DNA Cell Biol. 2018, 37, 154–159. [Google Scholar] [CrossRef]
- Ding, Y.; Chan, C.; Lawrence, C. RNA secondary structure prediction by centroids in a Boltzmann weighted ensemble. RNA 2005, 11, 1157–1166. [Google Scholar] [CrossRef] [PubMed]
- Ritz, J.; Martin, J.S.; Laederach, A. Evaluating our ability to predict the structural disruption of RNA by SNPs. BMC Genom. 2012, 13, S6. [Google Scholar] [CrossRef] [PubMed]
- Salari, R.; Kimchi-Sarfaty, C.; Gottesman, M.; Przytycka, T. Sensitive measurement of single-nucleotide polymor-phism-induced changes of RNA conformation: Application to disease studies. Nucleic Acids Res. 2013, 41, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Laederach, A.; Shcherbakova, I.; Jonikas, M.A.; Altman, R.B.; Brenowitz, M. Distinct contribution of electrostatics, initial conforma-tional ensemble, and macromolecular stability in RNA folding. Proc. Natl. Acad. Sci. USA 2007, 104, 7045–7050. [Google Scholar] [CrossRef] [PubMed]
- Glinsky, G.V. SNP-guided microRNA maps (MirMaps) of 16 common human disorders identify a clinically accessible therapy reversing transcriptional aberrations of nuclear import and inflammasome pathways. Cell Cycle 2008, 7, 3564–3576. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RT-PCR Results on Human Serum (n = 5) | DENV Serotype According to Sequence Analysis | Human-Derived DENV Samples | RT-PCR Results on Mosquito Tissues | Mosquito-Derived DENV Samples |
|---|---|---|---|---|
| + | DENV 1 | D1H_2019SL | +(5/12) | D1M1_2019SL-D1M5-2019SL |
| + | DENV 1 | D3H_2019SL | +(6/10) | D3M1_2019SL-D3M6_2019SL |
| + | DENV 3 | D4H_2019SL | +(7/11) | D4M1_2019SL-D4M7_2019SL |
| + | DENV 3 | D5H_2019SL | +(4/9) | D5M1_2019SL-D5M4_2019SL |
| + | DENV 3 | D6H_2019SL | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dayananda, P.D.; de Silva, H.; Fernando, L.; de Silva, B.G.D.N.K. Genetic Variation in the Domain II, 3′ Untranslated Region of Human and Mosquito Derived Dengue Virus Strains in Sri Lanka. Viruses 2021, 13, 421. https://doi.org/10.3390/v13030421
Dayananda PD, de Silva H, Fernando L, de Silva BGDNK. Genetic Variation in the Domain II, 3′ Untranslated Region of Human and Mosquito Derived Dengue Virus Strains in Sri Lanka. Viruses. 2021; 13(3):421. https://doi.org/10.3390/v13030421
Chicago/Turabian StyleDayananda, P. D., Harendra de Silva, LakKumar Fernando, and B. G. D. N. K. de Silva. 2021. "Genetic Variation in the Domain II, 3′ Untranslated Region of Human and Mosquito Derived Dengue Virus Strains in Sri Lanka" Viruses 13, no. 3: 421. https://doi.org/10.3390/v13030421
APA StyleDayananda, P. D., de Silva, H., Fernando, L., & de Silva, B. G. D. N. K. (2021). Genetic Variation in the Domain II, 3′ Untranslated Region of Human and Mosquito Derived Dengue Virus Strains in Sri Lanka. Viruses, 13(3), 421. https://doi.org/10.3390/v13030421