
Genetic Characteristics of Avian Influenza Virus Isolated from Wild Birds in South Korea, 2019–2020


Abstract
1. Introduction
2. Materials and Methods
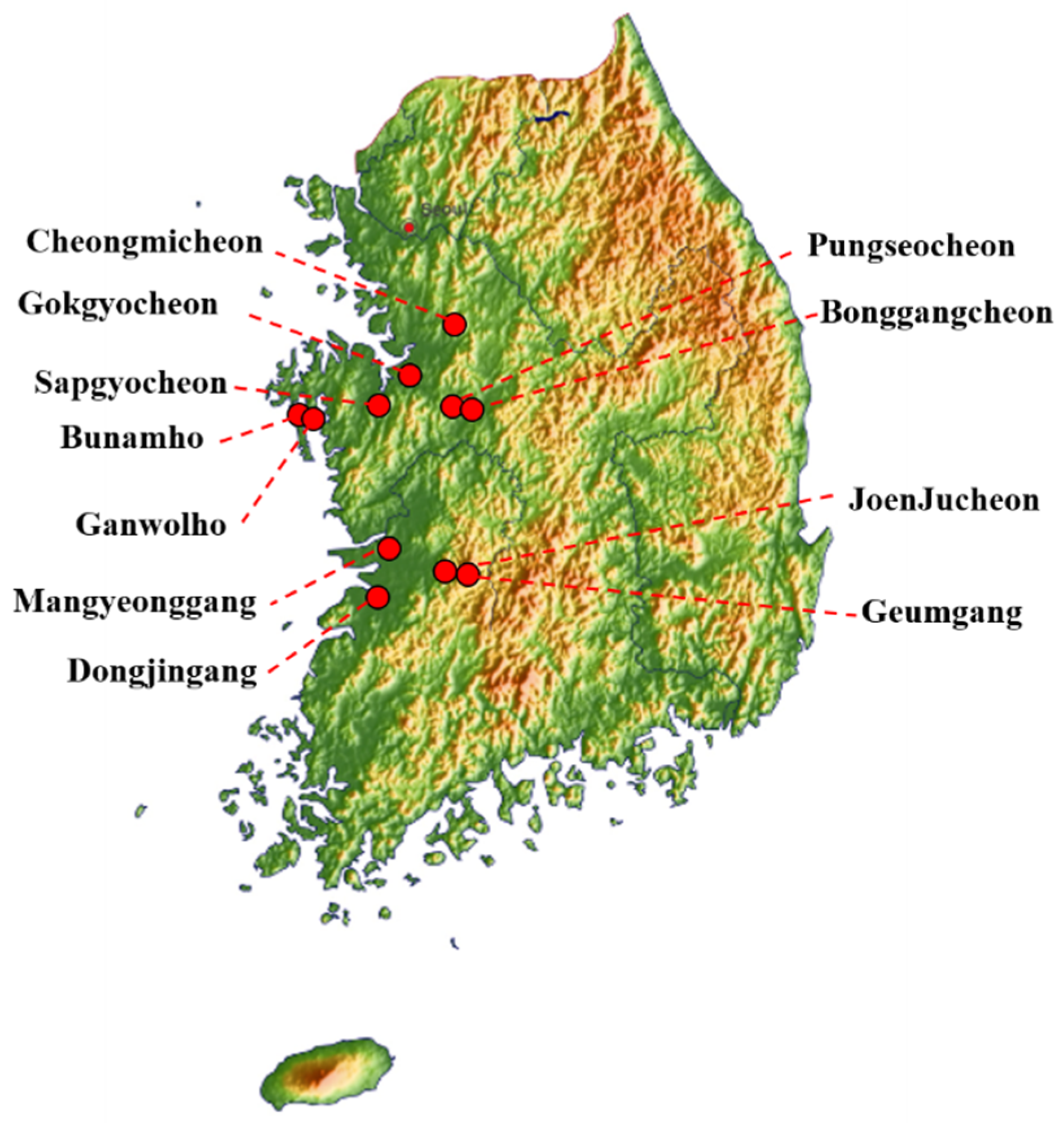
2.1. Sample Collection and Virus Isolation
2.2. Virus Isolation and Identification
2.3. Species Identification
2.4. Genetic Analysis of AIV Isolates
2.5. Pathogenicity in Mice
3. Results
3.1. Virus Isolation
3.2. Prevalence of AIVs
3.3. Genetic Characterization of AIV Isolates
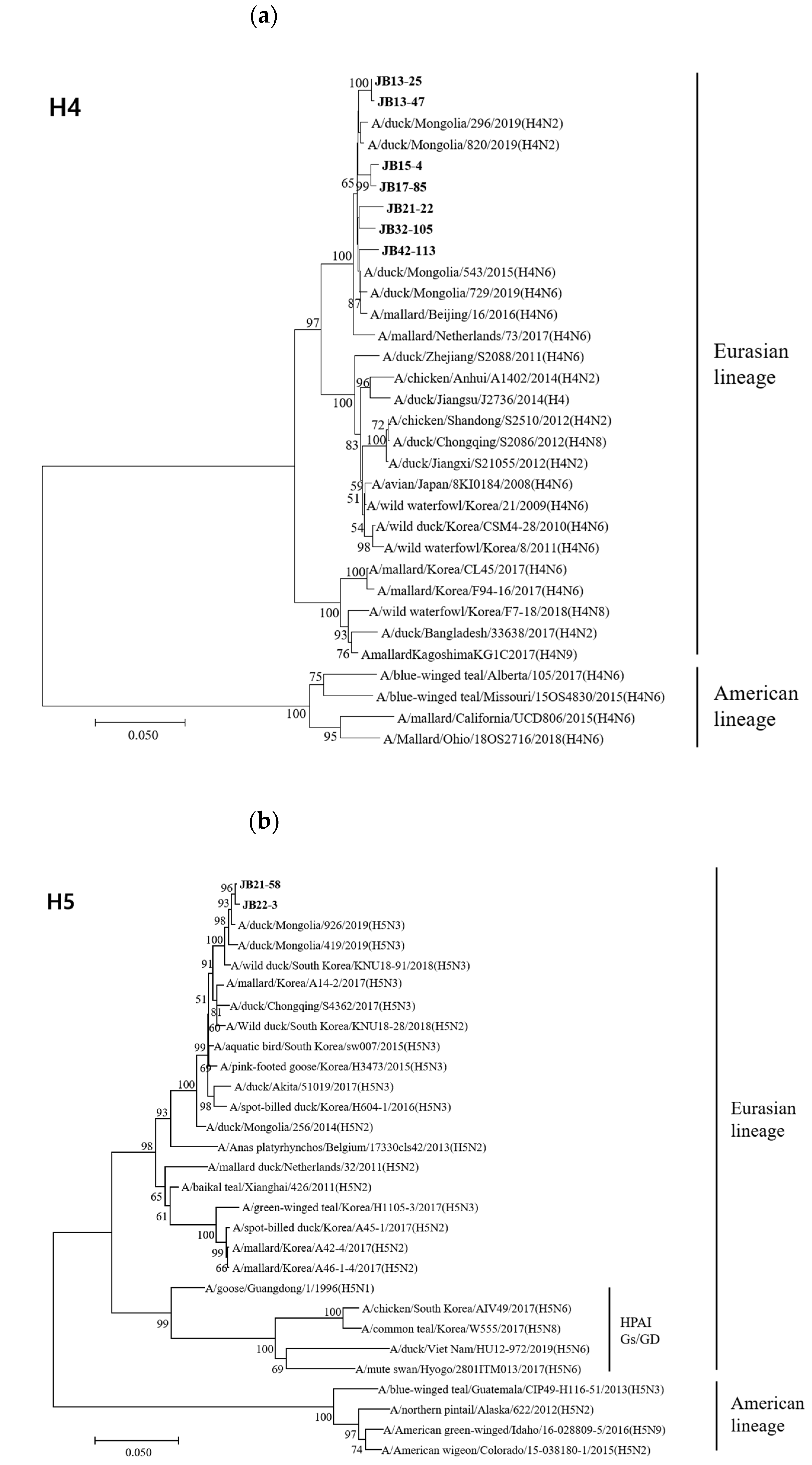
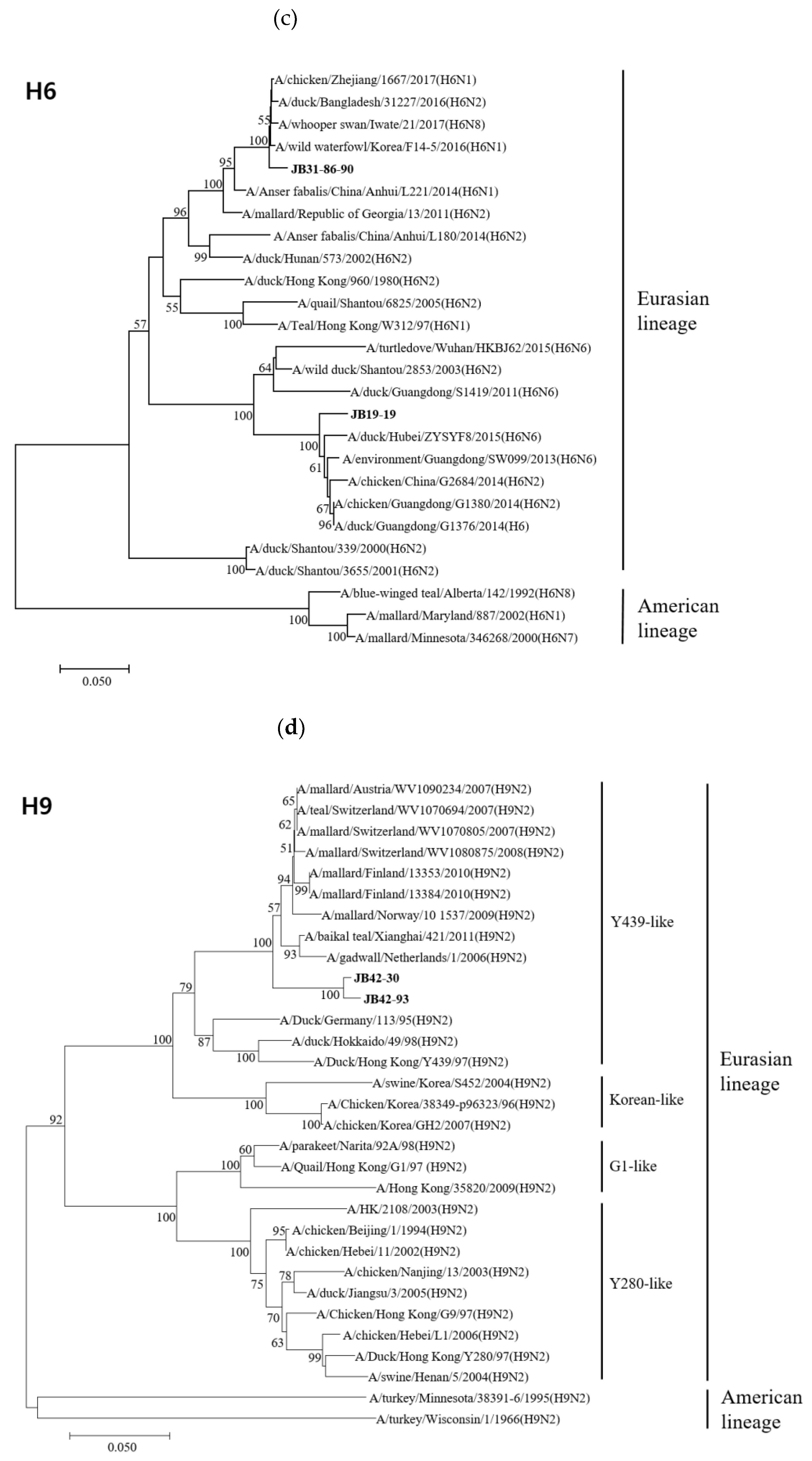
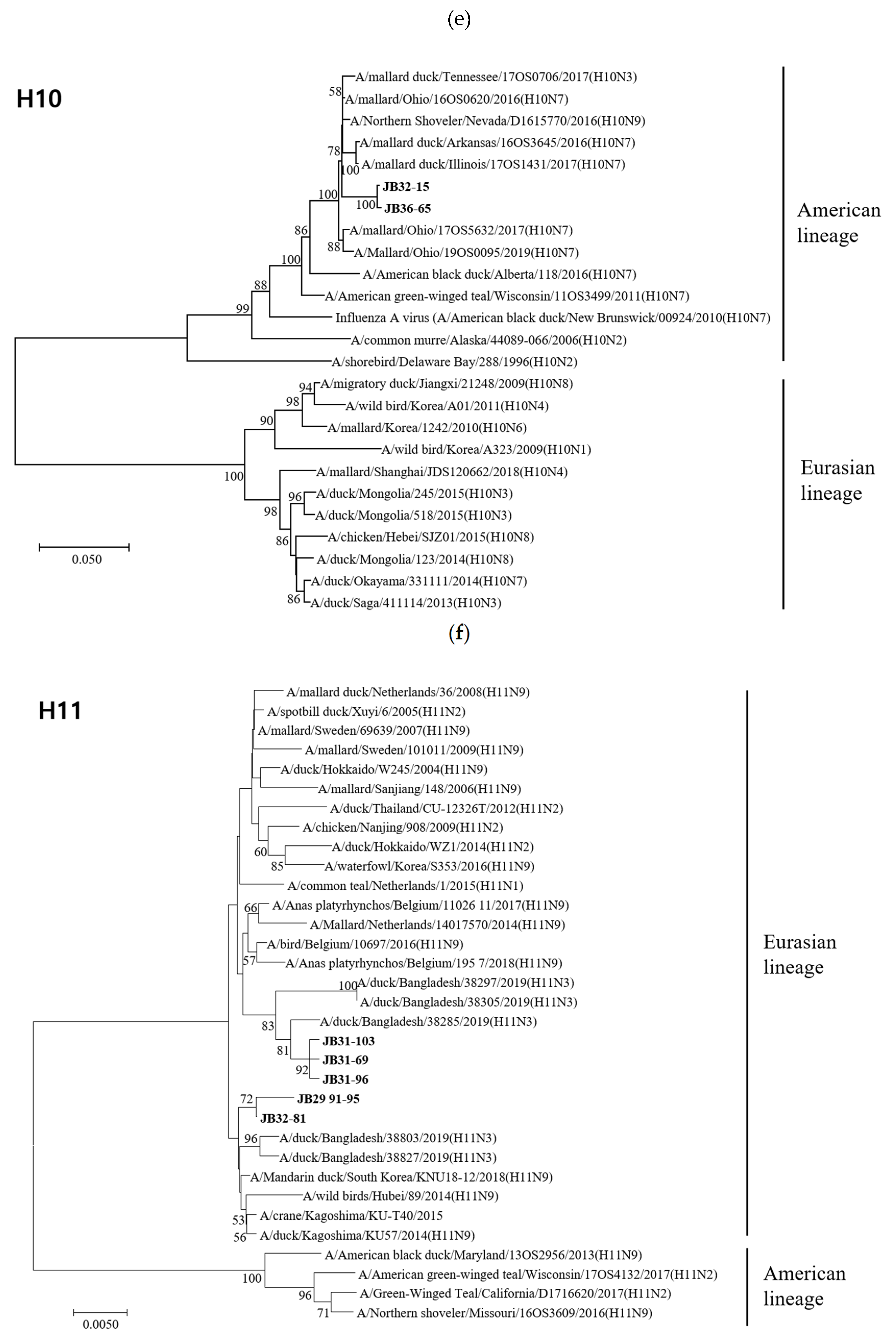
3.4. Phylogenetic Analyses
3.4.1. Hemagglutinin
3.4.2. Neuraminidase
3.4.3. Internal Genes
3.5. Pathogenicity in Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuchipudi, S.V.; Nissly, R.H. Novel Flu Viruses in Bats and Cattle: “Pushing the Envelope” of Influenza Infection. Vet. Sci. 2018, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Crawford, J.M.; Latimer, J.W.; Rivera-Cruz, E.; Perdue, M.L. Heterogeneity in the haemagglutinin gene and emergence of the highly pathogenic phenotype among recent H5N2 avian influenza viruses from Mexico. J. Gen. Virol. 1996, 77, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.K.; Seo, S.H.; Kim, J.A.; Webby, R.J.; Webster, R.G. Avian influenza viruses in Korean live poultry markets and their pathogenic potential. Virology 2005, 332, 529–537. [Google Scholar] [CrossRef]
- Lee, E.-K.; Lee, Y.-N.; Kye, S.-J.; Lewis, N.S.; Brown, I.H.; Sagong, M.; Heo, G.-B.; Kang, Y.-M.; Cho, H.-K.; Kang, H.-M.; et al. Characterization of a novel reassortant H5N6 highly pathogenic avian influenza virus clade 2.3.4.4 in Korea, 2017. Emerg. Microbes Infect. 2018, 7, 1–3. [Google Scholar] [CrossRef]
- Venkatesh, D.; Poen, M.J.; Bestebroer, T.M.; Scheuer, R.D.; Vuong, O.; Chkhaidze, M.; Machablishvili, A.; Mamuchadze, J.; Ninua, L.; Fedorova, N.B.; et al. Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Van Den Brand, J.M.A.; Verhagen, J.H.; Veldhuis Kroeze, E.J.B.; Van De Bildt, M.W.G.; Bodewes, R.; Herfst, S.; Richard, M.; Lexmond, P.; Bestebroer, T.M.; Fouchier, R.A.M.; et al. Wild ducks excrete highly pathogenic avian influenza virus H5N8 (2014–2015) without clinical or pathological evidence of disease. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef]
- Beerens, N.; Heutink, R.; Pritz-Verschuren, S.; Germeraad, E.A.; Bergervoet, S.A.; Harders, F.; Bossers, A.; Koch, G. Genetic relationship between poultry and wild bird viruses during the highly pathogenic avian influenza H5N6 epidemic in the Netherlands, 2017–2018. Transbound. Emerg. Dis. 2019, 66, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Cheng, K.; Gao, Y. Poultry Infection with Influenza Viruses of Wild Bird Origin, China, 2016. Emerg. Infect. Dis. 2018, 24, 1375–1377. [Google Scholar] [CrossRef]
- Yong, D.L.; Liu, Y.; Low, B.W.; Española, C.P.; Choi, C.-Y.; Kawakami, K. Migratory songbirds in the East Asian-Australasian Flyway: A review from a conservation perspective. Bird Conserv. Int. 2015, 25, 1–37. [Google Scholar] [CrossRef]
- Kim, G.S.; Kim, T.S.; Son, J.S.; Lai, V.D.; Park, J.E.; Wang, S.J.; Jheong, W.H.; Mo, I.P. The difference of detection rate of avian influenza virus in the wild bird surveillance using various methods. J. Vet. Sci. 2019, 20, e56. [Google Scholar] [CrossRef]
- Herfst, S.; Imai, M.; Kawaoka, Y.; Fouchier, R.A. Avian Influenza Virus Transmission to Mammals. Curr. Top. Microbiol. Immunol. 2014, 385, 137–155. [Google Scholar] [CrossRef]
- Russell, C.J.; Webster, R.G. The Genesis of a Pandemic Influenza Virus. Cell 2005, 123, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Hoffmann, D.; Henritzi, D.; Beer, M.; Harder, T.C. Riems influenza a typing array (RITA): An RT-qPCR-based low density array for subtyping avian and mammalian influenza a viruses. Sci. Rep. 2016, 6, 27211. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, H.J.; Lee, Y.J.; Kang, H.M.; Jeong, O.M.; Kim, M.C.; Kwon, J.S.; Kwon, J.H.; Kim, C.B.; Lee, J.B.; et al. DNA Barcoding Techniques for Avian Influenza Virus Surveillance in Migratory Bird Habitats. J. Wildl. Dis. 2010, 46, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef]
- Li, O.T.; Barr, I.; Leung, C.Y.; Chen, H.; Guan, Y.; Peiris, J.S.; Poon, L.L. Reliable universal RT-PCR assays for studying influenza polymerase subunit gene sequences from all 16 haemagglutinin subtypes. J. Virol. Methods 2007, 142, 218–222. [Google Scholar] [CrossRef]
- Yang, L.; Cheng, Y.; Zhao, X.; Wei, H.; Tan, M.; Li, X.; Zhu, W.; Huang, W.; Chen, W.; Liu, J.J.B.; et al. Mutations associated with egg adaptation of influenza A(H1N1)pdm09 virus in laboratory based surveillance in China, 2009–2016. Biosaf. Health 2019, 1, 41–45. [Google Scholar] [CrossRef]
- Park, Y.W.; Kim, Y.H.; Jung, H.U.; Jeong, O.S.; Hong, E.J.; Kim, H.; Lee, J.I. Comparison of antigenic mutation during egg and cell passage cultivation of H3N2 influenza virus. Clin. Exp. Vaccine Res. 2020, 9, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Long, J.S.; Mistry, B.; Haslam, S.M.; Barclay, W.S. Host and Viral Determinants of Influenza a Virus Species Specificity. Nat. Rev. Microbiol. 2019, 17, 67–81. [Google Scholar] [CrossRef]
- Zhu, W.; Zou, X.; Zhou, J.; Tang, J.; Shu, Y. Residues 41V and/or 210D in the NP protein enhance polymerase activities and potential replication of novel influenza (H7N9) viruses at low temperature. Virol. J. 2015, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ishaq, M.; Prudence, M.; Xi, X.; Hu, T.; Liu, Q.; Guo, D. Single Mutation at the Amino Acid Position 627 of Pb2 That Leads to Increased Virulence of an H5n1 Avian Influenza Virus During Adaptation in Mice Can Be Compensated by Multiple Muta-tions at Other Sites of Pb2. Virus Res. 2009, 144, 123–129. [Google Scholar] [CrossRef]
- Hulse-Post, D.J.; Franks, J.; Boyd, K.; Salomon, R.; Hoffmann, E.; Yen, H.L.; Webby, R.J.; Walker, D.; Nguyen, T.D.; Webster, R.G. Molecular Changes in the Polymerase Genes (PA and PB1) Associated with High Pathogenicity of H5N1 Influenza Virus in Mallard Ducks. J. Virol. 2007, 81, 8515–8524. [Google Scholar] [CrossRef] [PubMed]
- Schmolke, M.; Manicassamy, B.; Pena, L.; Sutton, T.; Hai, R.; Varga, Z.T.; Hale, B.G.; Steel, J.; Pérez, D.R.; García-Sastre, A. Differential Contribution of Pb1-F2 to the Virulence of Highly Pathogenic H5n1 Influenza a Virus in Mammalian and Avian Species. PLoS Pathog. 2011, 7, e1002186. [Google Scholar] [CrossRef] [PubMed]
- Suttie, A.; Deng, Y.M.; Greenhill, A.R.; Dussart, P.; Horwood, P.F.; Karlsson, E.A. Inventory of molecular markers affecting biological characteristics of avian influenza A viruses. Virus Genes 2019, 55, 739–768. [Google Scholar] [CrossRef] [PubMed]
- Taubenberger, J.K.; Reid, A.H.; Lourens, R.M.; Wang, R.; Jin, G.; Fanning, T.G. Characterization of the 1918 influenza virus polymerase genes. Nature 2005, 437, 889–893. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, G.; Dauber, B.; Wolff, T.; Planz, O.; Klenk, H.D.; Stech, J. The Viral Poly-merase Mediates Adaptation of an Avian Influenza Virus to a Mammalian Host. Proc. Natl. Acad. Sci. USA 2005, 102, 18590–18595. [Google Scholar] [CrossRef]
- Li, J.; Zu Dohna, H.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and Genetic Variation of Neuraminidase Stalk Deletions in Avian Influenza Viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Deng, G.; Song, J.; Tian, G.; Suo, Y.; Jiang, Y.; Guan, Y.; Bu, Z.; Kawaoka, Y.; Chen, H. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology 2009, 384, 28–32. [Google Scholar] [CrossRef]
- Jiao, P.; Tian, G.; Li, Y.; Deng, G.; Jiang, Y.; Liu, C.; Liu, W.; Bu, Z.; Kawaoka, Y.; Chen, H. A Single-Amino-Acid Substitution in the Ns1 Protein Changes the Pathogenicity of H5n1 Avian Influenza Viruses in Mice. J. Virol. 2008, 82, 1146–1154. [Google Scholar] [CrossRef]
- Li, M.; Wang, B. Homology modeling and examination of the effect of the D92E mutation on the H5N1 nonstructural protein NS1 effector domain. J. Mol. Model. 2007, 13, 1237–1244. [Google Scholar] [CrossRef]
- Gubareva, L.V.; Sleeman, K.; Guo, Z.; Yang, H.; Hodges, E.; Davis, C.T.; Baranovich, T.; Stevens, J. Drug Susceptibility Evaluation of an Influenza A(H7N9) Virus by Analyzing Recombinant Neuraminidase Proteins. J. Infect. Dis. 2017, 216, S566–S574. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.L.; Rayner, J.M.; Smith, G.J.; Wang, P.; Naipospos, T.S.; Zhang, J.; Yuen, K.Y.; Webster, R.G.; Peiris, J.S.; Guan, Y.; et al. Distribution of Amantadine-Resistant H5N1 Avian Influenza Variants in Asia. J. Infect. Dis. 2006, 193, 1626–1629. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-N.; Lee, D.-H.; Cheon, S.-H.; Park, Y.-R.; Baek, Y.-G.; Si, Y.-J.; Kye, S.-J.; Lee, E.-K.; Heo, G.-B.; Bae, Y.-C.; et al. Genetic characteristics and pathogenesis of H5 low pathogenic avian influenza viruses from wild birds and domestic ducks in South Korea. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Treanor, J.J.; Snyder, M.H.; London, W.T.; Murphy, B.R. The B allele of the NS gene of avian influenza viruses, but not the A allele, attenuates a human influenza a virus for squirrel monkeys. Virology 1989, 171, 1–9. [Google Scholar] [CrossRef]
- Kang, H.M.; Jeong, O.M.; Kim, M.C.; Kwon, J.S.; Paek, M.R.; Choi, J.G.; Lee, E.K.; Kim, Y.J.; Kwon, J.H.; Lee, Y.J. Surveillance of avian influenza virus in wild bird fecal samples from south korea, 2003–2008. J. Wildl. Dis. 2010, 46, 878–888. [Google Scholar] [CrossRef]
- Lee, K.; Yu, D.; Martínez-López, B.; Yoon, H.; Kang, S.I.; Hong, S.K.; Lee, I.; Kang, Y.; Jeong, W.; Lee, E. Fine-scale tracking of wild waterfowl and their impact on highly pathogenic avian influenza outbreaks in the Republic of Korea, 2014–2015. Sci. Rep. 2020, 10, 18631. [Google Scholar] [CrossRef]
- Hiono, T.; Okamatsu, M.; Matsuno, K.; Haga, A.; Iwata, R.; Nguyen, L.T.; Suzuki, M.; Kikutani, Y.; Kida, H.; Onuma, M.; et al. Characterization of H5N6 highly pathogenic avian influenza viruses isolated from wild and captive birds in the winter season of 2016-2017 in Northern Japan. Microbiol. Immunol. 2017, 61, 387–397. [Google Scholar] [CrossRef]
- Kwon, H.-I.; Kim, E.-H.; Kim, Y.-I.; Park, S.-J.; Si, Y.-J.; Lee, I.-W.; Nguyen, H.D.; Yu, K.M.; Yu, M.-A.; Jung, J.H.; et al. Comparison of the pathogenic potential of highly pathogenic avian influenza (HPAI) H5N6, and H5N8 viruses isolated in South Korea during the 2016–2017 winter season. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef]
- Brown, J.D.; Stallknecht, D.E. Wild Bird Surveillance for the Avian Influenza Virus. Avian Influenza Virus 2008, 436, 85–97. [Google Scholar] [CrossRef]
- Lee, E.-K.; Kang, H.-M.; Song, B.-M.; Lee, Y.-N.; Heo, G.-B.; Lee, H.-S.; Lee, Y.-J.; Kim, J.-H. Surveillance of avian influenza viruses in South Korea between 2012 and 2014. Virol. J. 2017, 14, 54. [Google Scholar] [CrossRef]
- Latorre-Margalef, N.; Tolf, C.; Grosbois, V.; Avril, A.; Bengtsson, D.; Wille, M.; Osterhaus, A.D.; Fouchier, R.A.; Olsen, B.; Waldenström, J. Long-term variation in influenza A virus prevalence and subtype diversity in migratory mallards in northern Europe. Proc. R. Soc. B Boil. Sci. 2014, 281, 20140098. [Google Scholar] [CrossRef] [PubMed]
- Diskin, E.R.; Friedman, K.; Krauss, S.; Nolting, J.M.; Poulson, R.L.; Slemons, R.D.; Stallknecht, D.E.; Webster, R.G.; Bowman, A.S. Subtype Diversity of Influenza A Virus in North American Waterfowl: A Multidecade Study. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Scott, A.; Hernandez-Jover, M.; Groves, P.; Toribio, J.-A. An overview of avian influenza in the context of the Australian commercial poultry industry. One Health 2020, 10, 100139. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Tang, W.; Li, X.; Hu, C.; Wu, D.; Wang, T.; He, G. Avian Influenza Virus Prevalence and Subtype Diversity in Wild Birds in Shanghai, China, 2016–2018. Viruses 2020, 12, 1031. [Google Scholar] [CrossRef]
- Oh, K.H.; Mo, J.S.; Bae, Y.J.; Lee, S.B.; Lai, V.D.; Wang, S.J.; Mo, I.P. Amino acid substitutions in low pathogenic avian influenza virus strains isolated from wild birds in Korea. Virus Genes 2018, 54, 397–405. [Google Scholar] [CrossRef]
- Toledo-Rueda, W.; Rosas-Murrieta, N.H.; Muñoz-Medina, J.E.; González-Bonilla, C.; Reyes-Leyva, J.; Santos-López, G. Antiviral resistance markers in influenza virus sequences in Mexico, 2000–2017. Infect. Drug Resist. 2018, 11, 1751–1756. [Google Scholar] [CrossRef]
- Park, Y.R.; Lee, Y.N.; Lee, D.H.; Baek, Y.G.; Si, Y.J.; Meeduangchanh, P.; Theppangna, W.; Douangngeun, B.; Kye, S.J.; Lee, M.H.; et al. Genetic and pathogenic characteristics of clade 2.3.2.1c H5N1 highly pathogenic avian influenza viruses isolated from poultry outbreaks in Laos during 2015–2018. Transbound. Emerg. Dis. 2019, 67, 947–955. [Google Scholar] [CrossRef]
- Lee, D.-H.; Song, C.-S.J.C. H9N2 avian influenza virus in Korea: Evolution and vaccination. Clin. Exp. Vaccine Res. 2013, 2, 26–33. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Sealy, J.E.; Iqbal, M.J.V. A Global Perspective on H9N2 Avian Influenza Virus. Viruses 2019, 11, 620. [Google Scholar] [CrossRef]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef] [PubMed]
- Butt, K.M.; Smith, G.J.D.; Chen, H.; Zhang, L.J.; Leung, Y.H.C.; Xu, K.M.; Lim, W.; Webster, R.G.; Yuen, K.Y.; Peiris, J.S.M.; et al. Human Infection with an Avian H9N2 Influenza A Virus in Hong Kong in 2003. J. Clin. Microbiol. 2005, 43, 5760–5767. [Google Scholar] [CrossRef]
- Claas, E.C.; Osterhaus, A.D.; van Beek, R.; De Jong, J.C.; Rimmelzwaan, G.F.; Senne, D.A.; Krauss, S.; Shortridge, K.F.; Webster, R.G. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet 1998, 351, 472–477. [Google Scholar] [CrossRef]
- Philippon, D.A.M.; Wu, P.; Cowling, B.J.; Lau, E.H.Y. Avian Influenza Human Infections at the Human-Animal Interface. J. Infect. Dis. 2020, 222, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhao, X.; Li, X.; Bo, H.; Li, D.; Liu, J.; Wang, D.J.B. Case report for human infection with a highly pathogenic avian influenza A(H5N6) virus in Beijing, China 2019. Biosaf. Health 2020, 2, 49–52. [Google Scholar] [CrossRef]
- Peiris, M.; Yuen, K.; Leung, C.; Chan, K.; Ip, P.; Lai, R.; Orr, W.; Shortridge, K. Human infection with influenza H9N2. Lancet 1999, 354, 916–917. [Google Scholar] [CrossRef]
- Song, W.; Qin, K. Human-infecting influenza A (H9N2) virus: A forgotten potential pandemic strain? Zoonoses Public Health 2020, 67, 203–212. [Google Scholar] [CrossRef]
- Gill, J.S.; Webby, R.; Gilchrist, M.J.; Gray, G.C. Avian Influenza among Waterfowl Hunters and Wildlife Professionals. Emerg. Infect. Dis. 2006, 12, 1284–1286. [Google Scholar] [CrossRef]
- Kayali, G.; Barbour, E.; Dbaibo, G.; Tabet, C.; Saade, M.; Shaib, H.A.; DeBeauchamp, J.; Webby, R.J. Evidence of Infection with H4 and H11 Avian Influenza Viruses among Lebanese Chicken Growers. PLoS ONE 2011, 6, e26818. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Jin, S.; Wang, T.; Sun, W.; Zhang, Y.; Li, F.; Zhao, M.; Sun, L.; Hu, X.; et al. H9N2 influenza virus spillover into wild birds from poultry in China bind to human-type receptors and transmit in mammals via respiratory droplets. Transbound. Emerg. Dis. 2021. [Google Scholar] [CrossRef]
- Jeong, S.; Lee, D.H.; Kwon, J.H.; Kim, Y.J.; Lee, S.H.; Cho, A.Y.; Kim, T.H.; Park, J.E.; Lee, S.I.; Song, C.S. Highly Pathogenic Avian Influenza Clade 2.3.4.4b Subtype H5N8 Virus Isolated from Mandarin Duck in South Korea, 2020. Viruses 2020, 12, 1389. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Primer | Primer Sequences | References |
|---|---|---|---|
| PB2 | Bm-PB2-1 | 5′-TATTGGTCTCAGGGAGCGAAAGCAGGTC-3′ | [15] |
| PB2-1250R | 5′-TCYTCYTGTGARAAYACCAT-3′ | [16] | |
| PB2-1105F | 5′-TAYGARGARTTCACAATGGT-3′ | ||
| Bm-PB2-2341R | 5′-ATATGGTCTCGTATTAGTAGAAACAAGGTCGTTT-3′ | [15] | |
| PB1 | Bm-PB1-1 | 5′-TATTCGTCTCAGGGAGCGAAAGCAGGCA-3′ | |
| PB1-800R | 5′-TCRAGYTTYTCACAKATGCTCC-3′ | customized primer | |
| PB1-560F | 5′-ARATACCNGCAGARATGCT-3′ | [16] | |
| PB1-1610R | 5′-ACTGTAACHCCAATGCTCAT-3′ | customized primer | |
| PB1-1124F | 5′-ARATACCNGCAGARATGCT-3′ | [16] | |
| Bm-PB1-2341R | 5′-ATATCGTCTCGTATTAGTAGAAACAAGGTACTT-3′ | [15] | |
| PA | Bm-PA-1 | 5′-TATTCGTCTCAGGGAGCGAAAGCAGGTAC-3′ | |
| PA-1498R | 5′-TNGTYCTRCAYTTGCTTATCAT-3′ | [16] | |
| PA-747F | 5′-CATTGAGGGCAAGCTTTC-3′ | ||
| Bm-PA-2233R | 5′-ATATCGTCTCGTATTAGTAGAAACAAGGTACTT-3′ | [15] | |
| HA | Bm-HA-1 | 5′-TATTCGTCTCAGGGAGCAAAAGCAGGGG-3′ | [15] |
| H4-1085R | 5′-GGCCTTGCCATCCATTCTCTAT-3′ | customized primer | |
| H4-865F | 5′-GGCTCATGTGTCAGTAAG-3′ | customized primer | |
| H11-771F | 5′-ACAGGCTGGACGATGAC-3′ | customized primer | |
| H11-881R | 5′-CACTT GTAGAGCATGATTC-3′ | customized primer | |
| H11-1100F | 5′-YTRATYAATGGWTGGTAYGG-3′ | customized primer | |
| H11-1166R | 5′-TGGTCTATCGCCTTCTGA-3′ | customized primer | |
| Bm-NS-890R | 5′-ATATCGTCTCGTATTAGTAGAAACAAGGGTGTTTT-3′ | [15] | |
| NP | Bm-NP-1 | 5′-TATTGGTCTCAGGGAGCAAAAGCAGGAGT-3′ | |
| Bm-NP-1565R | 5′-ATATCGTCTCGTATTAGTAGAAACAAGGGTATTTTT-3′ | ||
| NA | Ba-NA-1 | 5′-TATTGGTCTCAGGGAGCAAAAGCAGGAGT-3′ | |
| N2-848R | 5′-TCTCTGCARACACATCTGACGT-3′ | customized primer | |
| N3-689R | 5′- CATTCWGACTCYTGAGTTCT -3′ | customized primer | |
| N6-318F | 5′- ATGCAATAAGRATAGGDGA -3′ | customized primer | |
| N9-586R | 5′- TGCATATTGACATCCTGGAT-3′ | customized primer | |
| Ba-NA-1413R | 5′-ATATGGTCTCGTATTAGTAGAAACAAGGAGTTTTTT-3′ | [15] | |
| M | Bm-M-1 | 5′-TATTCGTCTCAGGGAGCAAAAGCAGGGTG-3′ | |
| Bm-M-1027R | 5′-ATATCGTCTCGTATTAGTAGAAACAAGGTAGTTTTT-3′ | ||
| NS | Bm-NS-1 | 5′-TATTCGTCTCAGGGAGCAAAAGCAGGGTG -3′ | |
| Bm-NS-890R | 5′-ATATCGTCTCGTATTAGTAGAAACAAGGGTGTTTT-3′ |
| Isolates | Abbreviation | Subtype | Location | Host |
|---|---|---|---|---|
| A/mallard/South Korea/JB12-8/2019(H3/10N3/8) | JB12-8 | H3/10M3/8 | Sapgyocheon | Anas platyrhynchos |
| A/spot-billed duck/South Korea/JB13-25/2019(H4N6) | JB13-25 | H4N6 | Pungseocheon | Anas poecilorhyncha |
| A/ spot-billed duck/ South Korea /JB13-47/2019(H4N6) | JB13-47 | H4N6 | Pungseocheon | Anas poecilorhyncha |
| A/ spot-billed duck/ South Korea /JB15-4/2019(H4N3) | JB15-4 | H4N3 | Gokgyocheon | Anas poecilorhyncha |
| A/ mallard/ South Korea/JB15-42/2019(H5/6N1/3) | JB15-42 | H5/6N1/3 | Gokgyocheon | Anas platyrhynchos |
| A/ mallard/ South Korea/JB17-85/2019(H4N6) | JB17-85 | H4N6 | Gokgyocheon | Anas platyrhynchos |
| A/mallard/South Korea/JB19-19/2019(H6N8) | JB19-19 | H6N8 | JoenJucheon | Anas platyrhynchos |
| A/mallard/South Korea/JB21-22/2019(H4N6) | JB21-22 | H4N6 | Bonggangcheon | Anas platyrhynchos |
| A/spot-billed duck/South Korea /JB21-48/2019(H6N1) | JB21-48 | H6N1 | Bonggangcheon | Anas poecilorhyncha |
| A/mallard/South Korea/JB21-58/2019(H5N3) | JB21-58 | H5N3 | Bonggangcheon | Anas platyrhynchos |
| A/mallard/South Korea/JB22-3/2019(H5N3) | JB22-3 | H5N3 | Cheongmicheon | Anas platyrhynchos |
| A/mallard/South Korea/JB29-40/2019(H3/10N2/7) | JB29-40 | H3/10N2/7 | Dongjingang | Anas platyrhynchos |
| A/mallard/South Korea/JB29-56/2019(H3N2/7) | JB29-56 | H3N2/7 | Dongjingang | Anas platyrhynchos |
| A/mallard/South Korea/JB29-91-95/2019(H11N2) | JB29-91-95 | H11N2 | Dongjingang | Anas platyrhynchos |
| A/mallard/South Korea/JB31-69/2019(H11N9) | JB31-69 | H11N9 | Mangyeonggang | Anas platyrhynchos |
| A/spot-billed duck/South Korea/JB31-86-90/2019(H6N2) | JB31-86-90 | H6N2 | Mangyeonggang | Anas poecilorhyncha |
| A/mallard/South Korea/JB31-96/2019(H11N9) | JB31-96 | H11N9 | Mangyeonggang | Anas platyrhynchos |
| A/mallard/South Korea/JB31-103/2019(H11N9) | JB31-103 | H11N9 | Mangyeonggang | Anas platyrhynchos |
| A/Eurasian teal/South Korea/JB32-15/2019(H10N7) | JB32-15 | H10N7 | Dongjingang | Anas crecca |
| A/mallard/South Korea/JB32-41-45/2019(H3/10N8) | JB32-41-45 | H3/10N8 | Dongjingang | Anas platyrhynchos |
| A/spot-billed duck/South Korea/JB32-81/2019(H11N2) | JB32-81 | H11N2 | Dongjingang | Anas poecilorhyncha |
| A/spot-billed duck/South Korea/JB32-105/2019(H11N2) | JB32-105 | H11N2 | Dongjingang | Anas poecilorhyncha |
| A/Greater White-fronted Goose/South Korea/JB36-16-20/2019(H10N2/7) | JB36-16-20 | H10N2/7 | Geumgang | Anser albifrons |
| A/Bean goose/South Korea/JB36-65/2019(H10N4) | JB36-65 | H10N4 | Geumgang | Anser fabalis |
| A/Falcated duck/South Korea/JB42-30/2020(H9N2) | JB42-30 | H9N2 | Mangyeonggang | Anas falcata |
| A/Falcated duck/South Korea/JB42-93/2020(H9N2) | JB42-93 | H9N2 | Mangyeonggang | Anas falcata |
| A/mallard/South Korea/JB42-113/2020(H4N6) | JB42-113 | H4N6 | Mangyeonggang | Anas platyrhynchos |
| A/mallard/South Korea/JB43-51-55/2020(H10N4) | JB43-51-55 | H10N4 | Mangyeonggang | Anas platyrhynchos |
| Subtype | N1 | N2 | N3 | N4 | N5 | N6 | N7 | N8 | N9 | Total |
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | - | - | - | - | - | - | - | - | - | 0 |
| H2 | - | - | - | - | - | - | - | - | - | 0 |
| H3 | - | (2) | (1) | - | - | - | (2) | (2) | - | (7) |
| H4 | - | 1 | 1 | - | - | 5 | - | - | - | 7 |
| H5 | (1) | - | 2(1) | - | - | - | - | - | - | 2(2) |
| H6 | 1(1) | 1 | (1) | - | - | - | - | 1 | - | 3(2) |
| H7 | - | - | - | - | - | - | - | - | - | 0 |
| H8 | - | - | - | - | - | - | - | - | - | 0 |
| H9 | - | 2 | - | - | - | - | - | - | - | 2 |
| H10 | - | (2) | (1) | 2 | - | - | 1(2) | (2) | - | 3(7) |
| H11 | - | 2 | - | - | - | - | - | - | 3 | 5 |
| H12 | - | - | - | - | - | - | - | - | - | 0 |
| H13 | - | - | - | - | - | - | - | - | - | 0 |
| H14 | - | - | - | - | - | - | - | - | - | 0 |
| H15 | - | - | - | - | - | - | - | - | - | 0 |
| H16 | - | - | - | - | - | - | - | - | - | 0 |
| Total | 1(2) | 6(4) | 3(3) | 2 | 0 | 5 | 1(4) | 1(4) | 3 |
| Name | HA | PB2 | PB1 | PB1-F2 | |||||||||
| Cleavage Site | E190D * | G225D * | Q226L * | G228S * | L89V † | G309D † | T339K † | E627K † | D701N † | H436Y † | D622G † | N66S † | |
| JB13-25 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| JB13-47 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| JB15-4 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB17-85 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB19-19 | PQIETR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| JB21-22 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB21-58 | PQRETR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB22-3 | PQRETR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB29-91-95 | PAIASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB31-69 | PAIASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB31-86-90 | PQIETR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| JB31-96 | PAIASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB31-103 | PAIASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB32-15 | EVVQGR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| JB32-81 | PAIASR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB32-105 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| JB36-65 | EVVQGR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB42-30 | PAASNR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB42-93 | PAASNR/G | E | G | Q | G | V | D | K | E | D | Y | G | S |
| JB42-113 | PEKASR/G | E | G | Q | G | V | D | K | E | D | Y | G | N |
| Name | PA | NP | NA | M1 | M2 | NS1 | |||||||
| N409S † | K615N † | N319K † | 54-72 deletion † | I117T § | R152K § | H274Y § | N30D † | T215A † | L26F § | S31N § | P42S † | D92E † | |
| JB13-25 | S | K | N | - | T | R | H | D | A | L | S | A | D |
| JB13-47 | S | K | N | - | T | R | H | D | A | L | S | A | D |
| JB15-4 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB17-85 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB19-19 | S | K | N | - | I | R | H | D | A | L | S | A | D |
| JB21-22 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB21-58 | S | K | N | - | T | R | H | D | A | L | S | A | D |
| JB22-3 | S | K | N | - | T | R | H | D | A | L | S | A | D |
| JB29-91-95 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB31-69 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB31-86-90 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB31-96 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB31-103 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB32-15 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB32-81 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB32-105 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB36-65 | S | K | N | - | I | R | H | D | A | L | S | A | D |
| JB42-30 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB42-93 | S | K | N | - | T | R | H | D | A | L | S | S | D |
| JB42-113 | N | K | N | - | T | R | H | D | A | L | S | A | D |
| Virus | Inoculum Titer (log10 EID50 per mL) | Lung Titer a (log10 EID50 per mL) | Seroconversion c (PI) | |||
|---|---|---|---|---|---|---|
| 1 dpi | 3 dpi | 5 dpi | 14 dpi | 14 dpi | ||
| JB31-69 (H11N9) | 6.5 | - b (3/3) | - b (1/3) | (0/3) | (0/3) | - d |
| JB32-81(H11N2) | 6.5 | 2.2 (3/3) | <1 (3/3) | <1 (3/3) | (0/3) | 1/3 (67) |
| JB21-58 (H5N3) | 6.5 | - b (2/3) | 1.9 (3/3) | - b (3/3) | (0/3) | 1/3 (74.1) |
| JB42-93 (H9N2) | 5.0 | - b (2/3) | 2.4 (3/3) | - b (3/3) | (0/3) | 0/3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Na, E.-J.; Kim, Y.-S.; Lee, S.-Y.; Kim, Y.-J.; Park, J.-S.; Oem, J.-K. Genetic Characteristics of Avian Influenza Virus Isolated from Wild Birds in South Korea, 2019–2020. Viruses 2021, 13, 381. https://doi.org/10.3390/v13030381
Na E-J, Kim Y-S, Lee S-Y, Kim Y-J, Park J-S, Oem J-K. Genetic Characteristics of Avian Influenza Virus Isolated from Wild Birds in South Korea, 2019–2020. Viruses. 2021; 13(3):381. https://doi.org/10.3390/v13030381
Chicago/Turabian StyleNa, Eun-Jee, Young-Sik Kim, Sook-Young Lee, Yoon-Ji Kim, Jun-Soo Park, and Jae-Ku Oem. 2021. "Genetic Characteristics of Avian Influenza Virus Isolated from Wild Birds in South Korea, 2019–2020" Viruses 13, no. 3: 381. https://doi.org/10.3390/v13030381
APA StyleNa, E.-J., Kim, Y.-S., Lee, S.-Y., Kim, Y.-J., Park, J.-S., & Oem, J.-K. (2021). Genetic Characteristics of Avian Influenza Virus Isolated from Wild Birds in South Korea, 2019–2020. Viruses, 13(3), 381. https://doi.org/10.3390/v13030381