
On the Prevalence and Potential Functionality of an Intrinsic Disorder in the MERS-CoV Proteome


 ,
,  , and
, and 
Abstract
1. Introduction
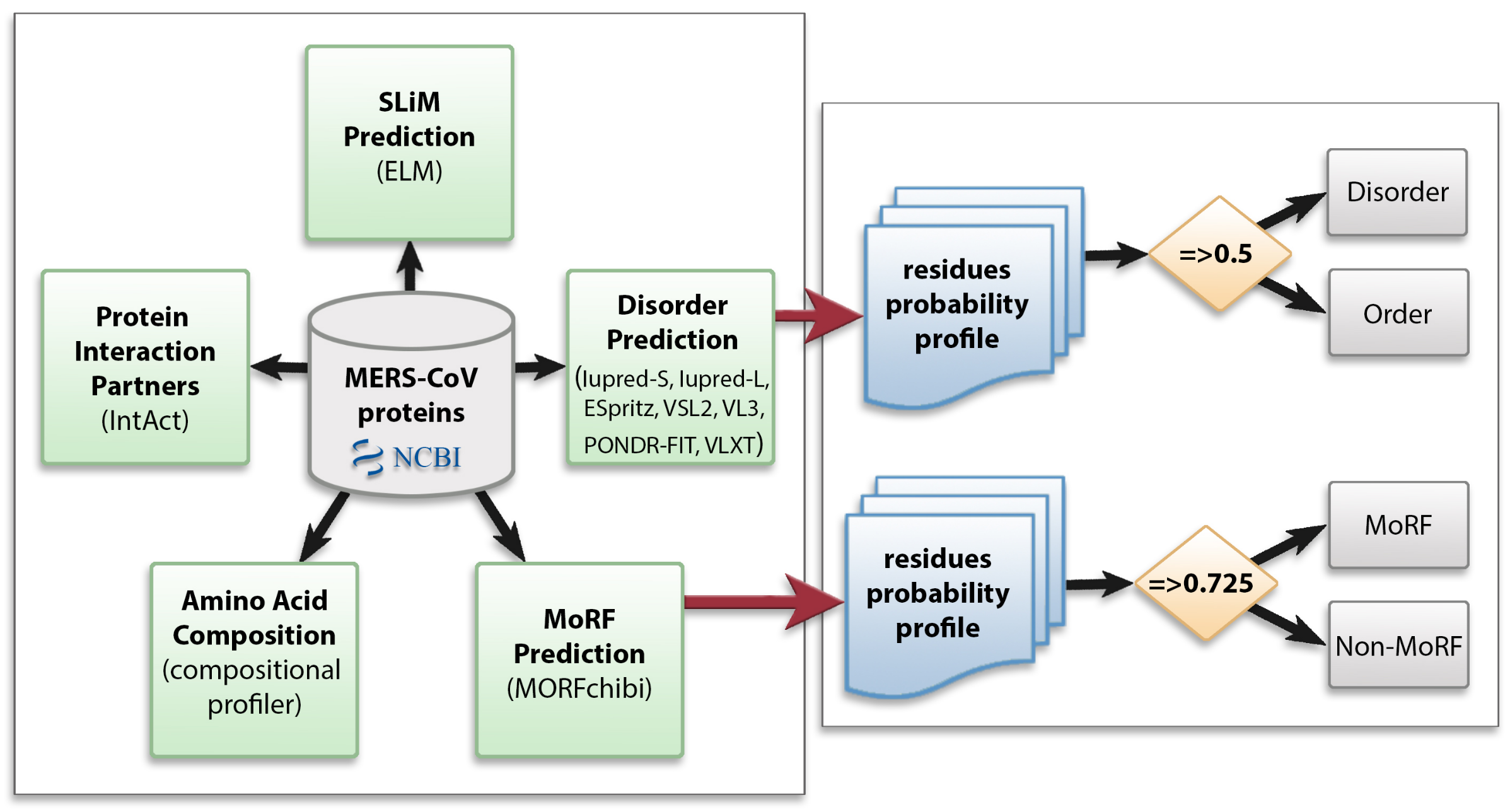
2. Materials and Methods
2.1. Data Collection
2.2. Protein Disorder Prediction
2.3. Amino Acid Compositional Profiling
2.4. Molecular Recognition Feature (MoRF) Prediction
2.5. Identification of Short Linear Motifs (SLiMs)
2.6. Interaction of MERS-CoV Proteins with Human Proteins
3. Results
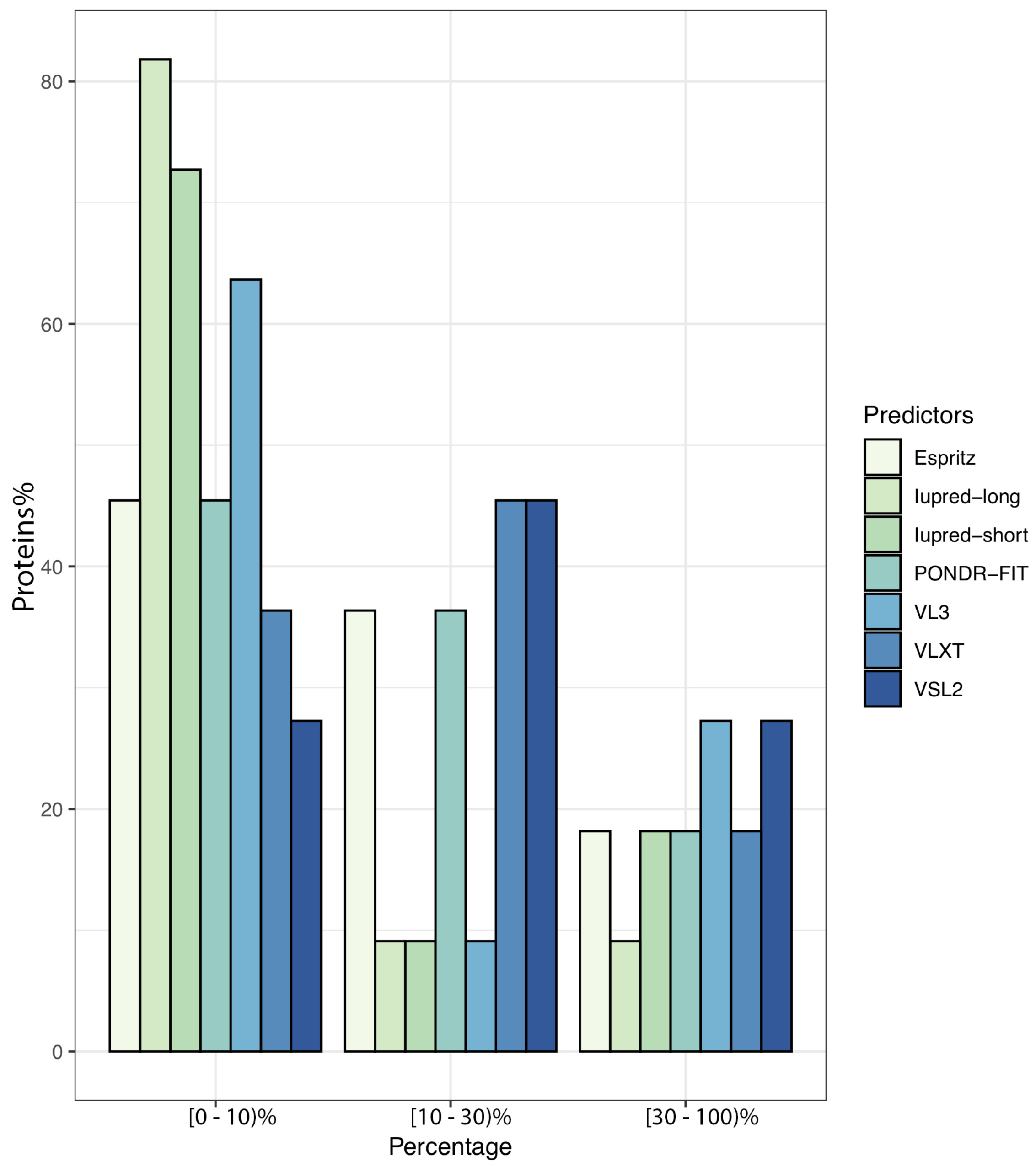
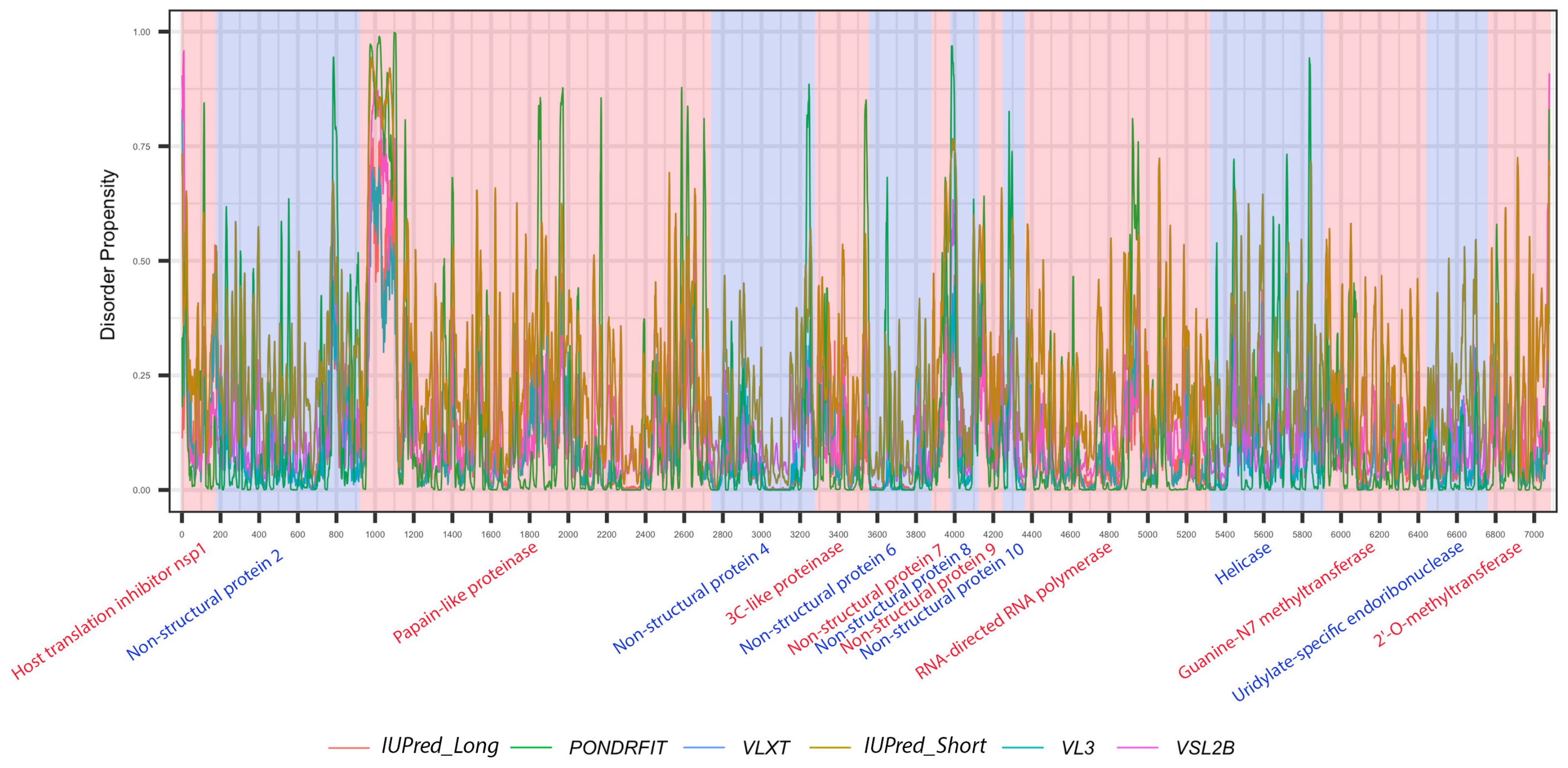
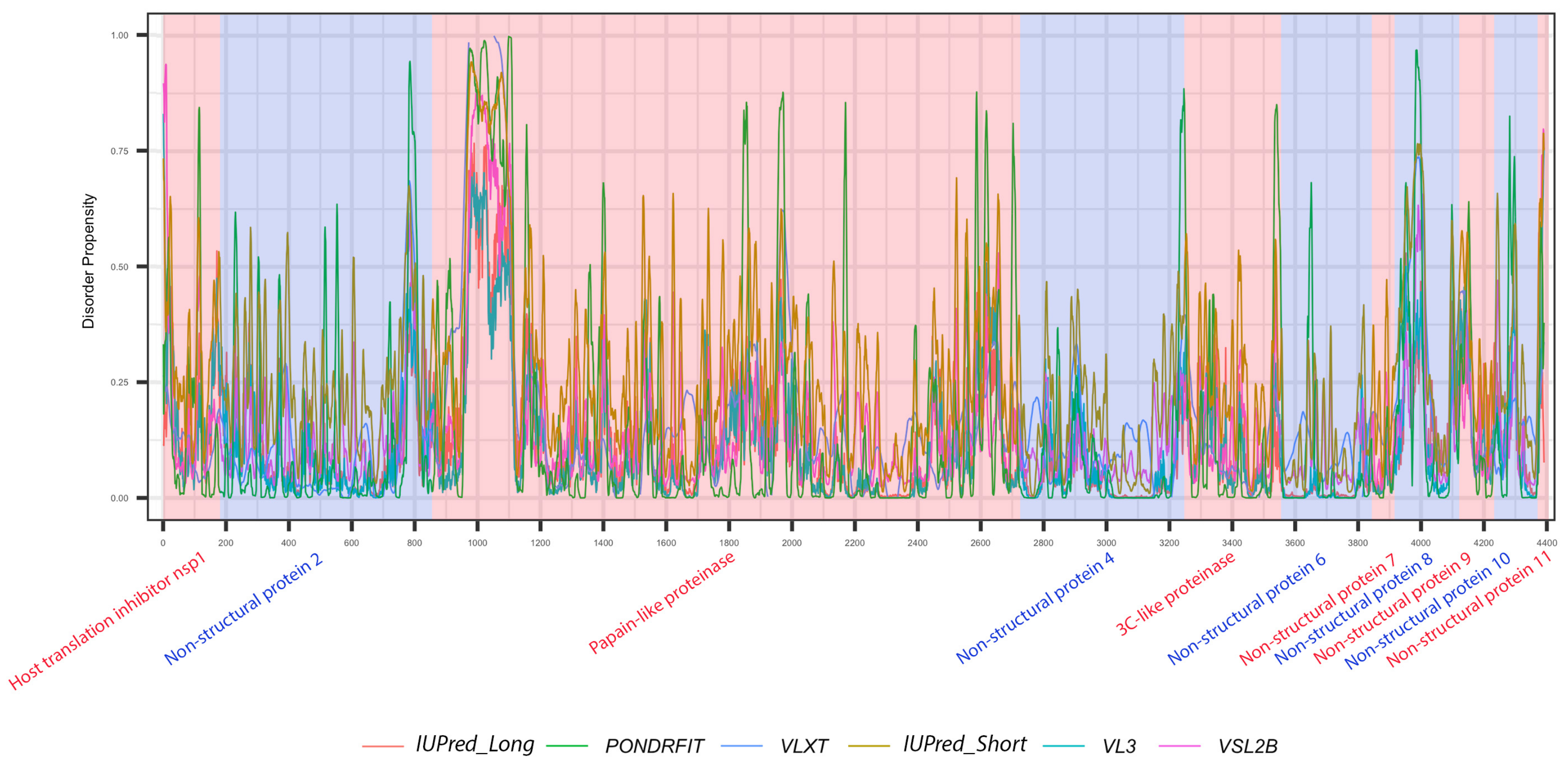
3.1. Overall Intrinsic Disorder in the MERS-CoV Proteome
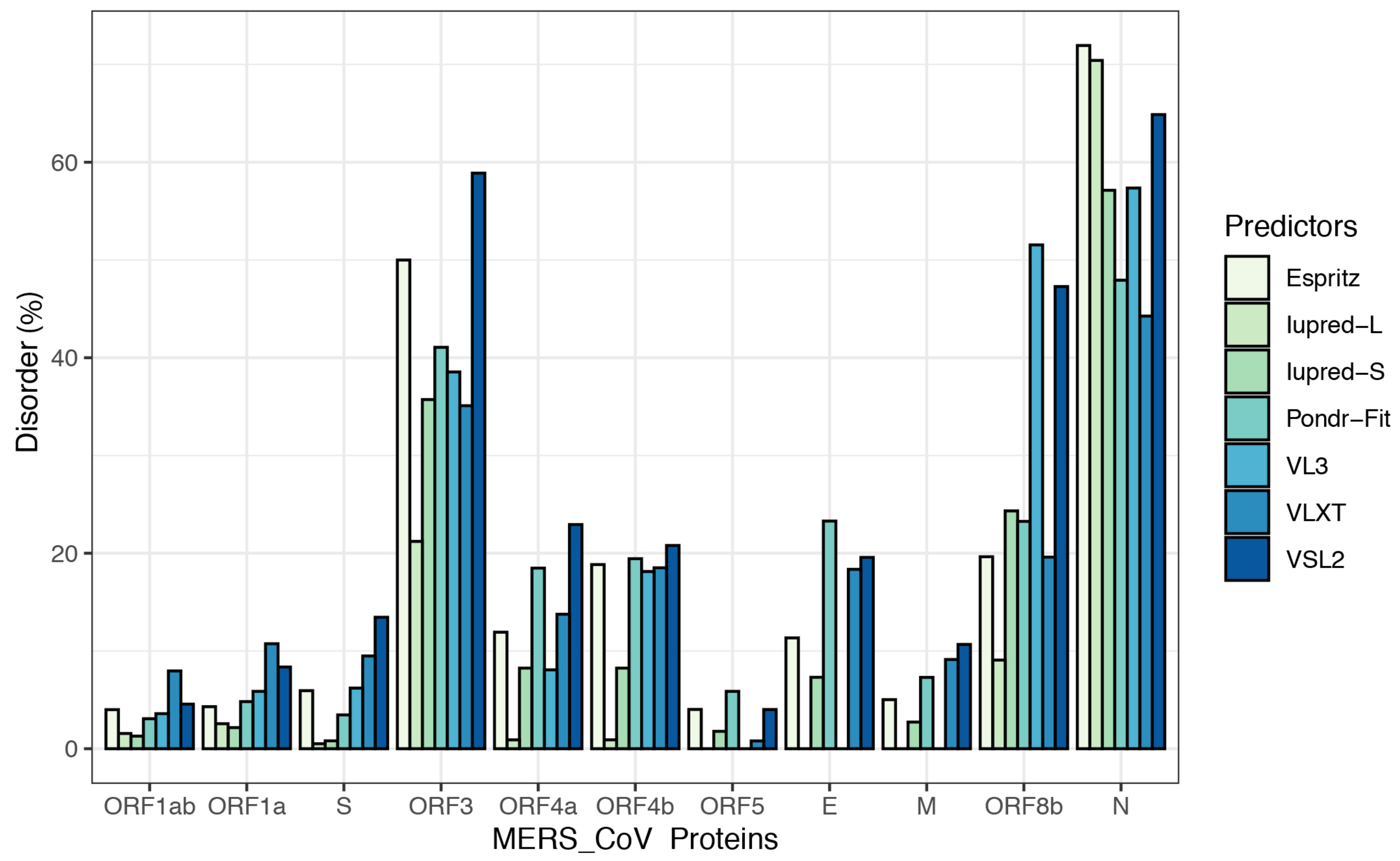
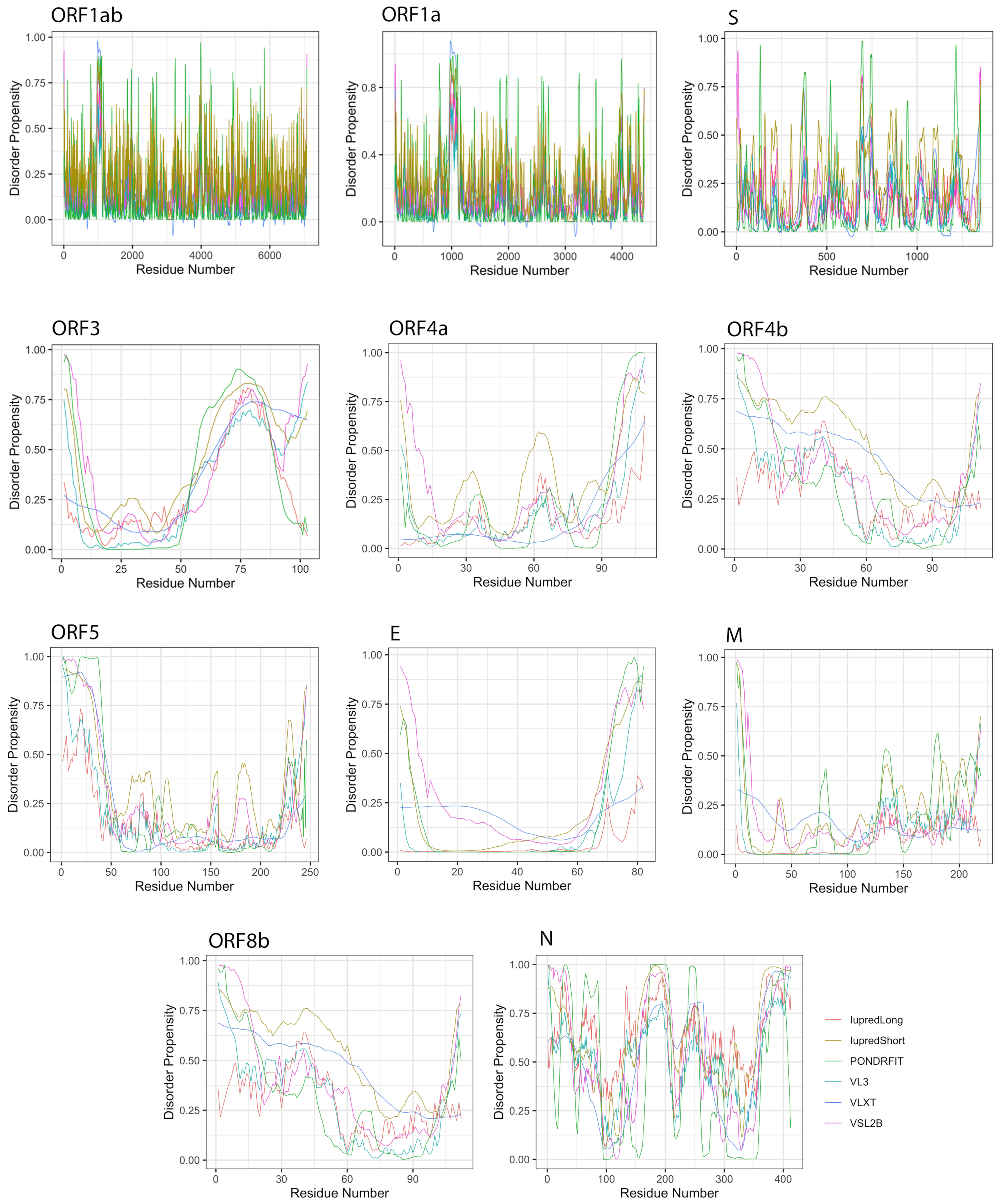
3.2. Intrinsic Disorder in MERS-CoV Structural Proteins
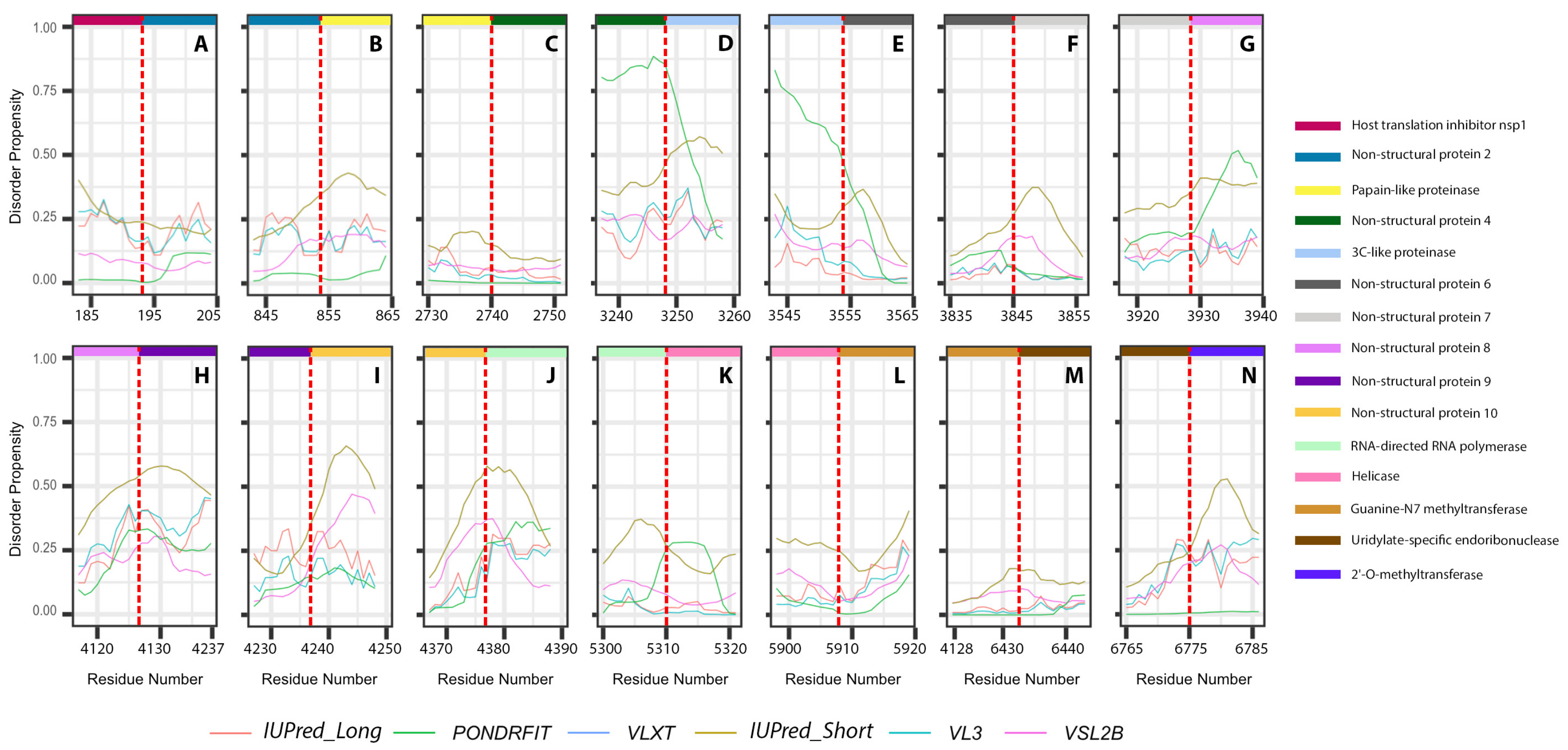
3.3. Intrinsic Disorder in MERS-CoV Non-Structural Proteins
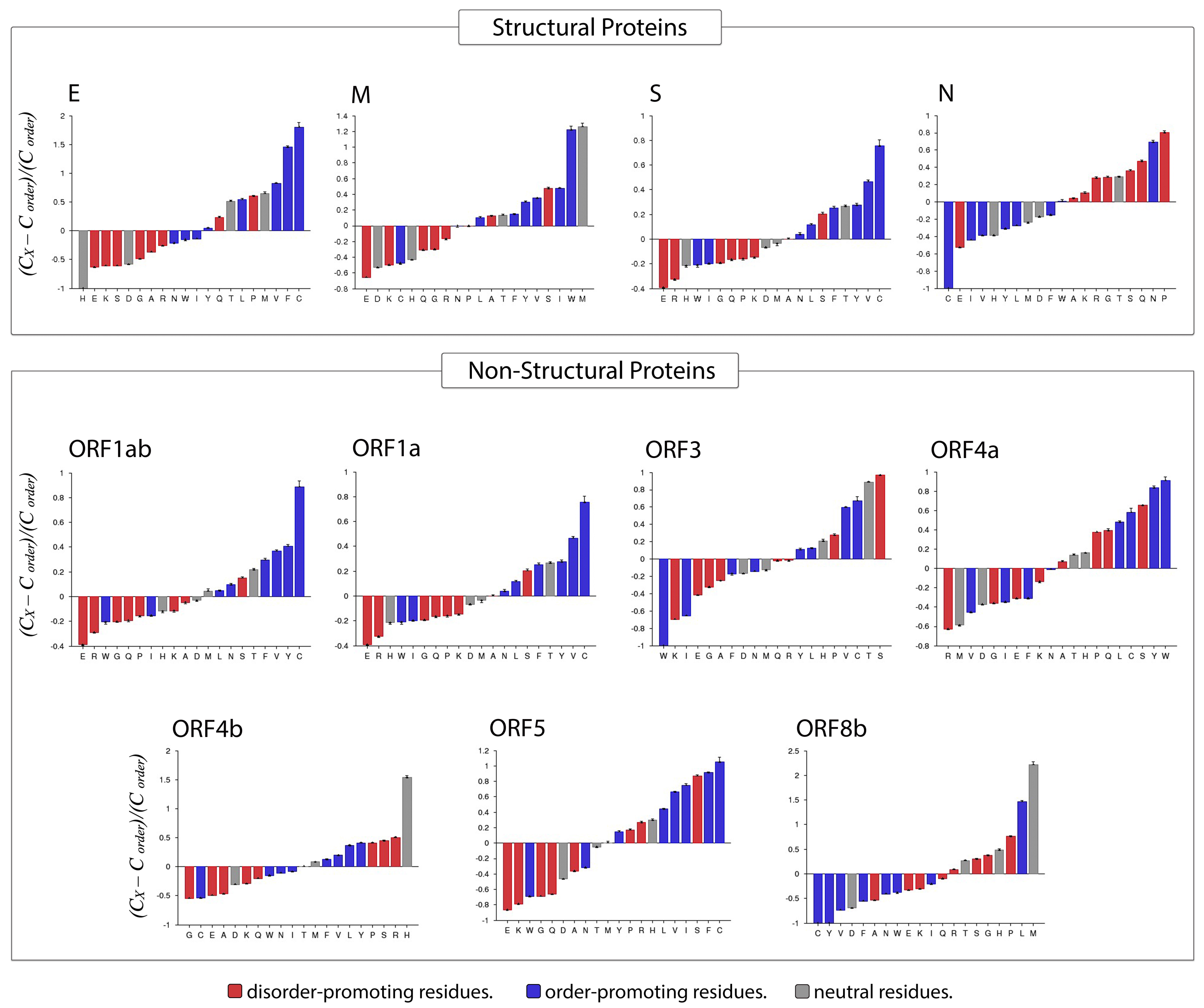
3.4. Amino Acid Compositional Profiling
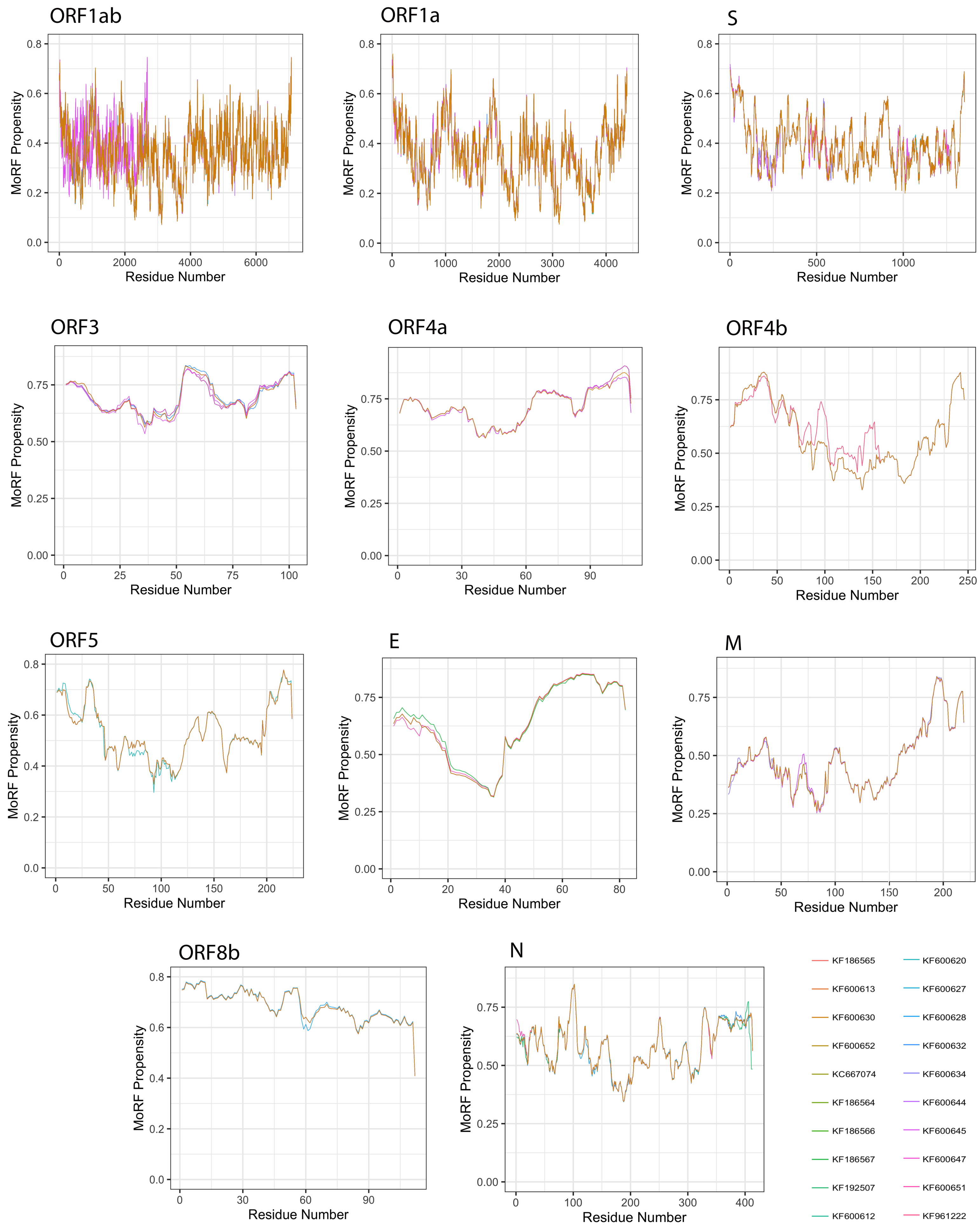
3.5. Analysis of Molecular Recognition Features (MoRFs)
3.6. Short Linear Motif (SLiM) Analysis
3.7. MERS-CoV Protein Interactions with Human Proteins
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al Sulayyim, H.J.; Khorshid, S.M.; Al Moummar, S.H. Demographic, clinical, and outcomes of confirmed cases of Middle East Respiratory Syndrome coronavirus (MERS-CoV) in Najran, Kingdom of Saudi Arabia (KSA); A retrospective record based study. J. Infect. Public Health 2020, 13, 1342–1346. [Google Scholar] [CrossRef]
- Al-Shomrani, B.M.; Manee, M.M.; Alharbi, S.N.; Altammami, M.A.; Alshehri, M.A.; Nassar, M.S.; Bakhrebah, M.A.; Al-Fageeh, M.B. Genomic Sequencing and Analysis of Eight Camel-Derived Middle East Respiratory Syndrome Coronavirus (MERS-CoV) Isolates in Saudi Arabia. Viruses 2020, 12, 611. [Google Scholar] [CrossRef]
- Zumla, A.; Chan, J.F.; Azhar, E.I.; Hui, D.S.; Yuen, K.Y. Coronaviruses—Drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef]
- Li, Y.H.; Hu, C.Y.; Wu, N.P.; Yao, H.P.; Li, L.J. Molecular characteristics, functions, and related pathogenicity of MERS-CoV proteins. Engineering 2019, 5, 940–947. [Google Scholar] [CrossRef]
- Jiaming, L.; Yanfeng, Y.; Yao, D.; Yawei, H.; Linlin, B.; Baoying, H.; Jinghua, Y.; Gao, G.F.; Chuan, Q.; Wenjie, T. The recombinant N-terminal domain of spike proteins is a potential vaccine against Middle East respiratory syndrome coronavirus (MERS-CoV) infection. Vaccine 2017, 35, 10–18. [Google Scholar] [CrossRef]
- Perrier, A.; Bonnin, A.; Desmarets, L.; Danneels, A.; Goffard, A.; Rouillé, Y.; Dubuisson, J.; Belouzard, S. The C-terminal domain of the MERS coronavirus M protein contains a trans-Golgi network localization signal. J. Biol. Chem. 2019, 294, 14406–14421. [Google Scholar] [CrossRef]
- Lin, S.M.; Lin, S.C.; Hsu, J.N.; Chang, C.k.; Chien, C.M.; Wang, Y.S.; Wu, H.Y.; Jeng, U.S.; Kehn-Hall, K.; Hou, M.H. Structure-based stabilization of non-native protein–protein interactions of coronavirus nucleocapsid proteins in antiviral drug design. J. Med. Chem. 2020, 63, 3131–3141. [Google Scholar] [CrossRef]
- Ward, J.J.; Sodhi, J.S.; McGuffin, L.J.; Buxton, B.F.; Jones, D.T. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 2004, 337, 635–645. [Google Scholar] [CrossRef]
- Frege, T.; Uversky, V.N. Intrinsically disordered proteins in the nucleus of human cells. Biochem. Biophys. Rep. 2015, 1, 33–51. [Google Scholar] [CrossRef]
- Alshehri, M.A.; Manee, M.M.; Al-Fageeh, M.B.; Al-Shomrani, B.M. Genomic Analysis of Intrinsically Disordered Proteins in the Genus Camelus. Int. J. Mol. Sci. 2020, 21, 4010. [Google Scholar] [CrossRef]
- Pietrosemoli, N.; García-Martín, J.A.; Solano, R.; Pazos, F. Genome-wide analysis of protein disorder in Arabidopsis thaliana: Implications for plant environmental adaptation. PLoS ONE 2013, 8, e55524. [Google Scholar] [CrossRef]
- Van Der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding protein non-folding. Biochim. Biophys. Acta-(Bba)-Proteins Proteom. 2010, 1804, 1231–1264. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder-based protein interactions and their modulators. Curr. Pharm. Des. 2013, 19, 4191–4213. [Google Scholar] [CrossRef]
- Xue, B.; Dunker, A.K.; Uversky, V.N. Orderly order in protein intrinsic disorder distribution: Disorder in 3500 proteomes from viruses and the three domains of life. J. Biomol. Struct. Dyn. 2012, 30, 137–149. [Google Scholar] [CrossRef]
- Fan, X.; Xue, B.; Dolan, P.T.; LaCount, D.J.; Kurgan, L.; Uversky, V.N. The intrinsic disorder status of the human hepatitis C virus proteome. Mol. Biosyst. 2014, 10, 1345–1363. [Google Scholar] [CrossRef]
- Xue, B.; Mizianty, M.J.; Kurgan, L.; Uversky, V.N. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell. Mol. Life Sci. 2012, 69, 1211–1259. [Google Scholar] [CrossRef]
- Uversky, V.N.; Roman, A.; Oldfield, C.J.; Dunker, A.K. Protein intrinsic disorder and human papillomaviruses: Increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J. Proteome Res. 2006, 5, 1829–1842. [Google Scholar] [CrossRef]
- Redwan, E.M.; AlJaddawi, A.A.; Uversky, V.N. Structural disorder in the proteome and interactome of Alkhurma virus (ALKV). Cell. Mol. Life Sci. 2019, 76, 577–608. [Google Scholar] [CrossRef]
- Chen, J.; Liu, X.; Chen, J. Targeting Intrinsically Disordered Proteins through Dynamic Interactions. Biomolecules 2020, 10, 743. [Google Scholar] [CrossRef]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef]
- Walsh, I.; Martin, A.J.; Di Domenico, T.; Tosatto, S.C. ESpritz: Accurate and fast prediction of protein disorder. Bioinformatics 2012, 28, 503–509. [Google Scholar] [CrossRef]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta-(Bba)-Proteins Proteom. 2010, 1804, 996–1010. [Google Scholar] [CrossRef]
- Davey, N.E.; Travé, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar] [CrossRef]
- Uyar, B.; Weatheritt, R.J.; Dinkel, H.; Davey, N.E.; Gibson, T.J. Proteome-wide analysis of human disease mutations in short linear motifs: Neglected players in cancer? Mol. Biosyst. 2014, 10, 2626–2642. [Google Scholar] [CrossRef]
- Peng, K.; Vucetic, S.; Radivojac, P.; Brown, C.J.; Dunker, A.K.; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol. 2005, 3, 35–60. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Obradovic, Z.; Peng, K.; Vucetic, S.; Radivojac, P.; Dunker, A.K. Exploiting heterogeneous sequence properties improves prediction of protein disorder. Proteins Struct. Funct. Bioinform. 2005, 61, 176–182. [Google Scholar] [CrossRef]
- Rajagopalan, K.; Mooney, S.M.; Parekh, N.; Getzenberg, R.H.; Kulkarni, P. A majority of the cancer/testis antigens are intrinsically disordered proteins. J. Cell. Biochem. 2011, 112, 3256–3267. [Google Scholar] [CrossRef]
- Lyngdoh, D.L.; Shukla, H.; Sonkar, A.; Anupam, R.; Tripathi, T. Portrait of the Intrinsically Disordered Side of the HTLV-1 Proteome. ACS Omega 2019, 4, 10003–10018. [Google Scholar] [CrossRef]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition Profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef]
- Malhis, N.; Jacobson, M.; Gsponer, J. MoRFchibi SYSTEM: Software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016, 44, W488–W493. [Google Scholar] [CrossRef]
- Kumar, M.; Gouw, M.; Michael, S.; Sámano-Sánchez, H.; Pancsa, R.; Glavina, J.; Diakogianni, A.; Valverde, J.A.; Bukirova, D.; Čalyševa, J.; et al. ELM—The eukaryotic linear motif resource in 2020. Nucleic Acids Res. 2020, 48, D296–D306. [Google Scholar] [CrossRef]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef]
- Fontana, A.; De Laureto, P.P.; Spolaore, B.; Frare, E.; Picotti, P.; Zambonin, M. Probing protein structure by limited proteolysis. Acta Biochim. Pol. 2004, 51, 299–321. [Google Scholar] [CrossRef]
- Chang, C.k.; Lo, S.C.; Wang, Y.S.; Hou, M.H. Recent insights into the development of therapeutics against coronavirus diseases by targeting N protein. Drug Discov. Today 2016, 21, 562–572. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Mishra, P.M.; Verma, N.C.; Rao, C.; Uversky, V.N.; Nandi, C.K. Intrinsically disordered proteins of viruses: Involvement in the mechanism of cell regulation and pathogenesis. Prog. Mol. Biol. Transl. Sci. 2020, 174, 1–78. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean Content of Disorder Residues (%) | Mean Proteins with at Least One LDR (%) | Average Length of LDRs (by Residues) | |
|---|---|---|---|
| IUPred-short | 3.94 | 36.36 | 43.1 |
| IUPred-long | 4.01 | 26.81 | 32.02 |
| ESpritz | 7.02 | 36.36 | 48.3 |
| VSL2 | 12.17 | 54.09 | 63.96 |
| PONDR-FIT | 6.03 | 35.90 | 53.37 |
| VLXT | 10.51 | 41.36 | 61.14 |
| VL3 | 6.91 | 54.09 | 62.05 |
| Average | 7.84 | 42.38 | 53.48 |
| Protein | PPIDshort | PPIDlong | PPIDEspritz | PPIDVSL2 | PPIDpondr-fit | PPIDVLXT | PPID VL3 | PPIDmean |
|---|---|---|---|---|---|---|---|---|
| ORF1ab | 1.29 | 1.56 | 4.00 | 4.56 | 3.07 | 7.96 | 3.59 | 3.72 |
| ORF1a | 2.16 | 2.56 | 4.30 | 8.36 | 4.82 | 10.74 | 5.87 | 5.54 |
| S | 0.81 | 0.51 | 5.94 | 13.45 | 3.46 | 9.49 | 6.2 | 5.69 |
| ORF3 | 35.72 | 21.21 | 49.99 | 58.88 | 41.06 | 35.09 | 38.54 | 40.07 |
| ORF4a | 8.25 | 0.91 | 11.92 | 22.93 | 18.48 | 13.76 | 8.07 | 12.05 |
| ORF4b | 8.25 | 0.91 | 18.84 | 20.79 | 19.44 | 18.51 | 18.13 | 14.98 |
| ORF5 | 1.78 | 0 | 4.02 | 4.01 | 5.87 | 0.8 | 0 | 2.35 |
| E | 7.31 | 0 | 11.34 | 19.57 | 23.29 | 18.35 | 0 | 11.41 |
| M | 2.73 | 0 | 5.02 | 10.67 | 7.3 | 9.13 | 0 | 4.98 |
| ORF8b | 24.33 | 9.07 | 19.64 | 47.28 | 23.26 | 19.6 | 51.54 | 27.82 |
| N | 57.13 | 70.41 | 71.94 | 64.87 | 47.929 | 44.26 | 57.36 | 59.13 |
| Protein | Length | MoRFs (%) | MoRFs Regions |
|---|---|---|---|
| ORF1ab | 7078 | 0.063 | 7074–7077 |
| ORF1a | 4391 | 0.101 | 12–15 |
| S | 1353 | 0 | 0 |
| ORF3 | 103 | 37.135 | 1–9 |
| 53–64 | |||
| 87–102 | |||
| ORF4a | 109 | 45.688 | 3–10 |
| 63–81 | |||
| 87–109 | |||
| ORF4b | 246 | 25.811 | 6–7 |
| 9–46 | |||
| 52–58 | |||
| 231–246 | |||
| ORF5 | 224 | 4.531 | 32–34 |
| 213–219 | |||
| E | 82 | 37.743 | 51–81 |
| M | 219 | 8.675 | 190–218 |
| ORF8b | 112 | 28.660 | 1–15 |
| 20 | |||
| 26–38 | |||
| 50–56 | |||
| N | 413 | 3.947 | 95–104 |
| 328–332 |
| Protein | Number of SLiM | Number of SLiM Instances | SLiM Name | SLiM Sequence | SLiM Location |
|---|---|---|---|---|---|
| ORF1ab | 137 | 2960 | DOC_PP2A_B56_1 | LNFVGEF | 484–490 |
| LTGLGES | 562–568 | ||||
| LDTCFEA | 655–661 | ||||
| YVIISE | 815–820 | ||||
| YTPIDE | 2880–2885 | ||||
| IATIKE | 5461–5466 | ||||
| LLLVWEA | 5473–5479 | ||||
| CCRIVE | 6216–6221 | ||||
| LGTIKE | 6987–6992 | ||||
| LIG_G3BP_FGDF_1 | YDFGDF | 4595−4600 | |||
| LIG_IRF3_LxIS_1 | VRAYLGIS | 2220–2227 | |||
| VDLVIS | 6899–6904 | ||||
| INELVIS | 7042–7048 | ||||
| LIG_NRP_CendR_1 | RKLR | 7075–7078 | |||
| KLR | 7076–7078 | ||||
| ORF1a | 122 | 1918 | DOC_PP2A_B56_1 | LNFVGEF | 484–490 |
| LTGLGES | 562–568 | ||||
| LDTCFEA | 655–661 | ||||
| YVIISE | 815–820 | ||||
| YTPIDE | 2880–2885 | ||||
| LIG_IRF3_LxIS_1 | VRAYLGIS | 2220–2227 | |||
| S | 84 | 660 | DOC_PP2A_B56_1 | FYCILE | 183–188 |
| LGNCVEY | 600–606 | ||||
| ORF3 | 23 | 48 | These residues are predicted in well folded region (globular protein domains) | ||
| ORF4a | 35 | 58 | These residues are predicted in well folded region (globular protein domains) | ||
| ORF4b | 54 | 102 | These residues are predicted in well folded region (globular protein domains) | ||
| ORF5 | 42 | 97 | These residues are predicted in well folded region (globular protein domains) | ||
| E | 16 | 23 | DOC_PP2A_B56_1 | LPFVQER | 2–8 |
| M | 43 | 103 | These residues are predicted in well folded region (globular protein domains) | ||
| ORF8 | 20 | 37 | These residues are predicted in well folded region (globular protein domains) | ||
| N | 51 | 151 | DOC_PP2A_B56_1 | WPQIAE | 293–298 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alshehri, M.A.; Manee, M.M.; Alqahtani, F.H.; Al-Shomrani, B.M.; Uversky, V.N. On the Prevalence and Potential Functionality of an Intrinsic Disorder in the MERS-CoV Proteome. Viruses 2021, 13, 339. https://doi.org/10.3390/v13020339
Alshehri MA, Manee MM, Alqahtani FH, Al-Shomrani BM, Uversky VN. On the Prevalence and Potential Functionality of an Intrinsic Disorder in the MERS-CoV Proteome. Viruses. 2021; 13(2):339. https://doi.org/10.3390/v13020339
Chicago/Turabian StyleAlshehri, Manal A., Manee M. Manee, Fahad H. Alqahtani, Badr M. Al-Shomrani, and Vladimir N. Uversky. 2021. "On the Prevalence and Potential Functionality of an Intrinsic Disorder in the MERS-CoV Proteome" Viruses 13, no. 2: 339. https://doi.org/10.3390/v13020339
APA StyleAlshehri, M. A., Manee, M. M., Alqahtani, F. H., Al-Shomrani, B. M., & Uversky, V. N. (2021). On the Prevalence and Potential Functionality of an Intrinsic Disorder in the MERS-CoV Proteome. Viruses, 13(2), 339. https://doi.org/10.3390/v13020339