2.3. General Method for Peptide Cyclization
Peptide cyclization was carried out, as described by Meuleman et al. [
33].
Cyclic peptide
6. Crude peptide
3 (0.35 g, 0.25 μmol, 1.0 equiv.) was treated with azido triazinane-tris(2-bromoethanone (TADB-N
3) [
34] (0.12 g, 0.30 mmol, 1.2 equiv.). Purification was done in batches of 30–40 mg crude dissolved in 3 mL buffer A:B (75:25,
v/v) while using a custom protocol: 27% buffer B in buffer A for 2 min., followed by a linear gradient of 27–38% buffer B in buffer A for 10 min., which afforded cyclic peptide 6 (0.12 g, 49 μmol, 20%). t
R = 17.4 min.; High Resolution(HR)MS: calculated
m/z for C
105H
150N
34O
34S
2: 1248.5326 ½[
M+2H]
2+; found 1248.5361; LRMS: calculated
m/z for C
105H
150N
34O
34S
2: 1248.53 ½[
M+2H]
2+; found 1249.58.
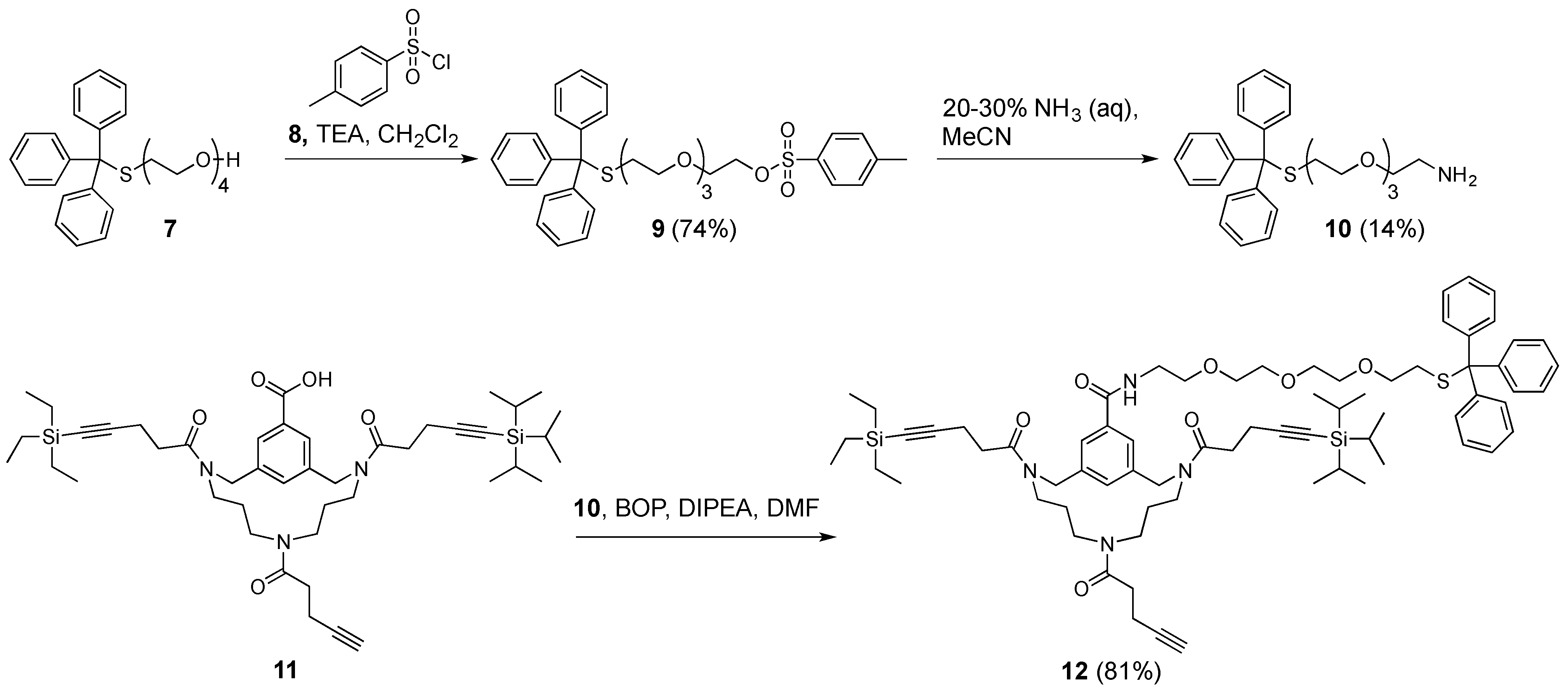
Tetraethylene glycol monotrityl thioether (
7). Synthesized, as described Meuleman et al. [
32].
Tetraethylene glycol monotrityl thioether p-toluenesulfonate (9). Tetraethylene glycol monotrityl thioether 7 (2.6 g, 5.8 mmol, 1.0 equiv.) was dissolved in anhydrous CH2Cl2 (50 mL). The resulting solution was cooled to 0 °C using an ice bath, followed by the addition of TEA (1.6 mL, 12 mmol, 2.1 equiv.). After 1 h, p-toluenesulfonyl chloride 8 (2.3 g, 12 mmol, 2.1 equiv.) was added. The resulting reaction mixture was stirred overnight at room temperature. Upon completion of the reaction, as determined by TLC (50% EtOAc in petroleum ether 40–60 °C), the mixture was washed with H2O (100 mL). Next, the aqueous layer was back-extracted with CH2Cl2 (2 × 100 mL). Subsequently, the combined organic layers were washed using brine (1 × 100 mL), dried over MgSO4, and then filtered. The filtrate was concentrated in vacuo and purified by automated flash column chromatography using a linear gradient (30–50% EtOAc in petroleum ether 40–60 °C over 10 column volumes). Pure fractions were combined and concentrated in vacuo, affording pure tetraethylene glycol monotrityl thioether ρ-toluenesulfonate 9 (2.6 g, 4.3 mmol, 74%) as a yellowish oil. Rf = 0.52 (50% EtOAc in petroleum ether 40–60 °C); tR = 46.6 min.; 1H-NMR (400 MHz, CDCl3): δ = 7.79 (d, 3JHH = 8.2 Hz, 2H, aryl o-H), 7.41 (m, 6H, trityl o-H), 7.32 (d, 3JHH = 8.2 Hz, 2H, aryl m-H), 7.28 (m, 6H, trityl m-H), 7.21 (m, 3H, trityl p-H), 4.14 (t, 3JHH = 4.8 Hz, 2H, TsOCH2), 3.66 (t, 3JHH = 4.8 Hz, 2H, TsOCH2CH2), 3.53 (m, 6H, 3 × CH2), 3.43 (m, 2H, CH2), 3.30 (t, 3JHH = 6.9 Hz, 2H, SCH2CH2), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH2), 2.43 (s, 3H, CH3) ppm; 13C-NMR (101 MHz, CDCl3): δ = 144.8 (aryl-C), 144.7 (aryl-C), 133.1 (aryl-C), 130.1 (aryl-C), 129.8 (aryl m-CH), 129.6 (trityl o-CH), 128.0 (aryl o-CH), 127.9 (trityl m-CH), 126.6 (trityl p-CH), 70.7 (CH2), 70.5 (CH2), 70.5 (CH2), 70.2 (CH2), 69.6 (SCH2CH2), 69.2 (TsOCH2), 68.7 (TsOCH2CH2), 66.6 (CS), 31.7 (SCH2), 21.6 (CH3) ppm; HRMS: calculated m/z for C34H38O6S2: 629.2008 [M+Na]1+; found 629.1996.
Triethylene glycol monotrityl thioether ethylamine (10). p-toluenesulfonate tetraethylene glycol monotrityl thioether 9 (2.6 g, 4.3 mmol, 1.0 equiv.) was dissolved in MeCN (100 mL), followed by the addition of aqueous 25% ammonia (50 mL). The resulting reaction mixture was stirred overnight at room temperature. Although the reaction was not complete according to TLC (20% MeOH in CH2Cl2), work-up was carried by the removal of MeCN in vacuo and extraction of the aqueous layer with CH2Cl2 that was supplemented with 1% TEA (3 × 50 mL). The combined CH2Cl2 layer were washed with brine (100 mL), dried over NaSO4, and then filtered. The filtrate was concentrated in vacuo, followed by purification using automated flash column chromatography using a linear gradient (0–10% MeOH in CH2Cl2 supplemented with 1% TEA over 10 column volumes). Pure triethylene glycol monotrityl thioether ethylamine 10 (0.26 g, 0.58 mmol, 14%) was obtained as a yellowish oil. Rf = 0.27 (10% MeOH in CH2Cl2 supplemented with TEA); tR = 34.4 min.; 1H-NMR (400 MHz, CDCl3): δ = 7.41 (m, 6H, trityl o-H), 7.27 (m, 6H, trityl m-H), 7.21 (m, 3H, trityl p-H), 3.61 (m, 4H, 2 × CH2), 3.57 (m, 2H, CH2), 3.50 (t, 3JHH = 5.2 Hz, 2H, CH2CH2NH3+), 3.45 (m, 2H, CH2), 3.31 (t, 3JHH = 6.9 Hz, 2H, SCH2CH2), 2.85 (t, 3JHH = 5.2 Hz, 2H, CH2NH3+), 2.43 (t, 3JHH = 6.9 Hz, 2H, SCH2), 1.91 (broad s, 3H, NH3+) ppm; 13C-NMR (101 MHz, CDCl3): δ = 144.8 (trityl-C), 129.6 (trityl o-CH), 127.9 (trityl m-CH), 126.7 (trityl p-CH), 73.1 (CH2CH2NH3+), 70.6 (CH2), 70.5 (CH2), 70.3 (CH2), 70.2 (CH2), 69.6 (SCH2CH2), 66.6 (CS), 41.7 (CH2NH3+), and 31.7 (SCH2) ppm; HRMS: calculated m/z for C27H33NO3S: 452.2259 [M+H]1+; found 452.2244.
Orthogonally protected trialkyne TAC-scaffold (
11). This compound was synthesized according to the earlier reported procedure of Werkhoven et al. [
31].
Triethylene glycol monotrityl thioether ethylamine that was equipped TAC-scaffold (12). TAC-scaffold 11 (42 mg, 53 μmol, 1.0 equiv.) was dissolved in DMF (8 mL). Next, benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) (30 mg, 68 μmol, 1.3 equiv.) was dissolved in DMF (3 × 2 mL) and subsequently added to the solution containing TAC-scaffold 25. Subsequently, triethylene glycol monotrityl thioether ethylamine 10 (51 mg, 0.11 mmol) was dissolved in DMF (3 × 2 mL) and added. Finally, DiPEA (37 μL, 0.21 mmol, 4.0 equiv.) was added and the reaction mixture was stirred for 1 h at room temperature. Upon completion of the reaction, as determined by LCMS, solvent was removed in vacuo. The residue was taken up in tBuOH/H2O (1/1, v/v) and then lyophilized. The crude yellowish oil was dissolved in CH2Cl2 and purified manually using silica gel chromatography (2% MeOH in CH2Cl2) and visualized with UV and potassium permanganate stain. Pure fractions were combined and concentrated in vacuo, the residue taken up in tBuOH/H2O (1/1, v/v) and lyophilized, affording pure triethylene glycol monotrityl thioether ethylamine equipped TAC-scaffold 12 (52 mg, 43 μmol, 81%) as a white powder. Rf = 0.52 (5% MeOH in CH2Cl2); tR = 49.6 min.; 1H-NMR (400 MHz, CDCl3): δ = 7.69 (m, 2H, aryl-H), 7.39 (m, 6H, trityl o-H), 7.34 (m, 1H, aryl-H), 7.27 (m, 6H, trityl m-H), 7.20 (m, 3H, trityl p-H), 6.84 (m, 1H, OCH2CH2NH), 4.61 (m, 4H, 2 × NCH2-aryl), 3.64 (m, 8H, 4 × CH2), 3.58 (m, 2H, OCH2CH2NH), 3.43 (m, 6H, NCH2CH2CH2N and OCH2CH2NH), 3.27 (t, 3JHH = 6.8 Hz, 2H, SCH2CH2), 2.96 (m, 4H, NCH2CH2CH2N), 2.64 (m, 8H, 2 × CH2CH2CCSi), 2.44 (m, 4H, CH2CH2CCH), 2.39 (t, 3JHH = 6.8 Hz, 2H, SCH2CH2), 1.95 (m, 1H, CCH), 1.45 (m, 4H, 2 × NCH2CH2CH2N), 1.05 (m, 21H, Si(CH(CH3)2)3), 0.97 (t, 3JHH = 7.9 Hz, 9H, Si(CH2CH3)3), 0.58 (q, 3JHH = 7.9 Hz, 6H, Si(CH2CH3)3) ppm; 13C-NMR (101 MHz, CDCl3): δ = 144.7 (trityl-C), 129.6 (trityl o-CH), 127.9 (trityl m-CH), 126.8 (aryl-CH) 126.6 (trityl p-CH), 126.2 (aryl-CH), 70.6 (CH2), 70.4 (OCH2CH2NH), 70.3 (CH2), 70.1 (OCH2CH2NH), 69.7 (CH2), 69.6 (SCH2CH2), 68.6 (CCH), 53.9 (NCH2-aryl), 51.9 (NCH2-aryl), 47.8 (NCH2CH2CH2N), 45.8 (NCH2CH2CH2N), 45.6 (NCH2CH2CH2N), 43.5 (NCH2CH2CH2N), 40.0 (CH2), 33.0 (CH2CH2CCSi), 32.9 (CH2CH2CCSi), 31.9 (CH2CH2CCH), 31.7 (SCH2CH2), 28.5 (NCH2CH2CH2N), 27.5 (NCH2CH2CH2N), 18.7 (Si(CH(CH3)2)3), 18.4 (CCH), 16.4 (CH2CH2CCSi), 16.4 (CH2CH2CCSi), 14.5 (CH2CH2CCH), 11.3 (Si(CH(CH3)2)3), 7.5 (Si(CH2CH3)3), and 4.4 (Si(CH2CH3)3) ppm; HRMS: calculated m/z for C72H100N4O7SSi2: 1221.6929 [M+H]1+; found 1221.6870; LRMS: calculated m/z for C72H100N4O7SSi2: 1221.69 [M+H]1+/1243.67 [M+Na]1+; found 1222.92/1244.17.
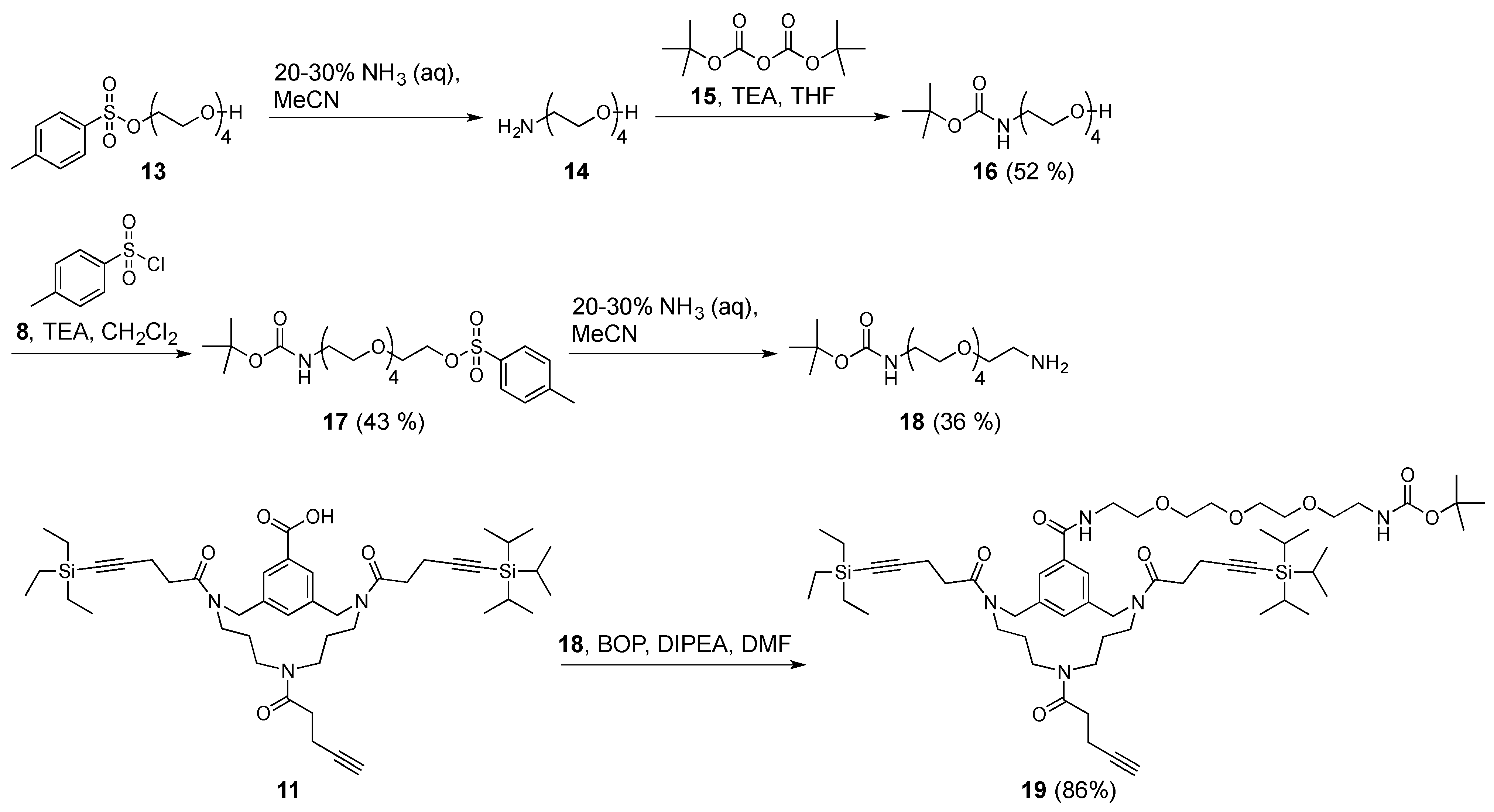
Tetraethylene glycol p-toluenesulfonate (
13). This compound was obtained, as described earlier by Meuleman et al. [
32].
Tetraethylene glycol Boc-amide (16). Tetraethylene glycol p-toluenesulfonate 13 (7.2 g, 21 mmol, 1.0 equiv.) was taken up in aqueous 25% ammonia (50 mL), and the reaction was stirred overnight at room temperature. The solvents were removed in vacuo and the residue re-dissolved in MeCN (50 mL) and fresh aqueous 25% ammonia (50 mL) was added because the reaction was not fully complete, as determined by TLC (100% EtOAc) and visualized with a ninhydrin stain. After stirring for an additional 8 h, the reaction was still not complete. Again, the solvents were removed in vacuo and the residue was now taken up in aqueous 25% ammonia (50 mL) and stirred overnight at room temperature. Finally, the next morning, the reaction was complete, as determined by TLC (100% EtOAc) and visualized with a ninhydrin stain. The solvent was removed in vacuo and the residue was co-evaporated with toluene (2×). Next, the thus obtained crude amine 14 was dissolved in anhydrous THF (50 mL), followed by the addition of di-tert-butyl dicarbonate 15 (8.2 g, 38 mmol, 1.8 equiv.). The reaction mixture was then cooled to 0 °C using an ice bath, followed by the addition of TEA (5.5 mL, 40 mmol, 1.9 equiv.). The reaction was stirred overnight, while slowly allowing to reach room temperature. The next morning, THF was removed in vacuo. The residue was dissolved in CH2Cl2 (50 mL) and then washed with H2O (3 × 100 mL). The aqueous layers were combined and back-extracted with CH2Cl2 (3 × 100 mL). The CH2Cl2 layers were combined and washed with brine (300 mL), dried over MgSO4, and then filtered. The filtrate was concentrated in vacuo, purified by automated flash column chromatography (100% EtOAc), and fractions evaluated by TLC and visualized by ninhydrin stain. Pure tetraethylene glycol Boc-amide 16 (3.1 g, 11 mmol, 52%) was obtained as a colourless oil. Rf = 0.70 (5% MeOH in CH2Cl2); 1H-NMR (400 MHz, CDCl3): δ = 5.62 (broad s, 1H, boc-NH), 3.69 (m, 4H, 2 × CH2), 3.62 (m, 8H, 4 × CH2), 3.52 (t, 3JHH = 5.3 Hz, 2H, boc-NHCH2CH2), 3.30 (q, 3JHH = 5.3 Hz, 2H, boc-NHCH2), 3.05 (broad s, 1H, OH), 1.42 (s, 9H, C(CH3)3) ppm; 13C-NMR (101 MHz, CDCl3): δ = 156.2 (tBuOOC), 78.9 (C(CH3)3), 72.6 (CH2), 70.6 (CH2), 70.5 (CH2), 70.3 (boc-NHCH2CH2), 70.1 (CH2), 61.7 (CH2), 40.4 (boc-NHCH2), and 28.4 (C(CH3)3) ppm; HRMS: calculated m/z for C13H27NO6: 316.1736 [M+Na]1+; found 316.1725.
Tetraethylene glycol Boc-amide p-toluenesulfonate (17). All of the steps were performed under N2 atmosphere. Boc-protected amino-tetraethylene glycol 16 (3.1 g, 11 mmol, 1.0 equiv.) and p-toluenesulfonyl chloride 8 (3.2 g, 17 mmol, 1.5 equiv.) were dissolved in anhydrous CH2Cl2 (50 mL). The resulting mixture was cooled to 0 °C using an ice bath, followed by the addition of TEA (2.3 mL, 17 mmol, 1.5 equiv.). The reaction was stirred overnight at room temperature. The next morning the reaction was not complete, as determined by TLC (100% EtOAc) and visualized with UV and ninhydrin stain. Nevertheless, the reaction mixture was worked-up by washing with H2O (3 × 50 mL). Subsequently, the combined aqueous layers were back-extracted with CH2Cl2 (2 × 100 mL). The combined CH2Cl2 layers were washed with brine (250 mL), dried over MgSO4, and then filtered. Next, the filtrate was concentrated in vacuo, purified by automated flash column chromatography using a linear gradient (50–60% EtOAc in petroleum ether 40–60 °C over 10 column volumes), and then visualized with UV and ninhydrin stain. Fractions containing pure product were combined and concentrated in vacuo, affording tetraethylene glycol Boc-amide p-toluenesulfonate 17 (2.1 g, 4.7 mmol, 43%) as a colourless oil. Rf = 0.55 (EtOAc); tR = 33.0 min.; 1H-NMR (400 MHz, CDCl3): δ = 7.79 (d, 3JHH = 8.2 Hz, 2H, aryl o-H), 7.33 (d, 3JHH = 8.2 Hz, 2H, aryl m-H), 4.98 (broad s, 1H, boc-NH), 4.15 (t, 3JHH = 4.8 Hz, 2H, TsOCH2), 3.69 (t, 3JHH = 4.8 Hz, 2H, TsOCH2CH2), 3.59 (m, 8H, 4 × CH2), 3.52 (t, 3JHH = 5.3 Hz, 2H, boc-NHCH2CH2), 3.29 (q, 3JHH = 5.3 Hz, 2H, boc-NHCH2), 2.44 (s, 3H, CH3), 1.43 (s, 9H, C(CH3)3) ppm; 13C-NMR (101 MHz, CDCl3): δ = 156.0 (tBuO(O)C), 144.8 (aryl-C), 133.1 (aryl-C), 129.8 (aryl m-CH), 128.0 (aryl o-CH), 79.2 (C(CH3)3), 70.8 (CH2), 70.6 (CH2), 70.5 (CH2), 70.2 (CH2), 70.2 (boc-NHCH2CH2), 69.2 (TsOCH2), 68.7 (TsOCH2CH2 (7)), 40.4 (boc-NHCH2), 28.4 (C(CH3)3), and 21.6 (CH3) ppm; HRMS: calculated m/z for C20H33NO8S: 470.1825 [M+Na]1+; found 470.1814.
Triethylene glycol Boc-amide ethylamine (18). p-toluenesulfonate tetraethylene glycol with a Boc-protected amine 17 (2.1 g, 4.7 mmol, 1.0 equiv.) was dissolved in MeCN (50 mL), followed by the addition of aqueous 28–30% ammonia (50 mL). The reaction was stirred overnight at room temperature. Because the reaction was not complete, the solvents were removed in vacuo, after which the residue was re-dissolved in MeCN (50 mL) and fresh aqueous 28–30% ammonia (50 mL) was added. Despite repeating this procedure twice, the reaction was not complete, as determined by TLC (5% MeOH in CH2Cl2 supplemented with TEA) and visualized with ninhydrin stain. Nevertheless, solvent was removed in vacuo and the residue was re-dissolved in CH2Cl2 (100 mL). The resulting solution was washed with aqueous 1.0 M NaOH (3 × 100 mL) and the resulting aqueous layer was back-extracted with CH2Cl2 (3 × 100 mL). The CH2Cl2 layers were combined and washed with brine (200 mL), dried over MgSO4, and then filtered. Next, the filtrate was concentrated in vacuo, purified by automated flash column chromatography using a linear gradient (0–5% MeOH in CH2Cl2 over 10 column volumes), and visualized with UV and ninhydrin stain. Fractions containing pure product were combined and concentrated in vacuo, affording pure triethylene glycol Boc-amide ethylamine 18 (0.49 g, 1.7 mmol, 36%) as a colourless oil. Rf = 0.41 (5% MeOH in CH2Cl2 supplemented with TEA); 1H-NMR (400 MHz, CDCl3): δ = 5.26 (broad s, 1H, boc-NH), 3.62 (m, 8H, 4 × CH2), 3.53 (m, 4H, boc-NHCH2CH2 and CH2CH2NH2), 3.29 (m, 2H, boc-NHCH2), 2.88 (m, 2H, CH2NH2), 2.43 (m, 2H, NH2), 1.43 (s, 9H, C(CH3)3) ppm; 13C-NMR (101 MHz, CDCl3): δ = 156.1 (tBuO(O)C), 79.1 (C(CH3)3), 72.7 (boc-NHCH2CH2 or CH2CH2NH2), 70.5 (CH2), 70.5 (CH2), 70.2 (boc-NHCH2CH2 or CH2CH2NH2), 41.5 (CH2NH2), 40.4 (boc-NHCH2), and 28.4 (C(CH3)3) ppm; HRMS: calculated m/z for C13H28N2O5: 315.1896 [M+Na]1+; found 315.1883.
Triethylene glycol Boc-amide ethylamine equipped TAC-scaffold (19). TAC-scaffold 11 (39 mg, 50 μmol, 1.0 equiv.) was dissolved in DMF (8 mL). Next, BOP (26 mg, 59 μmol, 1.2 equiv.) was dissolved in DMF (3 × 2 mL) and then added to the solution containing TAC-scaffold 11. Subsequently, triethylene glycol Boc-amide ethylamine 18 (29 mg, 100 μmol, 2.0 equiv.) was dissolved in DMF (3 × 2 mL) and added. Finally, DiPEA (35 μL, 0.20 mmol) was added and the reaction mixture was stirred for 1 h at room temperature. Upon completion of the reaction, as determined by LCMS, solvent was removed in vacuo (60 °C). The residue was taken up in tBuOH/H2O (1/1, v/v) and lyophilized. The crude yellowish oil was dissolved in CH2Cl2 and purified using automated flash column chromatography with a linear gradient (0–5% MeOH in CH2Cl2 over 10 column volumes) and then visualized with UV and ninhydrin stain. Pure fractions were combined and concentrated in vacuo (60 °C), the residue was taken up in tBuOH/H2O (1/1, v/v) and lyophilized, affording triethylene glycol Boc-amide ethylamine equipped TAC-scaffold 19 (46 mg, 43 μmol, 86%) as a white powder. Rf = 0.43 (5% MeOH in CH2Cl2); tR = 54.2 min.; 1H-NMR (400 MHz, CDCl3): δ = 7.73 (m, 2H, aryl-H), 7.28 (m, 1H, aryl-H), 6.90 (broad s, 1H, OCH2CH2NH), 5.11 (broad s, 1H, boc-NH), 4.64 (m, 4H, 2 × NCH2-aryl), 3.65 (m, 10H, 4 × CH2 and OCH2CH2NH), 3.52 (t, 3JHH = 5.2 Hz, 2H, boc-NHCH2CH2), 3.43 (m, 4H, NCH2CH2CH2N), 3.27 (q, 3JHH = 5.2 Hz, 2H, boc-NHCH2), 2.95 (m, 6H, NCH2CH2CH2N and OCH2CH2NH), 2.64 (m, 8H, 2 × CH2CH2CCSi), 2.45 (m, 4H, CH2CH2CCH), 1.95 (m, 1H, CCH), 1.58 (m, 4H, 2 × NCH2CH2CH2N), 1.43 (s, 9H, C(CH3)3), 1.06 (m, 21H, Si(CH(CH3)2)3), 0.98 (t, 3JHH = 7.9 Hz, 9H, Si(CH2CH3)3), 0.58 (q, 3JHH = 7.9 Hz, 6H, Si(CH2CH3)3) ppm; 13C-NMR (101 MHz, CDCl3): δ = 156.0 (tBuO(O)C)), 129.2 (aryl-CH), 126.8 (aryl-CH), 70.5 (CH2), 70.5 (CH2), 70.3 (CH2), 70.2 (boc-NHCH2CH2 or OCH2CH2NH), 70.2 (boc-NHCH2CH2 or OCH2CH2NH), 70.1 (CH2), 53.9 (NCH2-aryl), 51.9 (NCH2-aryl), 48.0 (NCH2CH2CH2N or OCH2CH2NH), 45.9 (NCH2CH2CH2N or OCH2CH2NH), 45.7 (NCH2CH2CH2N or OCH2CH2NH), 45.3 (NCH2CH2CH2N or OCH2CH2NH), 43.7 (NCH2CH2CH2N or OCH2CH2NH), 40.3 (boc-NHCH2), 39.8 (CH2), 33.0 (CH2CH2CCSi), 33.0 (CH2CH2CCSi), 32.0 (CH2CH2CCH), 28.4 (C(CH3)3), 28.0 (NCH2CH2CH2N), 28.0 (NCH2CH2CH2N), 18.6 (Si(CH(CH3)2)3), 16.3 (CH2CH2CCSi), 16.3 (CH2CH2CCSi), 14.5 (CH2CH2CCH), 11.3 (Si(CH(CH3)2)3), 7.5 (Si(CH2CH3)3), 4.5 (Si(CH2CH3)3) ppm; HRMS: calculated m/z for C58H95N5O9Si2: 1084.6566 [M+Na]1+; found 1084.6559; LRMS: calculated m/z for C58H95N5O9Si2:1062.67 [M+H]1+/1084.66 [M+Na]1+; Found m/z 1062.83/1084.75.
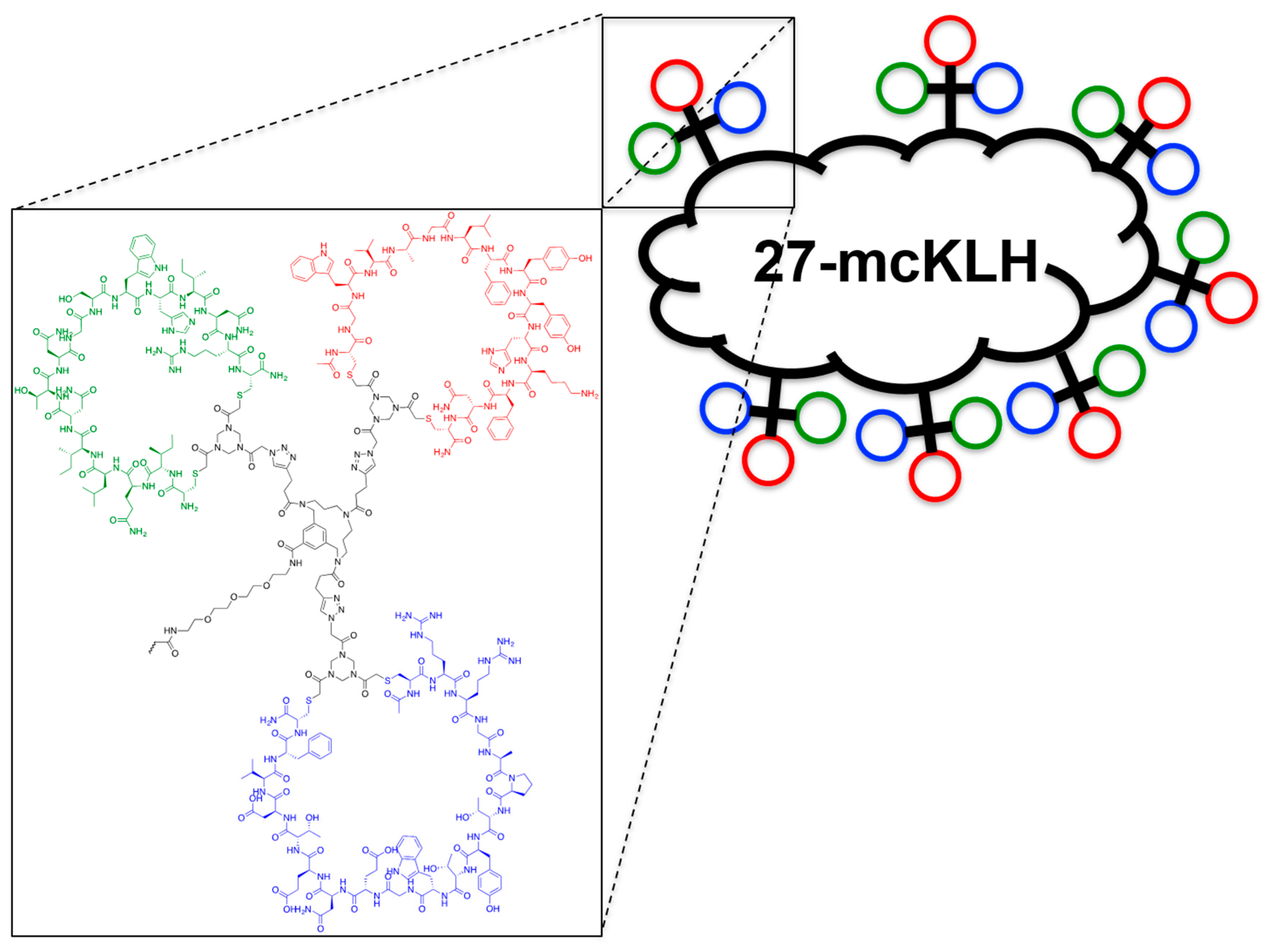
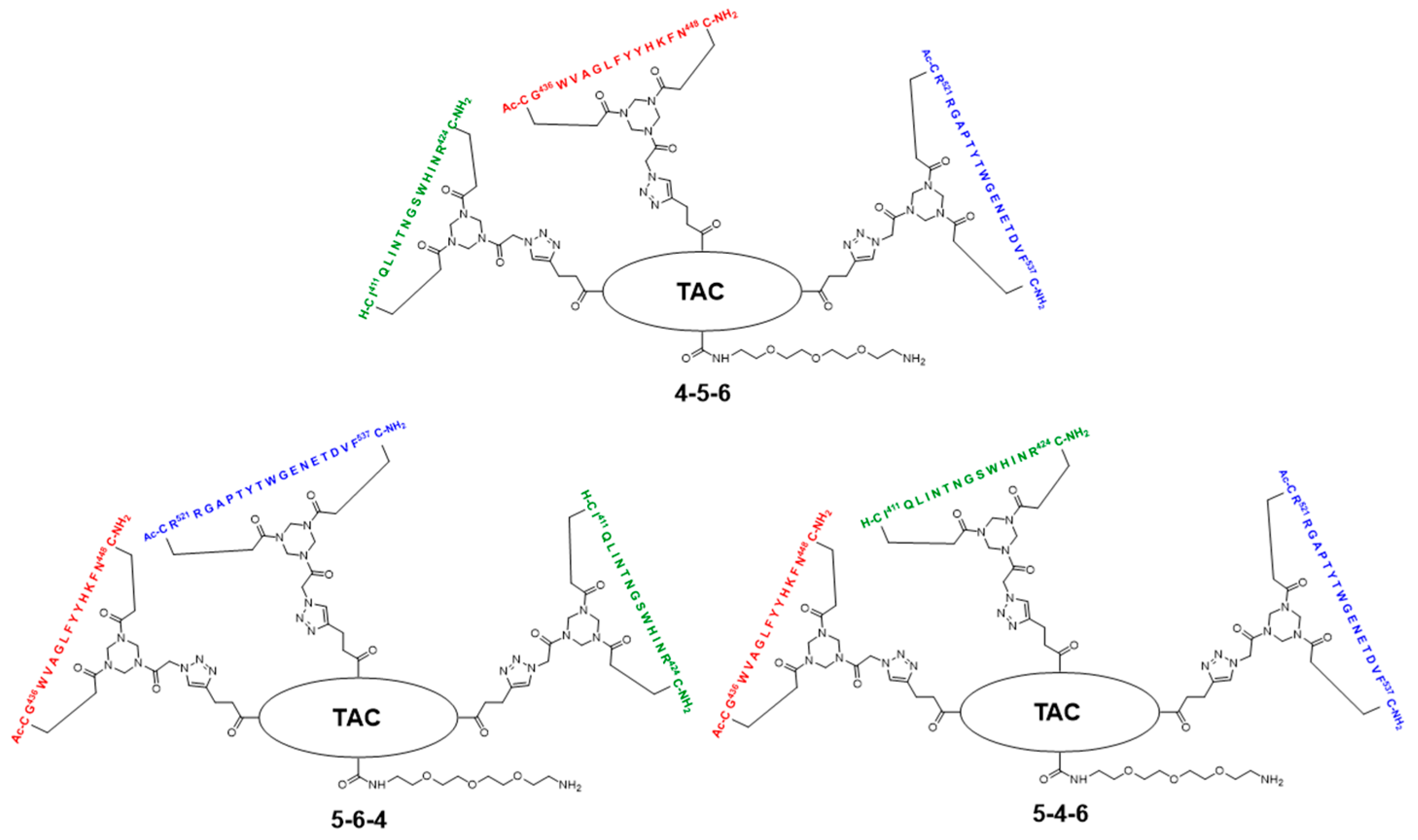
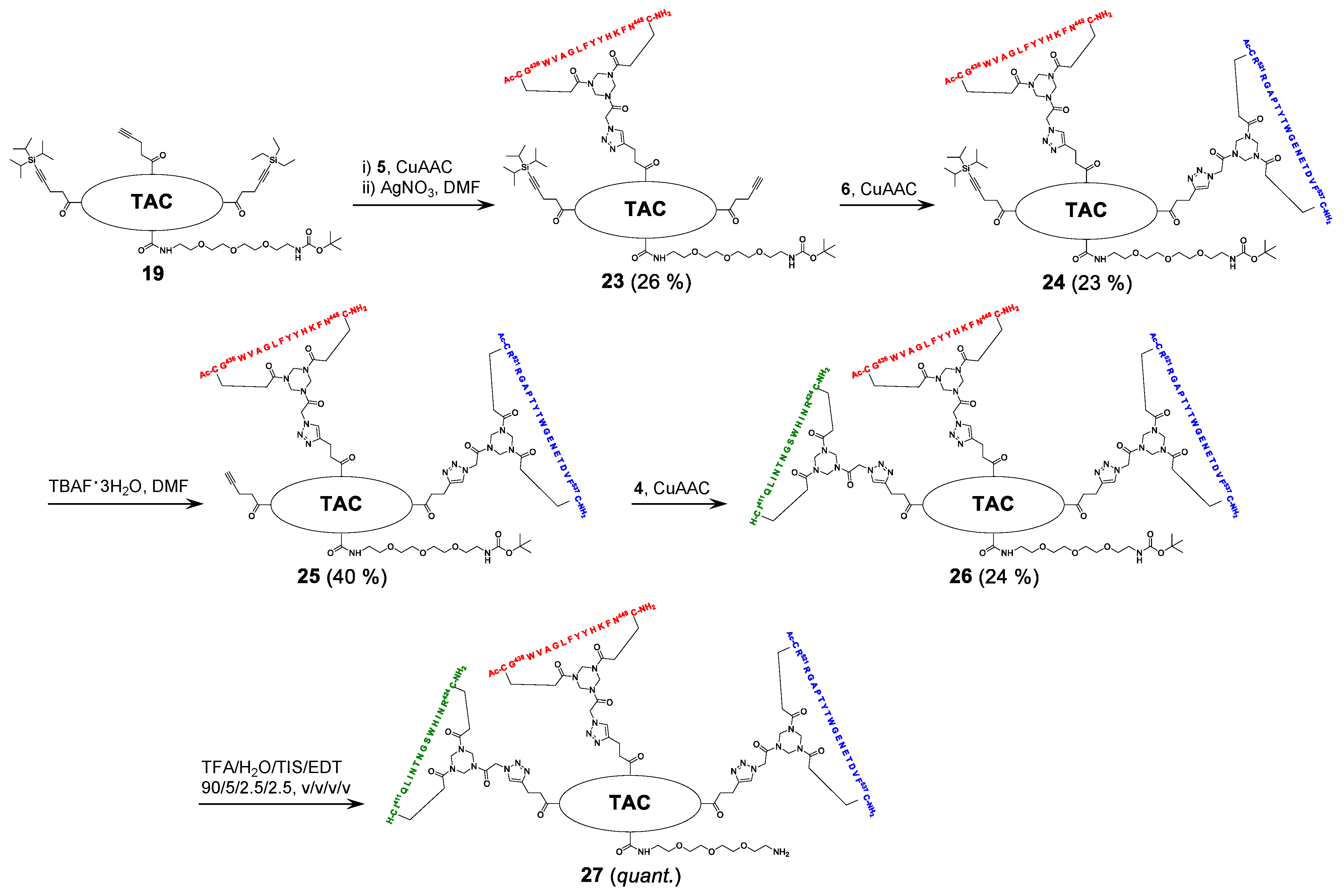
2.5. Assembly of the Discontinuous Epitope Mimic 27
The cycloaddition of the first cyclic peptide and subsequent triethylsilyl (TES) deprotection. Triethylene glycol Boc-amide ethylamine equipped TAC-scaffold 19 (66 mg, 62 μmol, 1.8 equiv.) was dissolved in DMF (600 μL) and added to cyclic peptide 5 (71 mg, 34 μmol, 1.0 equiv.). The flask-containing TAC-scaffold 19 was rinsed with additional DMF (500 μL), which was added to the reaction mixture to ensure the complete transfer of 19. Next, TBTA (15 mg, 28 μmol) was dissolved in DMF (161 μL), of which 100 μL (17 μmol, 0.50 equiv.) was added to the reaction mixture. Subsequently, aqueous solutions of CuSO4•5H2O (24 mg, 97 μmol, dissolved in 158 μL H2O), sodium L-ascorbate (68 mg, 0.34 mmol, dissolved in 202 μL H2O), and aminoguanidine•HCl (59 mg, 0.53 mmol, dissolved in 142 μL H2O) were prepared. The resulting aqueous solutions were mixed (CuSO4•5H2O (100 μL, 61 μmol, 1.8 equiv.), sodium L-ascorbate (200 μL, 0.34 mmol, 10 equiv.), and aminoguanidine•HCl (100 μL, 0.38 mmol, 11 equiv.)), and the resulting mix (400 μL) was added dropwise to the stirring reaction mixture. The reaction was kept under N2-atmosphere and monitored by LCMS. After 3 h, the reaction was complete, followed by the addition of Cuprisorb resin to capture the Cu-ions. The reaction mixture was filtered and the reaction vessel and filter were washed with extra DMF (5 × 2 mL). Next, DMF was removed in vacuo (60 °C) and the residue was dissolved in tBuOH/H2O/MeCN (1/1/1, v/v/v), and lyophilized.
The resulting crude product was dissolved in DMF (1 mL) and an aqueous solution of AgNO3 (0.25 g, 1.5 mmol, dissolved in 235 μL H2O) was prepared, of which 100 μL (0.62 mmol, 18 equiv.) was added. The reaction was stirred under N2-atmosphere for 1 h at room temperature and monitored by LCMS. After 1 h, the reaction was not complete and an additional 100 μL (0.62 mmol, 18 equiv.) was added. The reaction was complete after an additional hour of stirring, as was verified by LCMS. Next, additional DMF (900 μL) was added to the reaction mixture and an aqueous solution of NaCl (0.12 g, 2.1 mmol, dissolved in 510 μL H2O) was prepared, of which 300 μL (1.2 mmol, 35 equiv.) was added to the reaction mixture. After 15 min., the reaction mixture was collected, and the reaction vessel rinsed with DMF (3 × 2 mL). The resulting AgCl precipitate was removed by centrifugation (4500 rpm; 5 min.), the resulting precipitate was washed with DMF (5 mL), and it was centrifuged again. The supernatants were collected and pooled, followed by removal of DMF in vacuo (60 °C). The residue was dissolved in tBuOH/H2O/MeCN (1:1:1, v/v/v) and lyophilized.
The obtained crude product was dissolved in buffer 4 mL A/B (1/1, v/v), clarified by centrifugation (4500 rpm; 5 min.), and purified by preparative HPLC while using a customary protocol: 50% buffer B in buffer A for 5 min., followed by a linear gradient of 50–100% buffer B in buffer A for 50 min. Fractions containing pure product were identified by analytical HPLC, pooled, and lyophilized, which afforded pure scaffold molecular construct 23 (27 mg, 8.9 μmol, 26%). tR = 27.5 min.; HRMS: calculated m/z for C150H208N32O31S2Si: 1523.7525 ½[M+2H]2+; found 1523.7543; LRMS: calculated m/z for C150H208N32O31S2Si: 1523.75 ½[M+2H]2+/1016.17 1/3[M+3H]3+; found 1524.83/1016.92.
The cycloaddition of the second cyclic peptide. Cyclic peptide 6 (50 mg, 20 μmol, 1.5 equiv.) was dissolved in DMF (500 μL) and added to scaffold molecular construct 23 (40 mg, 13 μmol, 1.0 equiv.). The flask-containing cyclic peptide 6 was rinsed with additional DMF (500 μL), which was added to the reaction mixture to ensure complete transfer. Next, TBTA (9.3 mg, 18 μmol) was dissolved in DMF (265 μL), of which 100 μL (6.6 μmol, 0.51 equiv.) was added to the reaction mixture. Subsequently, aqueous solutions of CuSO4•5H2O (12 mg, 48 μmol, dissolved in 198 μL H2O), sodium L-ascorbate (52 mg, 0.26 mmol, dissolved in 200 μL H2O), and aminoguanidine•HCl (32 mg, 0.29 mmol, dissolved in 200 μL H2O) were prepared. The resulting aqueous solutions were mixed (CuSO4•5H2O (100 μL, 24 μmol, 1.8 equiv.), sodium L-ascorbate (100 μL, 0.13 mmol, 10.0 equiv.), and aminoguanidine•HCl (100 μL, 0.15 mmol, 11 equiv.)) and the resulting mix (300 μL) was added dropwise to the stirring reaction mixture. The reaction was kept under N2-atmosphere and monitored by LCMS. After 6 h, the reaction was complete; this was followed by the addition of Cuprisorb resin to capture the Cu-ions. The reaction mixture was filtered. The reaction vessel and filter were washed with extra DMF (5 × 2 mL). Next, DMF was removed in vacuo (60 °C), the residue was dissolved in tBuOH/H2O/MeCN (1/1/1, v/v/v), and then lyophilized.
The obtained crude product was dissolved in buffer 5 mL A/B (1/1, v/v), clarified by centrifugation (4500 rpm; 5 min.), and the resulting compound in the supernatant was purified by preparative HPLC using a customary protocol: 40% buffer B in buffer A for 5 min., followed by a linear gradient of 40–70% buffer B in buffer A for 60 min. Fractions containing pure product were identified by analytical HPLC, pooled, and lyophilized, which afforded pure scaffold molecular construct 24 (17 mg, 3.0 μmol, 23%). tR = 23.7 min.; HRMS: calculated m/z for C255H358N66O65S4Si: 1386.1425 1/4[M+4H]4+; found 1386.1481; LRMS: calculated m/z for C255H358N66O65S4Si: 1847.85 1/3[M+3H]3+/1386.14 1/4[M+4H]4+; found 1849.00/1387.17.
Triisopropylsilyl (TIPS) deprotection. Tetra-n-butylammonium fluoride (TBAF)•3H2O (13 mg, 41 μmol) was dissolved in DMF (133 μL), of which 100 μL (31 μmol, 10 equiv.) was used to dissolve the scaffold molecular construct 24 (17 mg, 3.0 μmol, 1.0 equiv.). The reaction was stirred under N2-atmosphere and closely monitored by LCMS. After 5 h, no conversion was observed and fresh TBAF•3H2O (14 mg, 44 μmol; dissolved in 152 μL DMF, of which 100 μL (29 μmol, 10 equiv.)) was added. The next day, the reaction progressed, but it was still not complete. Therefore, fresh TBAF•3H2O (24 mg, 76 μmol; dissolved in 255 μL DMF, of which 100 μL (29.8 μmol, 9.9 equiv.)) was added. The next morning, the reaction was complete, as verified by LCMS. The reaction mixture was collected and the reaction vessel was rinsed with 5 mL buffer A:B (1:1, v/v). Next, the reaction mixture was clarified by centrifugation (4500 rpm; 5 min.) and then purified by preparative HPLC using a customary protocol: 20% buffer B in buffer A for 5 min., followed by a linear gradient of 20–50% buffer B in buffer A for 40 min. Fractions containing pure product were identified by analytical HPLC, pooled, and lyophilized, which afforded pure scaffolded molecular construct 25 (6.6 mg, 1.2 μmol, 40%). tR = 20.4 min.; HRMS: calculated m/z for C246H338N66O65S4: 1347.1092 1/4[M+4H]4+; found 1347.1139; LRMS: calculated m/z for C246H338N66O65S4: 1795.81 1/3[M+3H]3+/1347.11 1/4[M+4H]4+; found 1797.17/1348.42.
The cycloaddition of the third cyclic peptide. Cyclic peptide 4 (4.4 mg, 2.1 μmol, 1.8 equiv.) was dissolved in DMF (500 μL) and added to scaffold molecular construct 25 (6.6 mg, 1.2 μmol, 1.0 equiv.). The flask-containing cyclic peptide 4 was rinsed with additional DMF (500 μL), which was added to the reaction mixture to ensure complete transfer. Next, TBTA (1.1 mg, 2.0 μmol) was dissolved in DMF (330 μL), of which 100 μL (0.60 μmol, 0.50 equiv.) was added to the reaction mixture. Subsequently, aqueous solutions of CuSO4•5H2O (3.0 mg, 12 μmol, dissolved in 555 μL H2O), sodium L-ascorbate (15 mg, 76 μmol, dissolved in 635 μL H2O), and aminoguanidine•HCl (14 mg, 0.13 mmol; dissolved in 950 μL H2O) were prepared. The resulting aqueous solutions were mixed ((CuSO4•5H2O (100 μL, 2.2 μmol, 1.8 equiv.), sodium L-ascorbate (100 μL, 12 μmol, 10 equiv.), and aminoguanidine•HCl (100 μL, 14 μmol, 12 equiv.)), and the resulting mix (300 μL) was added dropwise to the stirring reaction mixture. The reaction was kept under N2-atmosphere and monitored by LCMS. After 3 h, no more cyclic peptide 4 was observed in the reaction mixture but scaffold molecular construct 25 was still present. Therefore, additional cyclic peptide 4 (2.7 mg, 1.3 μmol, 1.1 equiv.) was dissolved in DMF (100 μL) and added. After 1 h, no improvement was observed by LCMS and fresh reagents were prepared. TBTA (1.1 mg, 2.0 μmol) was dissolved in DMF (330 μL), of which 100 μL (0.6 μmol, 0.5 equiv.) was added to the reaction mixture. Subsequently, solutions of CuSO4•5H2O (6.0 mg, 24 μmol; dissolved in 555 μL H2O), sodium L-ascorbate (7.9 mg, 40 μmol; dissolved in 166 μL H2O), and aminoguanidine•HCl (11 mg, 99 μmol, dissolved in 370 μL H2O) were prepared. The resulting solutions were mixed ((CuSO4•5H2O (50 μL, 2.2 μmol, 1.8 equiv.), sodium L-ascorbate (50 μL, 12 μmol, 10 equiv.), and aminoguanidine•HCl (100 μL, 13 μmol, 11.0 equiv.)) and the resulting mix (150 μL) was added dropwise to the stirring reaction mixture. The reaction was continued to stir under N2-atmosphere and monitored by LCMS. After 2 h, additional product was formed, but the reaction was not fully complete. Nevertheless, the reaction was worked-up by the addition of Cuprisorb resin to capture the Cu-species. The reaction mixture was filtered, and the reaction vessel and filter were washed with extra DMF (5 × 2 mL). Next, DMF was removed in vacuo (60 °C), the residue was dissolved in tBuOH/H2O/MeCN (1/1/1, v/v/v), and lyophilized.
The obtained crude product was suspended in buffer 3 mL A:B (1:1, v/v), clarified by centrifugation (4500 rpm; 5 min.), and the resulting compound in the supernatant was purified by preparative HPLC using a customary protocol: 0% buffer B in buffer A for 5 min., followed by a linear gradient of 0–50% buffer B in buffer A for 50 min. Fractions containing pure product were identified by analytical HPLC, pooled, and lyophilized, which afforded pure discontinuous epitope mimic 26 (2.2 mg, 0.29 μmol, 24%). tR = 19.6 min.; HRMS: calculated m/z for C334H475N99O90S6: 1501.8870 1/5[M+5H]5+; found 1501.8881; LRMS: calculated m/z for C334H475N99O90S6: 1877.11 1/4[M+4H]4+/1501.89 1/5[M+5H]5+/1251.74 1/6[M+6H]6+; found 1878.42/1503.00/1252.58.
The removal of Boc protective group of 26. Discontinuous epitope mimic 26 (2.2 mg, 0.29 μmol, 1.0 equiv.) was dissolved in 250 μL TFA/H2O/TIS/EDT (90/5/2.5/2.5, v/v/v/v) and the reaction mixture was stirred for 30 min. at room temperature. Next, the reaction mixture was added to Et2O (15 mL) to precipitate the product. The reaction vessel was rinsed (x3) with an additional 250 μL TFA/H2O/TIS/EDT (90/5/2.5/2.5, v/v/v/v), which was added to a separate volume of Et2O (15 mL) to collect all of the product. Precipitated product was obtained by centrifugation (4500 rpm; 5 min.), and washed twice using Et2O (15 mL), followed by centrifugation (4500 rpm; 5 min.). The obtained precipitated pellets were dissolved in tBuOH/H2O/MeCN (1/1/1, v/v/v), pooled, and lyophilized, which afforded pure discontinuous epitope mimic 27 (2.0 mg, 0.27 μmol, quant.). tR = 18.9 min.; HRMS: calculated m/z for C329H467N99O88S6: 1481.8765 1/5[M+5H]5+; found 1481.8699; LRMS: calculated m/z for C329H467N99O88S6: 1852.09 1/4[M+4H]4+/1481.88 1/5[M+5H]5+/1235.07 1/6[M+6H]6+; found 1853.42/1483.00/1236.17.
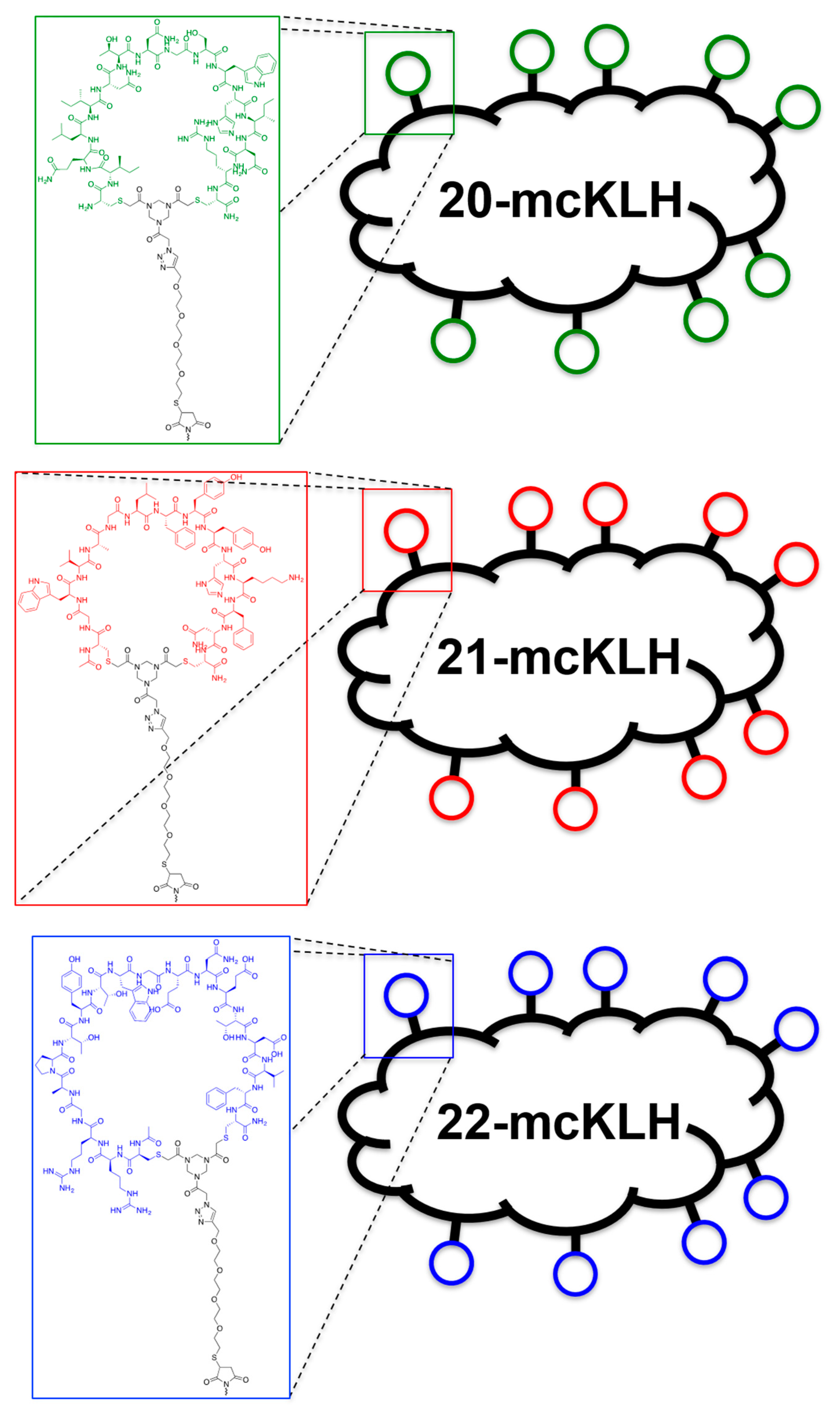
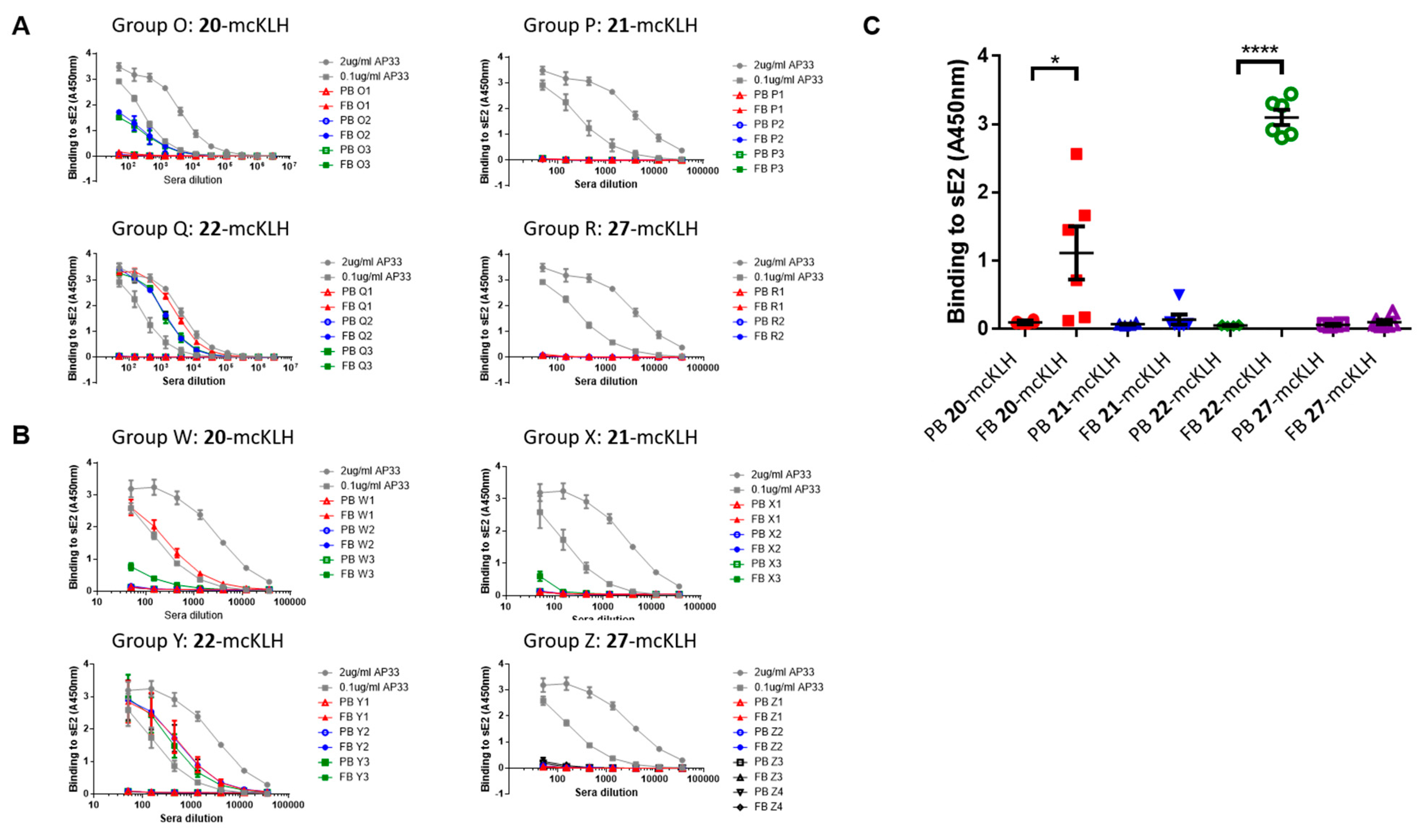
Conjugation of free thiol containing continuous epitope mimics (20, 21, and 22) on maleimide activated macriculture keyhole limpet hemocyanin (mcKLH). This procedure was performed, as instructed by the Imject® Maleimide Activated Carrier Protein Spin Kits purchased from Thermo Scientific.
One vial of the activated mcKLH was reconstituted per conjugation by adding 200 μL ultrapure H2O to obtain a 10 mg/mL translucent to whitish-blue solution. Next, 2.0 mg of continuous epitope mimic (20, 21, and 22) were separately dissolved in 500 μL conjugation buffer (83 mM sodium phosphate, 0.10 M EDTA, 0.90 M sodium chloride, 0.10 M sorbitol, and 0.02% sodium azide; pH 7.2). Immediately, each solution of continuous epitope mimic was mixed with one vial of activated mcKLH were mixed and incubated for 2 h at room temperature.
After conjugation, 10 mL of ultrapure H2O was added to one bottle of Imject purification buffer salts (83 mM sodium phosphate, 0.90 M sodium chloride, 0.10 M sorbitol; pH 7.2) per conjugation sample. One desalting column was drained by centrifugation (1000× g; 2 min.) per conjugation sample. The desalting columns were prepared by slowly adding 1 mL of purification buffer and draining by centrifugation (1000× g; 2 min.) for a total of four times. Each conjugation sample was collected and centrifuged (1000× g; 2 min.), the pellets were kept, and the resulting supernatants (700 μL) were carefully loaded on separate desalting columns. Each desalting column was placed in a clean collection tube and centrifuged (1000× g; 2 min.) to collect the conjugation samples. The collected conjugation samples were used to resuspend their corresponding pellets. The obtained continuous epitope-mcKLH conjugates (20-mcKLH, 21-mcKLH, and 22-mcKLH) were stored at −20 °C until immunization.
Conjugation of free amine containing discontinuous epitope mimic (27) on mcKLH by 1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) coupling. This procedure was performed, as instructed by the Imject® EDC Carrier Protein Spin Kits tha were purchased from Thermo Scientific.
One vial of mcKLH was reconstituted by adding 200 μL ultrapure H2O to obtain a 10 mg/mL translucent to a whitish-blue solution. Next, 2.0 mg of discontinuous epitope mimic 27 was dissolved in 450 μL Imject® EDC conjugation buffer (0.10 M MES, 0.90 M sodium chloride, 0.02% sodium azide; pH 4.7) and it was added to the mcKLH solution. After which, one vial of EDC (10 mg) was dissolved in 1 mL ultrapure H2O, of which 50 μL was immediately added to the epitope-mcKLH mixture. Subsequently, the mixture was incubated for 2 h at room temperature.
After the conjugation, 10 mL of ultrapure H2O was added to one bottle of Imject purification buffer salts (83 mM sodium phosphate, 0.90 M sodium chloride, 0.10 M sorbitol; pH 7.2). One desalting column was drained by centrifugation (1000× g; 2 min.) and was prepared by slowly adding 1 mL of purification buffer and draining by centrifugation (1000× g; 2 min.) for a total of four times. The conjugation sample was collected and centrifuged (1000× g; 2 min.), the pellet was kept, and the resulting supernatant (650 μL) was carefully loaded on the desalting column. The desalting column was placed in a clean collection tube and centrifuged (1000× g; 2 min.) to collect the conjugation sample. The collected conjugation sample was used to resuspend the obtained pellet and the resulting discontinuous epitope-mcKLH conjugate (27-mcKLH) was stored at −20 °C until immunization.
Immunization experiments. The mouse immunization experiments were approved by the University of Glasgow Animal Welfare and Ethical Board and they were carried out under the Home Office Project License P9722FD8E held by Prof. Arvind H. Patel at the MRC-University of Glasgow Centre for Virus Research.
Two independent immunization experiments were performed. Each experiment had four groups that contained three mice per group that were subcutaneously injected with epitope-mcKLH conjugate (20-mcKLH, 21-mcKLH, 22-mcKLH, or 27-mcKLH) mixed with Addavax (Invivogen) adjuvant (350 μL). Prime immunization was performed with 50 μg (131 μL) epitope-mcKLH conjugate (20-mcKLH, 21-mcKLH, 22-mcKLH, or 27-mcKLH). After two weeks, the prime immunization was followed up by one boost immunization of 10 μg (26 μL) epitope-mcKLH conjugate (20-mcKLH, 21-mcKLH, 22-mcKLH, or 27-mcKLH) every week for four weeks. The animals were sacrificed eight days after the last boost immunization and sera were collected for binding and neutralization assays.
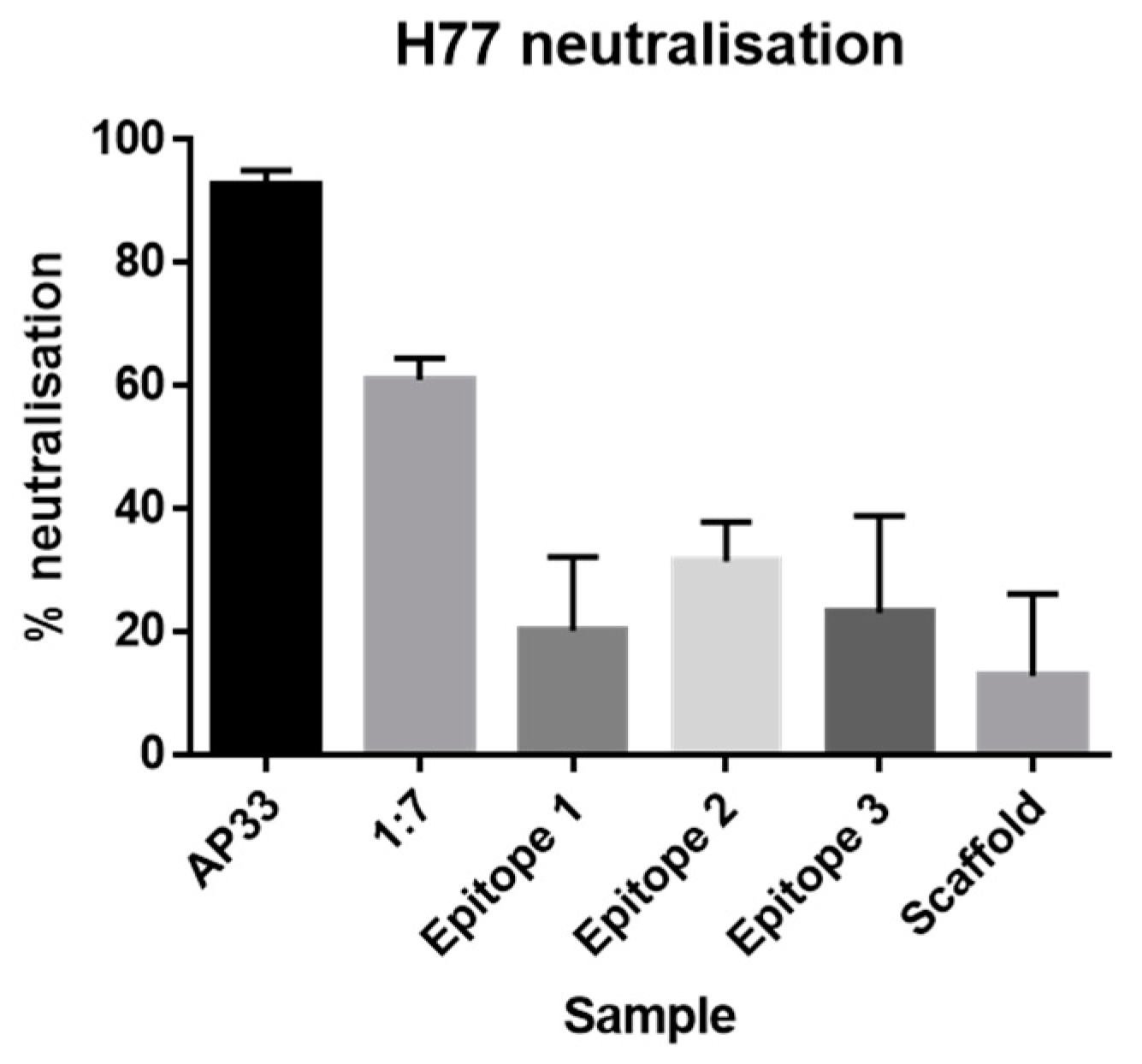
Monoclonal antibodies. The mAb AP33 [
21] and 1:7 [
35] used in this work were previously described.
General method for ELISA. Plate preparation: Immulon 2HB 96-well plates were coated with 1 μg/mL sE2 Gt1a H77 (purified from mammalian HEK-F cells) overnight at room temperature. After which, the wells were blocked overnight at room temperature with 200 μL 2% skimmed milk in PBST (phosphate buffered saline supplemented with 0.05% Tween®-20). Next, the wells were washed three times with PBST and then used immediately or stored at −20 °C until use.
Sera preparation: the obtained mice blood from the immunization experiment was incubated for 1 h at 37 °C and the tubes were flicked to dislodge the blood clot. Subsequently, the tubes were incubated at 4 °C for 2 h or overnight to enhance blood clotting. After which, the blood clots were centrifuged (10,000× g; 10 min) at 4 °C. Sera were collected from the blot clots and then transferred to a fresh tube. The centrifugation/transfer step was repeated if necessary.
ELISA: three-fold dilution series of sera were prepared starting from a 1:50 dilution (3 μL sera and 147 μL PBST) and then added to the plates (100 μL/well). Monoclonal antibody AP33 was included as a positive control (three-fold dilution at starting concentrations: 2.0 μg/mL and 0.1 μg/mL). The plates were incubated for 1–2 h at room temperature. After which, the plates were washed three times with PBST, before supplying 100 μL 1:3000 secondary α-mouse A4416 (Sigma) antibodies to each well. The plates were incubated for 1 h at room temperature and washed six times with PBST. The plates were developed using 100 μL 3, 3′, 5, 5′ tetramethylbenzidine (TMB) solution per well, obtained from Life Technologies, and incubated for 10 min. at room temperature. After which, development was stopped using 50 μL 0.5 M H2SO4 per well. Absorbance at 450 nm was measured on a Varioskan (Thermoscientific) instrument.
Cell lines. human hepatoma Huh-7 cells and HEK-293T cells were grown in Dulbecco’s modified Eagle’s medium that was supplemented with 10% fetal calf serum, 5% nonessential amino acids, Penicillin, Streptomycin, and 200 mM L-glutamine.
Generation of HCV pseudoparticles (HCVpp). HEK-293T cells were co-transfected with plasmids expressing MLV Gag-pol, the MLV transfer vector carrying firefly luciferase reporter and HCV E1E2 glycoprotein. After 72 h, the medium was harvested, filtered through a 0.45 µm membrane, and used as a source of HCVpp, as previously described [
36,
37].
Neutralization assays. IgG was purified from sera using the NAb™ Protein A/G spin columns (Thermofisher). Huh-7 cells were seeded at 4 × 103 cells per well in a 96-well plate. Three-fold dilution series of purified IgG were prepared starting from 100 μg/mL. Monoclonal antibodies AP33 and 1:7 were included as positive controls (three-fold dilution series starting from 50 μg/mL). 45 μL of Gt1a H77 HCVpp were added to each well and then incubated for 1 h at 37 °C. Next, Huh-7 cells were inoculated for 3 h at 37 °C. After which, the inoculum was removed and 120 μL of fresh media was added. The cells were lysed after an incubation period of 72 h at 37 °C post-inoculation. Infectivity and neutralization were assessed using the GloLysis Luciferase assay (Promega).


 and
and 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}