
Human Cytomegalovirus and Autoimmune Diseases: Where Are We?

 , , , , ,
, , , , ,  and
and 
Abstract
1. Introduction
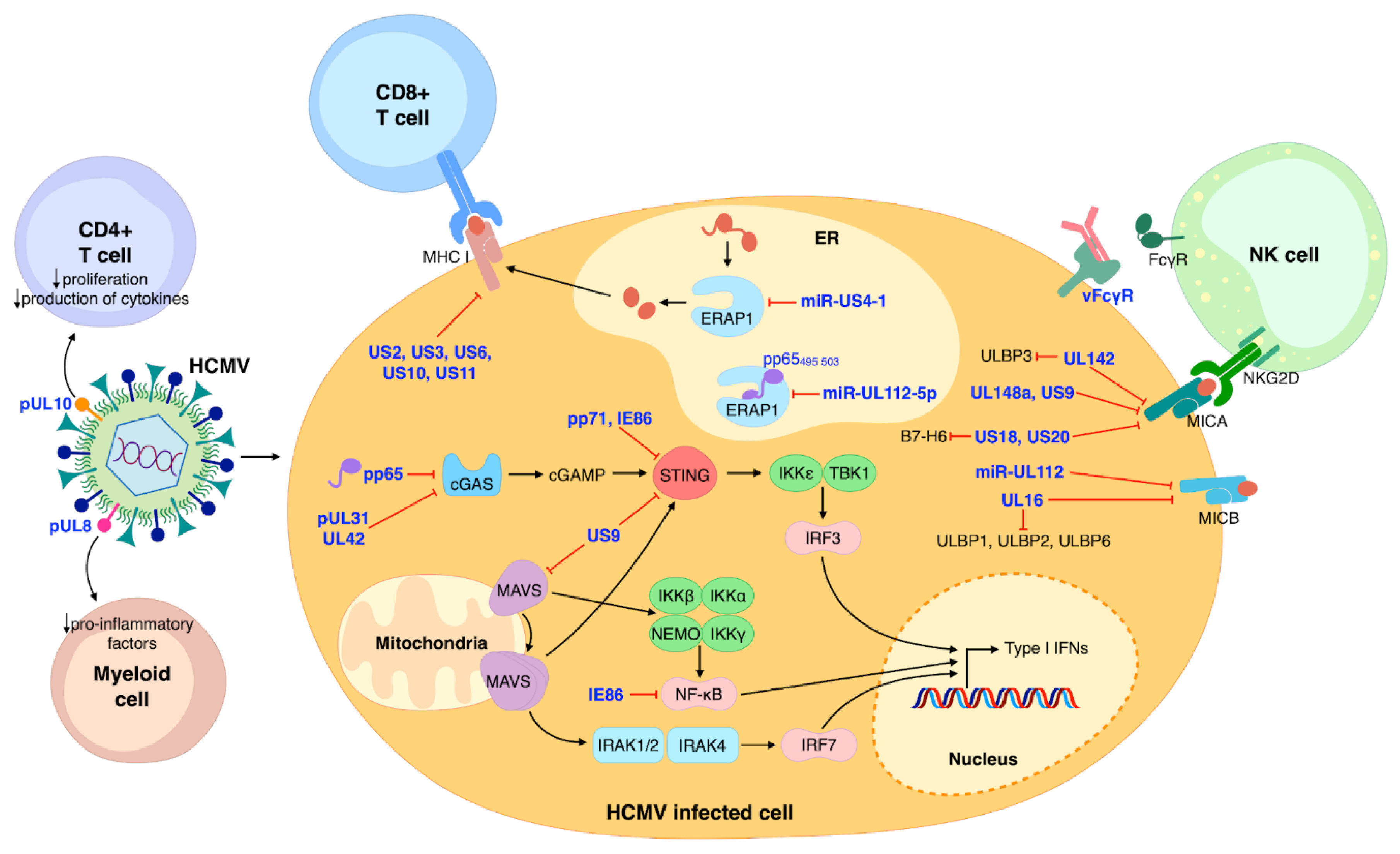
2. Modulation of the Immune System by HCMV
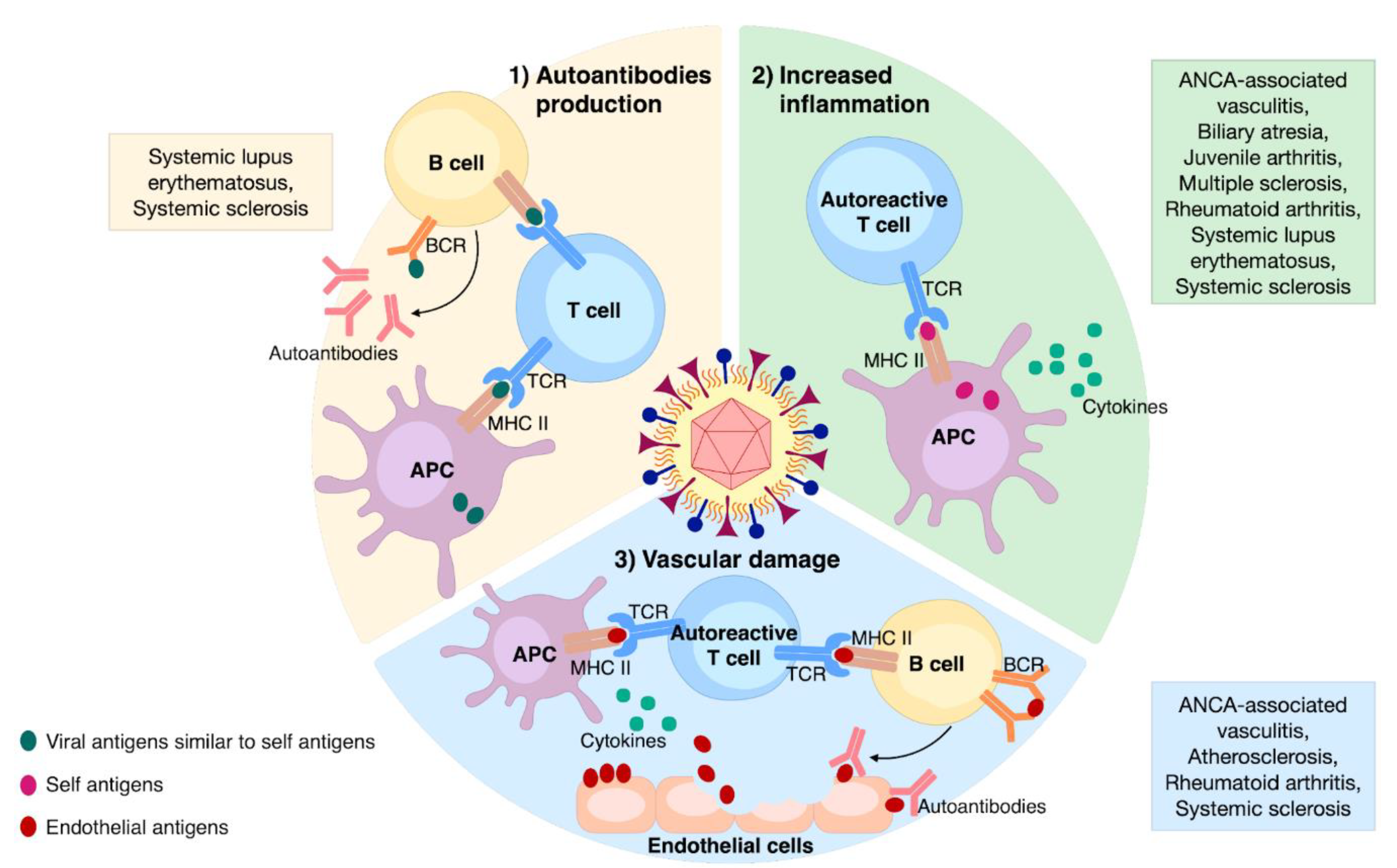
3. Documented Mechanisms of HCMV-Induced Autoimmunity
3.1. Autoantibodies Production
3.2. Enhanced Inflammation
3.3. Vascular Damage
4. The Main Autoimmune Diseases Associated with HCMV Infection
4.1. Rheumatologic Diseases
4.1.1. Systemic Lupus Erythematosus
4.1.2. Systemic Sclerosis
4.1.3. Rheumatoid Arthritis
4.2. Neurological Diseases
Multiple Sclerosis
4.3. Enteropathies
4.4. Metabolic Diseases
Type 1 Diabetes
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, F.-S.; Gershwin, M.E. Human Autoimmune Diseases: A Comprehensive Update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary Biliary Cirrhosis in Monozygotic and Dizygotic Twins: Genetics, Epigenetics, and Environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Perricone, C.; Versini, M.; Ben-Ami, D.; Gertel, S.; Watad, A.; Segel, M.J.; Ceccarelli, F.; Conti, F.; Cantarini, L.; Bogdanos, D.P.; et al. Smoke and Autoimmunity: The Fire behind the Disease. Autoimmun. Rev. 2016, 15, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C. Genetic and Environmental Risk Factors for Autoimmune Hepatitis. Clin. Liver Dis. 2019, 14, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Deitiker, P.; Atassi, M.Z. Non-MHC Genes Linked to Autoimmune Disease. Crit. Rev. Immunol. 2012, 32, 193–285. [Google Scholar] [CrossRef]
- Hu, X.; Daly, M. What Have We Learned from Six Years of GWAS in Autoimmune Diseases, and What Is Next? Curr. Opin. Immunol. 2012, 24, 571–575. [Google Scholar] [CrossRef]
- Fairweather, D.; Rose, N.R. Women and Autoimmune Diseases. Emerg. Infect. Dis. 2004, 10, 2005–2011. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.K.; Hein, M.Y.; Huang, S.-X.; Ma, M.; Shen, B.; Qian, S.-B.; Hengel, H.; et al. Decoding Human Cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the Transcriptional Landscape during Cytomegalovirus Latency with Single-Cell RNA Sequencing. mBio 2018, 9. [Google Scholar] [CrossRef]
- DiNardo, A.R.; Netea, M.G.; Musher, D.M. Postinfectious Epigenetic Immune Modifications—A Double-Edged Sword. N. Engl. J. Med. 2021, 384, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hummel, M.; Abecassis, M. Epigenetic Regulation of Cellular and Cytomegalovirus Genes during Myeloid Cell Development. Intern. Med. Rev. 2017, 3. [Google Scholar] [CrossRef]
- Kananen, L.; Nevalainen, T.; Jylhävä, J.; Marttila, S.; Hervonen, A.; Jylhä, M.; Hurme, M. Cytomegalovirus Infection Accelerates Epigenetic Aging. Exp. Gerontol. 2015, 72, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Surace, A.E.A.; Hedrich, C.M. The Role of Epigenetics in Autoimmune/Inflammatory Disease. Front. Immunol. 2019, 10, 1525. [Google Scholar] [CrossRef]
- Dunn, W.; Chou, C.; Li, H.; Hai, R.; Patterson, D.; Stolc, V.; Zhu, H.; Liu, F. Functional Profiling of a Human Cytomegalovirus Genome. Proc. Natl. Acad. Sci. USA 2003, 100, 14223–14228. [Google Scholar] [CrossRef]
- McSharry, B.P.; Avdic, S.; Slobedman, B. Human Cytomegalovirus Encoded Homologs of Cytokines, Chemokines and Their Receptors: Roles in Immunomodulation. Viruses 2012, 4, 2448–2470. [Google Scholar] [CrossRef]
- Halenius, A.; Hengel, H. Human Cytomegalovirus and Autoimmune Disease. Biomed. Res. Int. 2014, 2014, 472978. [Google Scholar] [CrossRef]
- Galitska, G.; Biolatti, M.; De Andrea, M.; Leone, A.; Coscia, A.; Bertolotti, L.; Ala, U.; Bertino, E.; Dell’Oste, V.; Landolfo, S. Biological Relevance of Cytomegalovirus Genetic Variability in Congenitally and Postnatally Infected Children. J. Clin. Virol. 2018, 108, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where Do We Stand after Decades of Studying Human Cytomegalovirus? Microorganisms 2020, 8, 685. [Google Scholar] [CrossRef]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of Innate and Adaptive Immunity by Cytomegaloviruses. Nat. Rev. Immunol. 2020, 20, 113–127. [Google Scholar] [CrossRef]
- Sinzger, C.; Schmidt, K.; Knapp, J.; Kahl, M.; Beck, R.; Waldman, J.; Hebart, H.; Einsele, H.; Jahn, G. Modification of Human Cytomegalovirus Tropism through Propagation in Vitro Is Associated with Changes in the Viral Genome. J. Gen. Virol. 1999, 80, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus Cell Tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar]
- Nguyen, C.C.; Kamil, J.P. Pathogen at the Gates: Human Cytomegalovirus Entry and Cell Tropism. Viruses 2018, 10, 704. [Google Scholar] [CrossRef]
- Gerna, G.; Kabanova, A.; Lilleri, D. Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines 2019, 7, 70. [Google Scholar] [CrossRef] [PubMed]
- Zuhair, M.; Smit, G.S.A.; Wallis, G.; Jabbar, F.; Smith, C.; Devleesschauwer, B.; Griffiths, P. Estimation of the Worldwide Seroprevalence of Cytomegalovirus: A Systematic Review and Meta-Analysis. Rev. Med. Virol. 2019, 29, e2034. [Google Scholar] [CrossRef] [PubMed]
- HoHsieh, A.; Wang, C.M.; Wu, Y.-J.J.; Chen, A.; Chang, M.-I.; Chen, J.-Y. B Cell Epitope of Human Cytomegalovirus Phosphoprotein 65 (HCMV Pp65) Induced Anti-DsDNA Antibody in BALB/c Mice. Arthritis Res. 2017, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Varani, S.; Mastroianni, A.; Frascaroli, G.; Tammik, C.; Rahbar, A.; Christensson, M.; Rossini, G.; Landini, M.P.; Söderberg-Nauclér, C. Generalized Wegener’s Granulomatosis in an Immunocompetent Adult after Cytomegalovirus Mononucleosis and Bacterial Urinary Tract Infection. Arthritis Rheum. 2009, 60, 1558–1562. [Google Scholar] [CrossRef]
- Giugni, T.D.; Söderberg, C.; Ham, D.J.; Bautista, R.M.; Hedlund, K.O.; Möller, E.; Zaia, J.A. Neutralization of Human Cytomegalovirus by Human CD13-Specific Antibodies. J. Infect. Dis. 1996, 173, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, C.; Sumitran-Karuppan, S.; Ljungman, P.; Moller, E. CD13-Specific Autoimmunity in Cytomegalovirus-Infected Immunocompromised Patients. Transplantation 1996, 61, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Mengarelli, A.; Minotti, C.; Palumbo, G.; Arcieri, P.; Gentile, G.; Iori, A.P.; Arcese, W.; Mandelli, F.; Avvisati, G. High Levels of Antiphospholipid Antibodies Are Associated with Cytomegalovirus Infection in Unrelated Bone Marrow and Cord Blood Allogeneic Stem Cell Transplantation. Br. J. Haematol. 2000, 108, 126–131. [Google Scholar] [CrossRef]
- Toyoda, M.; Galfayan, K.; Galera, O.A.; Petrosian, A.; Czer, L.S.; Jordan, S.C. Cytomegalovirus Infection Induces Anti-Endothelial Cell Antibodies in Cardiac and Renal Allograft Recipients. Transplant. Immunol. 1997, 5, 104–111. [Google Scholar] [CrossRef]
- Wager, O.; Räsänen, J.A.; Hagman, A.; Klemola, E. Mixed Cryoimmunoglobulinaemia in Infectious Mononucleois and Cytomegalovirus Mononucleosis. Int. Arch. Allergy Appl. Immunol. 1968, 34, 345–361. [Google Scholar] [CrossRef]
- Kantor, G.L.; Goldberg, L.S.; Johnson, B.L.; Derechin, M.M.; Barnett, E.V. Immunologic Abnormalities Induced by Postperfusion Cytomegalovirus Infection. Ann. Intern. Med. 1970, 73, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Lunardi, C.; Dolcino, M.; Peterlana, D.; Bason, C.; Navone, R.; Tamassia, N.; Beri, R.; Corrocher, R.; Puccetti, A. Antibodies against Human Cytomegalovirus in the Pathogenesis of Systemic Sclerosis: A Gene Array Approach. PLoS Med. 2006, 3, e2. [Google Scholar] [CrossRef] [PubMed]
- Streblow, D.N.; Orloff, S.L.; Nelson, J.A. Acceleration of Allograft Failure by Cytomegalovirus. Curr. Opin. Immunol. 2007, 19, 577–582. [Google Scholar] [CrossRef]
- Amaya-Amaya, J.; Montoya-Sánchez, L.; Rojas-Villarraga, A. Cardiovascular Involvement in Autoimmune Diseases. Biomed. Res. Int. 2014, 2014, 367359. [Google Scholar] [CrossRef] [PubMed]
- van de Berg, P.J.; Heutinck, K.M.; Raabe, R.; Minnee, R.C.; Young, S.L.; van Donselaar-van der Pant, K.A.; Bemelman, F.J.; van Lier, R.A.; ten Berge, I.J. Human Cytomegalovirus Induces Systemic Immune Activation Characterized by a Type 1 Cytokine Signature. J. Infect. Dis. 2010, 202, 690–699. [Google Scholar] [CrossRef]
- George, M.J.; Snydman, D.R.; Werner, B.G.; Griffith, J.; Falagas, M.E.; Dougherty, N.N.; Rubin, R.H. The Independent Role of Cytomegalovirus as a Risk Factor for Invasive Fungal Disease in Orthotopic Liver Transplant Recipients. Boston Center for Liver Transplantation CMVIG-Study Group. Cytogam, MedImmune, Inc. Gaithersburg, Maryland. Am. J. Med. 1997, 103, 106–113. [Google Scholar] [CrossRef]
- Forte, E.; Zhang, Z.; Thorp, E.B.; Hummel, M. Cytomegalovirus Latency and Reactivation: An Intricate Interplay With the Host Immune Response. Front. Cell Infect. Microbiol. 2020, 10, 130. [Google Scholar] [CrossRef]
- Dell’Oste, V.; Biolatti, M.; Galitska, G.; Griffante, G.; Gugliesi, F.; Pasquero, S.; Zingoni, A.; Cerboni, C.; De Andrea, M. Tuning the Orchestra: HCMV vs. Innate Immunity. Front. Microbiol. 2020, 11, 661. [Google Scholar] [CrossRef]
- Ye, L.; Qian, Y.; Yu, W.; Guo, G.; Wang, H.; Xue, X. Functional Profile of Human Cytomegalovirus Genes and Their Associated Diseases: A Review. Front. Microbiol. 2020, 11, 2104. [Google Scholar] [CrossRef]
- Manandhar, T.; Hò, G.-G.T.; Pump, W.C.; Blasczyk, R.; Bade-Doeding, C. Battle between Host Immune Cellular Responses and HCMV Immune Evasion. Int. J. Mol. Sci. 2019, 20, 3626. [Google Scholar] [CrossRef]
- Hewitt, E.W. The MHC Class I Antigen Presentation Pathway: Strategies for Viral Immune Evasion. Immunology 2003, 110, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Lee, S.; Shin, J.; Kim, Y.; Evnouchidou, I.; Kim, D.; Kim, Y.-K.; Kim, Y.-E.; Ahn, J.-H.; Riddell, S.R.; et al. Human Cytomegalovirus MicroRNA MiR-US4-1 Inhibits CD8(+) T Cell Responses by Targeting the Aminopeptidase ERAP1. Nat. Immunol. 2011, 12, 984–991. [Google Scholar] [CrossRef]
- Romania, P.; Cifaldi, L.; Pignoloni, B.; Starc, N.; D’Alicandro, V.; Melaiu, O.; Li Pira, G.; Giorda, E.; Carrozzo, R.; Bergvall, M.; et al. Identification of a Genetic Variation in ERAP1 Aminopeptidase That Prevents Human Cytomegalovirus MiR-UL112-5p-Mediated Immunoevasion. Cell Rep. 2017, 20, 846–853. [Google Scholar] [CrossRef]
- Pérez-Carmona, N.; Martínez-Vicente, P.; Farré, D.; Gabaev, I.; Messerle, M.; Engel, P.; Angulo, A. A Prominent Role of the Human Cytomegalovirus UL8 Glycoprotein in Restraining Proinflammatory Cytokine Production by Myeloid Cells at Late Times during Infection. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Bruno, L.; Cortese, M.; Monda, G.; Gentile, M.; Calò, S.; Schiavetti, F.; Zedda, L.; Cattaneo, E.; Piccioli, D.; Schaefer, M.; et al. Human Cytomegalovirus PUL10 Interacts with Leukocytes and Impairs TCR-Mediated T-Cell Activation. Immunol Cell Biol. 2016, 94, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Galitska, G.; Biolatti, M.; Griffante, G.; Gugliesi, F.; Pasquero, S.; Dell’Oste, V.; Landolfo, S. Catch Me If You Can: The Arms Race between Human Cytomegalovirus and the Innate Immune System. Future Virol. 2019, 14, 247–263. [Google Scholar] [CrossRef]
- Biolatti, M.; Gugliesi, F.; Dell’Oste, V.; Landolfo, S. Modulation of the Innate Immune Response by Human Cytomegalovirus. Infect. Genet. Evol. 2018, 64, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Biolatti, M.; Dell’Oste, V.; Pautasso, S.; Gugliesi, F.; von Einem, J.; Krapp, C.; Jakobsen, M.R.; Borgogna, C.; Gariglio, M.; De Andrea, M.; et al. Human Cytomegalovirus Tegument Protein Pp65 (PUL83) Dampens Type I Interferon Production by Inactivating the DNA Sensor CGAS without Affecting STING. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-F.; Zou, H.-M.; Liao, B.-W.; Zhang, H.-Y.; Yang, Y.; Fu, Y.-Z.; Wang, S.-Y.; Luo, M.-H.; Wang, Y.-Y. Human Cytomegalovirus Protein UL31 Inhibits DNA Sensing of CGAS to Mediate Immune Evasion. Cell Host Microbe 2018, 24, 69–80.e4. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Su, S.; Gao, Y.-Q.; Wang, P.-P.; Huang, Z.-F.; Hu, M.-M.; Luo, W.-W.; Li, S.; Luo, M.-H.; Wang, Y.-Y.; et al. Human Cytomegalovirus Tegument Protein UL82 Inhibits STING-Mediated Signaling to Evade Antiviral Immunity. Cell Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.-Z.; Guo, Y.; Zou, H.-M.; Su, S.; Wang, S.-Y.; Yang, Q.; Luo, M.-H.; Wang, Y.-Y. Human Cytomegalovirus Protein UL42 Antagonizes CGAS/MITA-Mediated Innate Antiviral Response. PLoS Pathog. 2019, 15, e1007691. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human Cytomegalovirus-Encoded US9 Targets MAVS and STING Signaling to Evade Type I Interferon Immune Responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.T.; Bresnahan, W.A. Human Cytomegalovirus IE86 Attenuates Virus- and Tumor Necrosis Factor Alpha-Induced NFkappaB-Dependent Gene Expression. J. Virol. 2006, 80, 10763–10771. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Kim, Y.-E.; Stinski, M.F.; Ahn, J.-H.; Song, Y.-J. Human Cytomegalovirus IE2 86 KDa Protein Induces STING Degradation and Inhibits CGAMP-Mediated IFN-β Induction. Front. Microbiol. 2017, 8, 1854. [Google Scholar] [CrossRef]
- Zingoni, A.; Molfetta, R.; Fionda, C.; Soriani, A.; Paolini, R.; Cippitelli, M.; Cerboni, C.; Santoni, A. NKG2D and Its Ligands: “One for All, All for One”. Front. Immunol. 2018, 9, 476. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.C.; Lanier, L.L. NK Cells in Host Responses to Viral Infections. Curr. Opin. Immunol. 2017, 44, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Vlahava, V.-M.; Forbes, S.K.; Fielding, C.A.; Stanton, R.J.; Wang, E.C.Y. HCMV-Encoded NK Modulators: Lessons From in Vitro and in Vivo Genetic Variation. Front. Immunol. 2018, 9, 2214. [Google Scholar] [CrossRef]
- Schmiedel, D.; Mandelboim, O. Disarming Cellular Alarm Systems-Manipulation of Stress-Induced NKG2D Ligands by Human Herpesviruses. Front. Immunol. 2017, 8, 390. [Google Scholar] [CrossRef]
- Cosman, D.; Müllberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, Novel MHC Class I-Related Molecules, Bind to CMV Glycoprotein UL16 and Stimulate NK Cytotoxicity through the NKG2D Receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Kubin, M.; Cassiano, L.; Chalupny, J.; Chin, W.; Cosman, D.; Fanslow, W.; Müllberg, J.; Rousseau, A.M.; Ulrich, D.; Armitage, R. ULBP1, 2, 3: Novel MHC Class I-Related Molecules That Bind to Human Cytomegalovirus Glycoprotein UL16, Activate NK Cells. Eur. J. Immunol. 2001, 31, 1428–1437. [Google Scholar] [CrossRef]
- Rölle, A.; Mousavi-Jazi, M.; Eriksson, M.; Odeberg, J.; Söderberg-Nauclér, C.; Cosman, D.; Kärre, K.; Cerboni, C. Effects of Human Cytomegalovirus Infection on Ligands for the Activating NKG2D Receptor of NK Cells: Up-Regulation of UL16-Binding Protein (ULBP)1 and ULBP2 Is Counteracted by the Viral UL16 Protein. J. Immunol. 2003, 171, 902–908. [Google Scholar] [CrossRef]
- Eagle, R.A.; Traherne, J.A.; Hair, J.R.; Jafferji, I.; Trowsdale, J. ULBP6/RAET1L Is an Additional Human NKG2D Ligand. Eur. J. Immunol. 2009, 39, 3207–3216. [Google Scholar] [CrossRef] [PubMed]
- Ashiru, O.; Bennett, N.J.; Boyle, L.H.; Thomas, M.; Trowsdale, J.; Wills, M.R. NKG2D Ligand MICA Is Retained in the Cis-Golgi Apparatus by Human Cytomegalovirus Protein UL142. J. Virol. 2009, 83, 12345–12354. [Google Scholar] [CrossRef]
- Bennett, N.J.; Ashiru, O.; Morgan, F.J.E.; Pang, Y.; Okecha, G.; Eagle, R.A.; Trowsdale, J.; Sissons, J.G.P.; Wills, M.R. Intracellular Sequestration of the NKG2D Ligand ULBP3 by Human Cytomegalovirus. J. Immunol. 2010, 185, 1093–1102. [Google Scholar] [CrossRef]
- Fielding, C.A.; Weekes, M.P.; Nobre, L.V.; Ruckova, E.; Wilkie, G.S.; Paulo, J.A.; Chang, C.; Suárez, N.M.; Davies, J.A.; Antrobus, R.; et al. Control of Immune Ligands by Members of a Cytomegalovirus Gene Expansion Suppresses Natural Killer Cell Activation. Elife 2017, 6. [Google Scholar] [CrossRef]
- Charpak-Amikam, Y.; Kubsch, T.; Seidel, E.; Oiknine-Djian, E.; Cavaletto, N.; Yamin, R.; Schmiedel, D.; Wolf, D.; Gribaudo, G.; Messerle, M.; et al. Human Cytomegalovirus Escapes Immune Recognition by NK Cells through the Downregulation of B7-H6 by the Viral Genes US18 and US20. Sci. Rep. 2017, 7, 8661. [Google Scholar] [CrossRef]
- Jenks, J.A.; Goodwin, M.L.; Permar, S.R. The Roles of Host and Viral Antibody Fc Receptors in Herpes Simplex Virus (HSV) and Human Cytomegalovirus (HCMV) Infections and Immunity. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Atalay, R.; Zimmermann, A.; Wagner, M.; Borst, E.; Benz, C.; Messerle, M.; Hengel, H. Identification and Expression of Human Cytomegalovirus Transcription Units Coding for Two Distinct Fcγ Receptor Homologs. J. Virol. 2002, 76, 8596–8608. [Google Scholar] [CrossRef]
- Corrales-Aguilar, E.; Trilling, M.; Hunold, K.; Fiedler, M.; Le, V.T.K.; Reinhard, H.; Ehrhardt, K.; Mercé-Maldonado, E.; Aliyev, E.; Zimmermann, A.; et al. Human Cytomegalovirus Fcγ Binding Proteins Gp34 and Gp68 Antagonize Fcγ Receptors I, II and III. PLoS Pathog. 2014, 10, e1004131. [Google Scholar] [CrossRef]
- Cortese, M.; Calò, S.; D’Aurizio, R.; Lilja, A.; Pacchiani, N.; Merola, M. Recombinant Human Cytomegalovirus (HCMV) RL13 Binds Human Immunoglobulin G Fc. PLoS ONE 2012, 7, e50166. [Google Scholar] [CrossRef]
- Sprague, E.R.; Reinhard, H.; Cheung, E.J.; Farley, A.H.; Trujillo, R.D.; Hengel, H.; Bjorkman, P.J. The Human Cytomegalovirus Fc Receptor Gp68 Binds the Fc CH2-CH3 Interface of Immunoglobulin G. J. Virol. 2008, 82, 3490–3499. [Google Scholar] [CrossRef]
- Perrier, S.; Serre, A.F.; Dubost, J.J.; Beaujon, G.; Plazonnet, M.P.; Albuisson, E.; Sauvezie, B. Increased Serum Levels of Interleukin 10 in Sjögren’s Syndrome; Correlation with Increased IgG1. J. Rheumatol. 2000, 27, 935–939. [Google Scholar]
- Elazeem, M.I.A.; Mohammed, R.A.; Abdallah, N.H. Correlation of Serum Interleukin-10 Level with Disease Activity and Severity in Systemic Lupus Erythematosus. Egypt Rheumatol. Rehabil. 2018, 45, 25–33. [Google Scholar] [CrossRef]
- Manolova, I.; Ivanova, M.; Stanilova, S. Gene Polymorphisms of Immunoregulatory Cytokines IL-10 and TGF-Β1 in Systemic Lupus Erythematosus. Genes Autoimmun. Intracell. Signal. Microbiome Contrib. 2013. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Ciccacci, C.; Priori, R.; Latini, A.; Picarelli, G.; Arienzo, F.; Novelli, G.; Valesini, G.; Perricone, C.; Borgiani, P. STAT4, TRAF3IP2, IL10, and HCP5 Polymorphisms in Sjögren’s Syndrome: Association with Disease Susceptibility and Clinical Aspects. Available online: https://www.hindawi.com/journals/jir/2019/7682827/ (accessed on 2 February 2021).
- Ciccacci, C.; Perricone, C.; Ceccarelli, F.; Rufini, S.; Fusco, D.D.; Alessandri, C.; Spinelli, F.R.; Cipriano, E.; Novelli, G.; Valesini, G.; et al. A Multilocus Genetic Study in a Cohort of Italian SLE Patients Confirms the Association with STAT4 Gene and Describes a New Association with HCP5 Gene. PLoS ONE 2014, 9, e111991. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.D.; Lee, W.J.; Kong, K.A.; Woo, J.H.; Choi, S.J.; Lee, Y.H.; Song, G.G. Association of STAT4 Polymorphism with Rheumatoid Arthritis and Systemic Lupus Erythematosus: A Meta-Analysis. Mol. Biol. Rep. 2010, 37, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, N.; Rönnblom, L. Interferon-α Enhances the IL-12-Induced STAT4 Activation Selectively in Carriers of the STAT4 SLE Risk Allele Rs7574865[T]. Ann. Rheum. Dis. 2019, 78, 429–431. [Google Scholar] [CrossRef]
- Nagata, K.; Hayashi, K. Epstein-Barr Virus Reactivation-Induced Immunoglobulin Production: Significance on Autoimmunity. Microorganisms 2020, 8, 1875. [Google Scholar] [CrossRef]
- Muryoi, T.; Kasturi, K.N.; Kafina, M.J.; Cram, D.S.; Harrison, L.C.; Sasaki, T.; Bona, C.A. Antitopoisomerase I Monoclonal Autoantibodies from Scleroderma Patients and Tight Skin Mouse Interact with Similar Epitopes. J. Exp. Med. 1992, 175, 1103–1109. [Google Scholar] [CrossRef]
- Lunardi, C.; Bason, C.; Navone, R.; Millo, E.; Damonte, G.; Corrocher, R.; Puccetti, A. Systemic Sclerosis Immunoglobulin G Autoantibodies Bind the Human Cytomegalovirus Late Protein UL94 and Induce Apoptosis in Human Endothelial Cells. Nat. Med. 2000, 6, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Pan, M.-R.; Chen, D.-Y.; Lan, J.-L. Human Cytomegalovirus Pp65 Lower Matrix Protein: A Humoral Immunogen for Systemic Lupus Erythematosus Patients and Autoantibody Accelerator for NZB/W F1 Mice. Clin. Exp. Immunol. 2006, 143, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Mu, R.; Gao, Y.-P.; Dong, J.; Zhu, L.; Ma, Y.; Li, Y.-H.; Zhang, H.-Q.; Han, D.; Zhang, Y.; et al. A Cytomegalovirus Peptide-Specific Antibody Alters Natural Killer Cell Homeostasis and Is Shared in Several Autoimmune Diseases. Cell Host Microbe 2016, 19, 400–408. [Google Scholar] [CrossRef]
- Hsieh, A.-H.; Jhou, Y.-J.; Liang, C.-T.; Chang, M.; Wang, S.-L. Fragment of Tegument Protein Pp65 of Human Cytomegalovirus Induces Autoantibodies in BALB/c Mice. Arthritis Res. 2011, 13, R162. [Google Scholar] [CrossRef] [PubMed]
- Neo, J.Y.J.; Wee, S.Y.K.; Bonne, I.; Tay, S.H.; Raida, M.; Jovanovic, V.; Fairhurst, A.-M.; Lu, J.; Hanson, B.J.; MacAry, P.A. Characterisation of a Human Antibody That Potentially Links Cytomegalovirus Infection with Systemic Lupus Erythematosus. Sci. Rep. 2019, 9, 9998. [Google Scholar] [CrossRef]
- Pak, C.Y.; Cha, C.Y.; Rajotte, R.V.; McArthur, R.G.; Yoon, J.W. Human Pancreatic Islet Cell Specific 38 Kilodalton Autoantigen Identified by Cytomegalovirus-Induced Monoclonal Islet Cell Autoantibody. Diabetologia 1990, 33, 569–572. [Google Scholar] [CrossRef]
- Curtis, H.A.; Singh, T.; Newkirk, M.M. Recombinant Cytomegalovirus Glycoprotein GB (UL55) Induces an Autoantibody Response to the U1-70 KDa Small Nuclear Ribonucleoprotein. Eur. J. Immunol. 1999, 29, 3643–3653. [Google Scholar] [CrossRef]
- Varani, S.; Cederarv, M.; Feld, S.; Tammik, C.; Frascaroli, G.; Landini, M.P.; Söderberg-Nauclér, C. Human Cytomegalovirus Differentially Controls B Cell and T Cell Responses through Effects on Plasmacytoid Dendritic Cells. J. Immunol. 2007, 179, 7767–7776. [Google Scholar] [CrossRef]
- Xu, H.; Dong, P.; Ma, X.; Song, D.; Xue, D.; Xu, R.; Lu, H.; He, X. B Cell-Activating Factor Regulates the Survival of B Lymphocytes Infected with Human Cytomegalovirus. Immunol. Lett. 2017, 187, 1–6. [Google Scholar] [CrossRef]
- van Leeuwen, E.M.M.; Remmerswaal, E.B.M.; Vossen, M.T.M.; Rowshani, A.T.; Wertheim-van Dillen, P.M.E.; van Lier, R.A.W.; ten Berge, I.J.M. Emergence of a CD4+CD28- Granzyme B+, Cytomegalovirus-Specific T Cell Subset after Recovery of Primary Cytomegalovirus Infection. J. Immunol. 2004, 173, 1834–1841. [Google Scholar] [CrossRef]
- Fasth, A.E.R.; Dastmalchi, M.; Rahbar, A.; Salomonsson, S.; Pandya, J.M.; Lindroos, E.; Nennesmo, I.; Malmberg, K.-J.; Söderberg-Nauclér, C.; Trollmo, C.; et al. T Cell Infiltrates in the Muscles of Patients with Dermatomyositis and Polymyositis Are Dominated by CD28null T Cells. J. Immunol. 2009, 183, 4792–4799. [Google Scholar] [CrossRef] [PubMed]
- Gerli, R.; Schillaci, G.; Giordano, A.; Bocci, E.B.; Bistoni, O.; Vaudo, G.; Marchesi, S.; Pirro, M.; Ragni, F.; Shoenfeld, Y.; et al. CD4+CD28− T Lymphocytes Contribute to Early Atherosclerotic Damage in Rheumatoid Arthritis Patients. Circulation 2004, 109, 2744–2748. [Google Scholar] [CrossRef]
- Bano, A.; Pera, A.; Almoukayed, A.; Clarke, T.H.S.; Kirmani, S.; Davies, K.A.; Kern, F. CD28null CD4 T-Cell Expansions in Autoimmune Disease Suggest a Link with Cytomegalovirus Infection. F1000Res 2019, 8, 327. [Google Scholar] [CrossRef]
- Chanouzas, D.; Sagmeister, M.; Faustini, S.; Nightingale, P.; Richter, A.; Ferro, C.J.; Morgan, M.D.; Moss, P.; Harper, L. Subclinical Reactivation of Cytomegalovirus Drives CD4+CD28null T-Cell Expansion and Impaired Immune Response to Pneumococcal Vaccination in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. J. Infect. Dis. 2019, 219, 234–244. [Google Scholar] [CrossRef]
- Wu, C.-S.; Chyuan, I.-T.; Chiu, Y.-L.; Chen, W.-L.; Shen, C.-Y.; Hsu, P.-N. Preserved Specific Anti-Viral T-Cell Response but Associated with Decreased Lupus Activity in SLE Patients with Cytomegalovirus Infection. Rheumatology 2020, 59, 3340–3349. [Google Scholar] [CrossRef] [PubMed]
- Zabalza, A.; Vera, A.; Alari-Pahissa, E.; Munteis, E.; Moreira, A.; Yélamos, J.; Llop, M.; López-Botet, M.; Martínez-Rodríguez, J.E. Impact of Cytomegalovirus Infection on B Cell Differentiation and Cytokine Production in Multiple Sclerosis. J. Neuroinflammation 2020, 17, 161. [Google Scholar] [CrossRef]
- Almanzar, G.; Schmalzing, M.; Trippen, R.; Höfner, K.; Weißbrich, B.; Geissinger, E.; Meyer, T.; Liese, J.; Tony, H.-P.; Prelog, M. Significant IFNγ Responses of CD8+ T Cells in CMV-Seropositive Individuals with Autoimmune Arthritis. J. Clin. Virol. 2016, 77, 77–84. [Google Scholar] [CrossRef]
- Rothe, K.; Quandt, D.; Schubert, K.; Rossol, M.; Klingner, M.; Jasinski-Bergner, S.; Scholz, R.; Seliger, B.; Pierer, M.; Baerwald, C.; et al. Latent Cytomegalovirus Infection in Rheumatoid Arthritis and Increased Frequencies of Cytolytic LIR-1+CD8+ T Cells. Arthritis Rheumatol. 2016, 68, 337–346. [Google Scholar] [CrossRef] [PubMed]
- Janahi, E.M.A.; Das, S.; Bhattacharya, S.N.; Haque, S.; Akhter, N.; Jawed, A.; Wahid, M.; Mandal, R.K.; Lohani, M.; Areeshi, M.Y.; et al. Cytomegalovirus Aggravates the Autoimmune Phenomenon in Systemic Autoimmune Diseases. Microb. Pathog. 2018, 120, 132–139. [Google Scholar] [CrossRef]
- Arcangeletti, M.-C.; Maccari, C.; Vescovini, R.; Volpi, R.; Giuggioli, D.; Sighinolfi, G.; De Conto, F.; Chezzi, C.; Calderaro, A.; Ferri, C. A Paradigmatic Interplay between Human Cytomegalovirus and Host Immune System: Possible Involvement of Viral Antigen-Driven CD8+ T Cell Responses in Systemic Sclerosis. Viruses 2018, 10, 508. [Google Scholar] [CrossRef]
- Arcangeletti, M.-C.; D’Accolti, M.; Maccari, C.; Soffritti, I.; Conto, F.D.; Chezzi, C.; Calderaro, A.; Ferri, C.; Caselli, E. Impact of Human Cytomegalovirus and Human Herpesvirus 6 Infection on the Expression of Factors Associated with Cell Fibrosis and Apoptosis: Clues for Implication in Systemic Sclerosis Development. Int. J. Mol. Sci. 2020, 21, 6397. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Ye, S.; Xie, S.; Ye, L.; Lin, C.; Yang, M.; Shi, X.; Wang, F.; Li, B.; Li, M.; et al. The Cytomegalovirus Protein US31 Induces Inflammation through Mono-Macrophages in Systemic Lupus Erythematosus by Promoting NF-ΚB2 Activation. Cell Death Dis. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic, J.; Popovic, B.; Milovanovic, M.; Kvestak, D.; Arsenijevic, A.; Stojanovic, B.; Tanaskovic, I.; Krmpotic, A.; Arsenijevic, N.; Jonjic, S.; et al. Murine Cytomegalovirus Infection Induces Susceptibility to EAE in Resistant BALB/c Mice. Front. Immunol. 2017, 8, 192. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wen, J.; Xiao, Y.; Wang, J.; Pan, W.; Zhou, Y.; Zhang, X.; Guan, W.; Chen, Y.; Zhou, K.; Wang, Y.; et al. Low Doses of CMV Induce Autoimmune-Mediated and Inflammatory Responses in Bile Duct Epithelia of Regulatory T Cell-Depleted Neonatal Mice. Lab. Investig. 2015, 95, 180–192. [Google Scholar] [CrossRef][Green Version]
- Zitti, B.; Bryceson, Y.T. Natural Killer Cells in Inflammation and Autoimmunity. Cytokine Growth Factor Rev. 2018, 42, 37–46. [Google Scholar] [CrossRef]
- Martínez-Rodríguez, J.E.; Cobo-Calvo, A.; Villar, L.M.; Munteis, E.; Blanco, Y.; Rasal, R.; Vera, A.; Muntasell, A.; Alvarez-Lafuente, R.; Saiz, A.; et al. Adaptive Natural Killer Cell Response to Cytomegalovirus and Disability Progression in Multiple Sclerosis. Mult. Scler. 2016, 22, 741–752. [Google Scholar] [CrossRef]
- McCarthy, N.E.; Eberl, M. Human Γδ T-Cell Control of Mucosal Immunity and Inflammation. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Tometten, I.; Felgentreff, K.; Hönig, M.; Hauck, F.; Albert, M.H.; Niehues, T.; Perez, R.; Ghosh, S.; Picard, C.; Stary, J.; et al. Increased Proportions of Γδ T Lymphocytes in Atypical SCID Associate with Disease Manifestations. Clin. Immunol. 2019, 201, 30–34. [Google Scholar] [CrossRef]
- Lunardi, C.; Bason, C.; Corrocher, R.; Puccetti, A. Induction of Endothelial Cell Damage by HCMV Molecular Mimicry. Trends Immunol. 2005, 26, 19–24. [Google Scholar] [CrossRef]
- Bason, C.; Corrocher, R.; Lunardi, C.; Puccetti, P.; Olivieri, O.; Girelli, D.; Navone, R.; Beri, R.; Millo, E.; Margonato, A.; et al. Interaction of Antibodies against Cytomegalovirus with Heat-Shock Protein 60 in Pathogenesis of Atherosclerosis. Lancet 2003, 362, 1971–1977. [Google Scholar] [CrossRef]
- Dolcino, M.; Puccetti, A.; Barbieri, A.; Bason, C.; Tinazzi, E.; Ottria, A.; Patuzzo, G.; Martinelli, N.; Lunardi, C. Infections and Autoimmunity: Role of Human Cytomegalovirus in Autoimmune Endothelial Cell Damage. Lupus 2015, 24, 419–432. [Google Scholar] [CrossRef]
- Broadley, I.; Pera, A.; Morrow, G.; Davies, K.A.; Kern, F. Expansions of Cytotoxic CD4+CD28- T Cells Drive Excess Cardiovascular Mortality in Rheumatoid Arthritis and Other Chronic Inflammatory Conditions and Are Triggered by CMV Infection. Front. Immunol. 2017, 8, 195. [Google Scholar] [CrossRef] [PubMed]
- Pera, A.; Broadley, I.; Davies, K.A.; Kern, F. Cytomegalovirus as a Driver of Excess Cardiovascular Mortality in Rheumatoid Arthritis: A Red Herring or a Smoking Gun? Circ. Res. 2017, 120, 274–277. [Google Scholar] [CrossRef]
- Di Battista, M.; Marcucci, E.; Elefante, E.; Tripoli, A.; Governato, G.; Zucchi, D.; Tani, C.; Alunno, A. One Year in Review 2018: Systemic Lupus Erythematosus. Clin. Exp. Rheumatol. 2018, 36, 763–777. [Google Scholar] [PubMed]
- Luo, S.; Long, H.; Lu, Q. Recent Advances in Understanding Pathogenesis and Therapeutic Strategies of Systemic Lupus Erythematosus. Int. Immunopharmacol. 2020, 89, 107028. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Mercado, A.E.; Vilá-Pérez, S. Cytomegalovirus as a Trigger for Systemic Lupus Erythematosus. J. Clin. Rheumatol. 2010, 16, 335–337. [Google Scholar] [CrossRef]
- Bendiksen, S.; Van Ghelue, M.; Rekvig, O.P.; Gutteberg, T.; Haga, H.J.; Moens, U. A Longitudinal Study of Human Cytomegalovirus Serology and Viruria Fails to Detect Active Viral Infection in 20 Systemic Lupus Erythematosus Patients. Lupus 2000, 9, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Rider, J.R.; Ollier, W.E.; Lock, R.J.; Brookes, S.T.; Pamphilon, D.H. Human Cytomegalovirus Infection and Systemic Lupus Erythematosus. Clin. Exp. Rheumatol. 1997, 15, 405–409. [Google Scholar]
- Takizawa, Y.; Inokuma, S.; Tanaka, Y.; Saito, K.; Atsumi, T.; Hirakata, M.; Kameda, H.; Hirohata, S.; Kondo, H.; Kumagai, S.; et al. Clinical Characteristics of Cytomegalovirus Infection in Rheumatic Diseases: Multicentre Survey in a Large Patient Population. Rheumatology 2008, 47, 1373–1378. [Google Scholar] [CrossRef]
- Newkirk, M.M.; van Venrooij, W.J.; Marshall, G.S. Autoimmune Response to U1 Small Nuclear Ribonucleoprotein (U1 SnRNP) Associated with Cytomegalovirus Infection. Arthritis Res. 2001, 3, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Su, B.Y.-J.; Su, C.-Y.; Yu, S.-F.; Chen, C.-J. Incidental Discovery of High Systemic Lupus Erythematosus Disease Activity Associated with Cytomegalovirus Viral Activity. Med. Microbiol. Immunol. 2007, 196, 165–170. [Google Scholar] [CrossRef] [PubMed]
- McClain, M.T.; Poole, B.D.; Bruner, B.F.; Kaufman, K.M.; Harley, J.B.; James, J.A. An Altered Immune Response to Epstein-Barr Nuclear Antigen 1 in Pediatric Systemic Lupus Erythematosus. Arthritis Rheum. 2006, 54, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Parks, C.G.; Cooper, G.S.; Hudson, L.L.; Dooley, M.A.; Treadwell, E.L.; St.Clair, E.W.; Gilkeson, G.S.; Pandey, J.P. Association of Epstein-Barr Virus with Systemic Lupus Erythematosus: Effect Modification by Race, Age, and Cytotoxic T Lymphocyte–Associated Antigen 4 Genotype. Arthritis Rheum. 2005, 52, 1148–1159. [Google Scholar] [CrossRef]
- James, J.A.; Neas, B.R.; Moser, K.L.; Hall, T.; Bruner, G.R.; Sestak, A.L.; Harley, J.B. Systemic Lupus Erythematosus in Adults Is Associated with Previous Epstein-Barr Virus Exposure. Arthritis Rheum. 2001, 44, 1122–1126. [Google Scholar] [CrossRef]
- Harley, J.B.; James, J.A. Epstein-Barr Virus Infection Induces Lupus Autoimmunity. Bull. Nyu Hosp. Jt Dis. 2006, 64, 45–50. [Google Scholar] [PubMed]
- Baboonian, C.; Venables, P.J.; Booth, J.; Williams, D.G.; Roffe, L.M.; Maini, R.N. Virus Infection Induces Redistribution and Membrane Localization of the Nuclear Antigen La (SS-B): A Possible Mechanism for Autoimmunity. Clin. Exp. Immunol. 1989, 78, 454–459. [Google Scholar]
- Bouza, E.; Moya, J.G.; Muñoz, P. Infections in Systemic Lupus Erythematosus and Rheumatoid Arthritis. Infect. Dis. Clin. North. Am. 2001, 15, 335–361. [Google Scholar] [CrossRef]
- Bertsias, G.; Ioannidis, J.P.A.; Boletis, J.; Bombardieri, S.; Cervera, R.; Dostal, C.; Font, J.; Gilboe, I.M.; Houssiau, F.; Huizinga, T.; et al. EULAR Recommendations for the Management of Systemic Lupus Erythematosus. Report of a Task Force of the EULAR Standing Committee for International Clinical Studies Including Therapeutics. Ann. Rheum. Dis. 2008, 67, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Shi, X.; Gan, F.; Li, X.; Zhang, W.; Li, M.; Hou, Y.; Zhang, X.; Zhao, Y.; Zeng, X.; et al. Death Causes and Pathogens Analysis of Systemic Lupus Erythematosus during the Past 26 Years. Clin. Rheumatol. 2014, 33, 57–63. [Google Scholar] [CrossRef]
- Choo, H.M.C.; Cher, W.Q.; Kwan, Y.H.; Fong, W.W.S. Risk Factors for Cytomegalovirus Disease in Systemic Lupus Erythematosus (SLE): A Systematic Review. Adv. Rheumatol. 2019, 59, 12. [Google Scholar] [CrossRef]
- Xue, Y.; Jiang, L.; Wan, W.-G.; Chen, Y.-M.; Zhang, J.; Zhang, Z.-C. Cytomegalovirus Pneumonia in Patients with Rheumatic Diseases After Immunosuppressive Therapy: A Single Center Study in China. Chin. Med. J. 2016, 129, 267–273. [Google Scholar] [CrossRef]
- Qin, L.; Qiu, Z.; Hsieh, E.; Geng, T.; Zhao, J.; Zeng, X.; Wan, L.; Xie, J.; Ramendra, R.; Routy, J.P.; et al. Association between Lymphocyte Subsets and Cytomegalovirus Infection Status among Patients with Systemic Lupus Erythematosus: A Pilot Study. Medicine 2019, 98, e16997. [Google Scholar] [CrossRef] [PubMed]
- Elhai, M.; Meune, C.; Avouac, J.; Kahan, A.; Allanore, Y. Trends in Mortality in Patients with Systemic Sclerosis over 40 Years: A Systematic Review and Meta-Analysis of Cohort Studies. Rheumatology 2012, 51, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Marie, I.; Gehanno, J.-F. Environmental Risk Factors of Systemic Sclerosis. Semin. Immunopathol. 2015, 37, 463–473. [Google Scholar] [CrossRef]
- Gyftaki-Venieri, D.A.; Abraham, D.J.; Ponticos, M. Insights into Myofibroblasts and Their Activation in Scleroderma: Opportunities for Therapy? Curr. Opin. Rheumatol. 2018, 30, 581–587. [Google Scholar] [CrossRef]
- Harvey, G.R.; McHugh, N.J. Serologic Abnormalities in Systemic Sclerosis. Curr. Opin. Rheumatol. 1999, 11, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Radić, M.; Martinović Kaliterna, D.; Radić, J. Infectious Disease as Aetiological Factor in the Pathogenesis of Systemic Sclerosis. Neth. J. Med. 2010, 68, 348–353. [Google Scholar]
- Randone, S.B.; Guiducci, S.; Cerinic, M.M. Systemic Sclerosis and Infections. Autoimmun. Rev. 2008, 8, 36–40. [Google Scholar] [CrossRef]
- Bilgin, H.; Kocabaş, H.; Keşli, R. The Prevalence of Infectious Agents in Patients with Systemic Sclerosis. Turk. J. Med. Sci. 2015, 45, 1192–1197. [Google Scholar] [CrossRef] [PubMed]
- Trombetta, A.C.; Tomatis, V.; Alessandri, E.; Paolino, S.; Pizzorni, C.; Ghio, M.; Ruaro, B.; Patane, M.; Gotelli, E.; Goegan, F.; et al. AB0723 Seroprevalence of Epstein-Barr Virus and Cytomegalovirus in Systemic Sclerosis Patients: Preliminary Results. Ann. Rheum. Dis. 2018, 77, 1500–1501. [Google Scholar] [CrossRef]
- Esen, B.A.; Yılmaz, G.; Uzun, S.; Ozdamar, M.; Aksözek, A.; Kamalı, S.; Türkoğlu, S.; Gül, A.; Ocal, L.; Aral, O.; et al. Serologic Response to Epstein-Barr Virus Antigens in Patients with Systemic Lupus Erythematosus: A Controlled Study. Rheumatol. Int. 2012, 32, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Neidhart, M.; Kuchen, S.; Distler, O.; Brühlmann, P.; Michel, B.A.; Gay, R.E.; Gay, S. Increased Serum Levels of Antibodies against Human Cytomegalovirus and Prevalence of Autoantibodies in Systemic Sclerosis. Arthritis Rheum. 1999, 42, 389–392. [Google Scholar] [CrossRef]
- Pandey, J.P. Immunoglobulin GM Genes and IgG Antibodies to Cytomegalovirus in Patients with Systemic Sclerosis. Clin. Exp. Rheumatol. 2004, 22, S35–S37. [Google Scholar]
- Arnson, Y.; Amital, H.; Guiducci, S.; Matucci-Cerinic, M.; Valentini, G.; Barzilai, O.; Maya, R.; Shoenfeld, Y. The Role of Infections in the Immunopathogensis of Systemic Sclerosis--Evidence from Serological Studies. Ann. N. Y. Acad. Sci. 2009, 1173, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, J.H.; Shaw, P.X.; Nguyen, M.D.; Medsger, T.A.; Wright, T.M.; Metcalf, J.S.; Leroy, E.C. Evidence of Activation of 2 Herpesviruses, Epstein-Barr Virus and Cytomegalovirus, in Systemic Sclerosis and Normal Skins. J. Rheumatol. 2000, 27, 821–823. [Google Scholar] [PubMed]
- Jarvis, M.A.; Nelson, J.A. Human Cytomegalovirus Persistence and Latency in Endothelial Cells and Macrophages. Curr. Opin. Microbiol. 2002, 5, 403–407. [Google Scholar] [CrossRef]
- Efthymiou, G.; Dardiotis, E.; Liaskos, C.; Marou, E.; Scheper, T.; Meyer, W.; Daponte, A.; Daoussis, D.; Hadjigeorgiou, G.; Bogdanos, D.P.; et al. A Comprehensive Analysis of Antigen-Specific Antibody Responses against Human Cytomegalovirus in Patients with Systemic Sclerosis. Clin. Immunol. 2019, 207, 87–96. [Google Scholar] [CrossRef]
- Balandraud, N.; Roudier, J. Epstein-Barr Virus and Rheumatoid Arthritis. Jt. Bone Spine 2018, 85, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Musiani, M.; Zerbini, M.; Ferri, S.; Plazzi, M.; Gentilomi, G.; La Placa, M. Comparison of the Immune Response to Epstein-Barr Virus and Cytomegalovirus in Sera and Synovial Fluids of Patients with Rheumatoid Arthritis. Ann. Rheum. Dis. 1987, 46, 837–842. [Google Scholar] [CrossRef]
- Bassyouni, R.H.; Dwedar, R.A.; Ezzat, E.M.; Marzaban, R.N.; Nassr, M.H.; Rashid, L. Elevated Cytomegalovirus and Epstein-Barr Virus Burden in Rheumatoid Arthritis: A True Pathogenic Role or Just a Coincidence. Egypt. Rheumatol. 2019, 41, 255–259. [Google Scholar] [CrossRef]
- Pierer, M.; Rothe, K.; Quandt, D.; Schulz, A.; Rossol, M.; Scholz, R.; Baerwald, C.; Wagner, U. Association of Anticytomegalovirus Seropositivity with More Severe Joint Destruction and More Frequent Joint Surgery in Rheumatoid Arthritis. Arthritis Rheum. 2012, 64, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Lafuente, R.; Fernández-Gutiérrez, B.; de Miguel, S.; Jover, J.A.; Rollin, R.; Loza, E.; Clemente, D.; Lamas, J.R. Potential Relationship between Herpes Viruses and Rheumatoid Arthritis: Analysis with Quantitative Real Time Polymerase Chain Reaction. Ann. Rheum. Dis. 2005, 64, 1357–1359. [Google Scholar] [CrossRef] [PubMed]
- Mourgues, C.; Henquell, C.; Tatar, Z.; Pereira, B.; Nourisson, C.; Tournadre, A.; Soubrier, M.; Couderc, M. Monitoring of Epstein-Barr Virus (EBV)/Cytomegalovirus (CMV)/Varicella-Zoster Virus (VZV) Load in Patients Receiving Tocilizumab for Rheumatoid Arthritis. Jt. Bone Spine 2016, 83, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Mehraein, Y.; Lennerz, C.; Ehlhardt, S.; Remberger, K.; Ojak, A.; Zang, K.D. Latent Epstein-Barr Virus (EBV) Infection and Cytomegalovirus (CMV) Infection in Synovial Tissue of Autoimmune Chronic Arthritis Determined by RNA- and DNA-in Situ Hybridization. Mod. Pathol. 2004, 17, 781–789. [Google Scholar] [CrossRef] [PubMed]
- Garrod, A.B. On Gout and Rheumatism. The Differential Diagnosis, and the Nature of the so-Called Rheumatic Gout. Med. Chir. Trans. 1854, 37, 181–220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Silman, A.J.; Pearson, J.E. Epidemiology and Genetics of Rheumatoid Arthritis. Arthritis Res. 2002, 4 (Suppl. 3), S265–S272. [Google Scholar] [CrossRef]
- Wolfe, A.M.; Kellgren, J.H.; Masi, A.T. The Epidemiology of Rheumatoid Arthritis: A Review. II. Incidence and Diagnostic Criteria. Bull. Rheum Dis. 1968, 19, 524–529. [Google Scholar] [PubMed]
- Firestein, G.S. Evolving Concepts of Rheumatoid Arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Cojocaru, M.; Cojocaru, I.M.; Silosi, I.; Vrabie, C.D.; Tanasescu, R. Extra-Articular Manifestations in Rheumatoid Arthritis. Maedica 2010, 5, 286–291. [Google Scholar]
- Taylor, P.; Gartemann, J.; Hsieh, J.; Creeden, J. A Systematic Review of Serum Biomarkers Anti-Cyclic Citrullinated Peptide and Rheumatoid Factor as Tests for Rheumatoid Arthritis. Autoimmune Dis. 2011, 2011, 815038. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, K.; Sugiyama, D.; Kogata, Y.; Tsuji, G.; Nakazawa, T.; Kawano, S.; Saigo, K.; Morinobu, A.; Koshiba, M.; Kuntz, K.M.; et al. Meta-Analysis: Diagnostic Accuracy of Anti-Cyclic Citrullinated Peptide Antibody and Rheumatoid Factor for Rheumatoid Arthritis. Ann. Intern. Med. 2007, 146, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Bizzaro, N.; Bartoloni, E.; Morozzi, G.; Manganelli, S.; Riccieri, V.; Sabatini, P.; Filippini, M.; Tampoia, M.; Afeltra, A.; Sebastiani, G.; et al. Anti-Cyclic Citrullinated Peptide Antibody Titer Predicts Time to Rheumatoid Arthritis Onset in Patients with Undifferentiated Arthritis: Results from a 2-Year Prospective Study. Arthritis Res. 2013, 15, R16. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; van Venrooij, W.J. Citrullinated Proteins: Sparks That May Ignite the Fire in Rheumatoid Arthritis. Arthritis Res. 2004, 6, 107–111. [Google Scholar] [CrossRef]
- Acharya, N.K.; Nagele, E.P.; Han, M.; Coretti, N.J.; DeMarshall, C.; Kosciuk, M.C.; Boulos, P.A.; Nagele, R.G. Neuronal PAD4 Expression and Protein Citrullination: Possible Role in Production of Autoantibodies Associated with Neurodegenerative Disease. J. Autoimmun. 2012, 38, 369–380. [Google Scholar] [CrossRef]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine Deiminase Inhibition Disrupts NET Formation and Protects against Kidney, Skin and Vascular Disease in Lupus-Prone MRL/Lpr Mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Yang, L.; Tan, D.; Piao, H. Myelin Basic Protein Citrullination in Multiple Sclerosis: A Potential Therapeutic Target for the Pathology. Neurochem. Res. 2016, 41, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Yuzhalin, A.E. Citrullination in Cancer. Cancer Res. 2019, 79, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional Haplotypes of PADI4, Encoding Citrullinating Enzyme Peptidylarginine Deiminase 4, Are Associated with Rheumatoid Arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Kuhn, K.A.; Kulik, L.; Tomooka, B.; Braschler, K.J.; Arend, W.P.; Robinson, W.H.; Holers, V.M. Antibodies against Citrullinated Proteins Enhance Tissue Injury in Experimental Autoimmune Arthritis. J. Clin. Investig. 2006, 116, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, C.; Sebbag, M.; Clavel, C.; Chapuy-Regaud, S.; Al Badine, R.; Méchin, M.-C.; Vincent, C.; Nachat, R.; Yamada, M.; Takahara, H.; et al. Peptidyl Arginine Deiminase Type 2 (PAD-2) and PAD-4 but Not PAD-1, PAD-3, and PAD-6 Are Expressed in Rheumatoid Arthritis Synovium in Close Association with Tissue Inflammation. Arthritis Rheum. 2007, 56, 3541–3553. [Google Scholar] [CrossRef]
- Willis, V.C.; Gizinski, A.M.; Banda, N.K.; Causey, C.P.; Knuckley, B.; Cordova, K.N.; Luo, Y.; Levitt, B.; Glogowska, M.; Chandra, P.; et al. N-α-Benzoyl-N5-(2-Chloro-1-Iminoethyl)-L-Ornithine Amide, a Protein Arginine Deiminase Inhibitor, Reduces the Severity of Murine Collagen-Induced Arthritis. J. Immunol. 2011, 186, 4396–4404. [Google Scholar] [CrossRef]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A.-K.; Rantapää-Dahlqvist, S.; et al. A New Model for an Etiology of Rheumatoid Arthritis: Smoking May Trigger HLA-DR (Shared Epitope)-Restricted Immune Reactions to Autoantigens Modified by Citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.S.; Shetty, R.R.; Mistry, K.J.; Chogle, A.R. Detection of Viral Citrullinated Peptide Antibodies Directed Against EBV or VCP: In Early Rheumatoid Arthritis Patients of Indian Origin. J. Lab. Physicians 2010, 2, 93–99. [Google Scholar] [CrossRef]
- Kraal, L.J.N.; Nijland, M.L.; Germar, K.L.; Baeten, D.L.P.; ten Berge, I.J.M.; Fehres, C.M. Anti-Citrullinated Protein Antibody Response after Primary EBV Infection in Kidney Transplant Patients. PLoS ONE 2018, 13, e0197219. [Google Scholar] [CrossRef]
- Trier, N.H.; Holm, B.E.; Heiden, J.; Slot, O.; Locht, H.; Lindegaard, H.; Svendsen, A.; Nielsen, C.T.; Jacobsen, S.; Theander, E.; et al. Antibodies to a Strain-Specific Citrullinated Epstein-Barr Virus Peptide Diagnoses Rheumatoid Arthritis. Sci. Rep. 2018, 8, 3684. [Google Scholar] [CrossRef] [PubMed]
- Casanova, V.; Sousa, F.H.; Shakamuri, P.; Svoboda, P.; Buch, C.; D’Acremont, M.; Christophorou, M.A.; Pohl, J.; Stevens, C.; Barlow, P.G. Citrullination Alters the Antiviral and Immunomodulatory Activities of the Human Cathelicidin LL-37 During Rhinovirus Infection. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.M.; Knutson, K.L.; Skinner, J.A.; Strausbauch, M.A.; Crowson, C.S.; Therneau, T.M.; Wettstein, P.J.; Matteson, E.L.; Gabriel, S.E. A Profile of Immune Response to Herpesvirus Is Associated with Radiographic Joint Damage in Rheumatoid Arthritis. Arthritis Res. 2012, 14, R24. [Google Scholar] [CrossRef]
- Martens, P.B.; Goronzy, J.J.; Schaid, D.; Weyand, C.M. Expansion of Unusual CD4+ T Cells in Severe Rheumatoid Arthritis. Arthritis Rheum. 1997, 40, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, A.; Ostanek, L.; Brzosko, I.; Brzosko, M.; Masiuk, M.; Machalinski, B.; Gawronska-Szklarz, B. The Expansion of CD4+CD28− T Cells in Patients with Rheumatoid Arthritis. Arthritis Res. 2003, 5, R210–R213. [Google Scholar] [CrossRef]
- Namekawa, T.; Wagner, U.G.; Goronzy, J.J.; Weyand, C.M. Functional Subsets of CD4 T Cells in Rheumatoid Synovitis. Arthritis Rheum. 1998, 41, 2108–2116. [Google Scholar] [CrossRef]
- Hooper, M.; Kallas, E.G.; Coffin, D.; Campbell, D.; Evans, T.G.; Looney, R.J. Cytomegalovirus Seropositivity Is Associated with the Expansion of CD4+CD28− and CD8+CD28− T Cells in Rheumatoid Arthritis. J. Rheumatol. 1999, 26, 1452–1457. [Google Scholar]
- Lamprecht, P.; Gross, W.L. Wegener’s Granulomatosis. Herz 2004, 29, 47–56. [Google Scholar] [CrossRef]
- Hamerman, D.; Gresser, I.; Smith, C. Isolation of Cytomegalovirus from Synovial Cells of a Patient with Rheumatoid Arthritis. J. Rheumatol. 1982, 9, 658–664. [Google Scholar] [PubMed]
- Einsele, H.; Steidle, M.; Müller, C.A.; Fritz, P.; Zacher, J.; Schmidt, H.; Saal, J.G. Demonstration of Cytomegalovirus (CMV) DNA and Anti-CMV Response in the Synovial Membrane and Serum of Patients with Rheumatoid Arthritis. J. Rheumatol. 1992, 19, 677–681. [Google Scholar]
- Murayama, T.; Jisaki, F.; Ayata, M.; Sakamuro, D.; Hironaka, T.; Hirai, K.; Tsuchiya, N.; Ito, K.; Furukawa, T. Cytomegalovirus Genomes Demonstrated by Polymerase Chain Reaction in Synovial Fluid from Rheumatoid Arthritis Patients. Clin. Exp. Rheumatol. 1992, 10, 161–164. [Google Scholar]
- Xu, X.; Estekizadeh, A.; Davoudi, B.; Varani, S.; Malmström, V.; Rahbar, A.; Söderberg-Nauclér, C. Detection of Human Cytomegalovirus in Synovial Neutrophils Obtained from Patients with Rheumatoid Arthritis. Scand. J. Rheumatol. 2020, 1–6. [Google Scholar] [CrossRef]
- Melnick, J.L.; Hu, C.; Burek, J.; Adam, E.; DeBakey, M.E. Cytomegalovirus DNA in Arterial Walls of Patients with Atherosclerosis. J. Med. Virol. 1994, 42, 170–174. [Google Scholar] [CrossRef]
- Xenaki, E.; Hassoulas, J.; Apostolakis, S.; Sourvinos, G.; Spandidos, D.A. Detection of Cytomegalovirus in Atherosclerotic Plaques and Nonatherosclerotic Arteries. Angiology 2009, 60, 504–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Peng, G.; Bai, J.; He, B.; Huang, K.; Hu, X.; Liu, D. Cytomegalovirus Infection and Relative Risk of Cardiovascular Disease (Ischemic Heart Disease, Stroke, and Cardiovascular Death): A Meta-Analysis of Prospective Studies Up to 2016. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Shenk, T.; Alwine, J.C. Human Cytomegalovirus: Coordinating Cellular Stress, Signaling, and Metabolic Pathways. Annu. Rev. Virol. 2014, 1, 355–374. [Google Scholar] [CrossRef]
- Northfield, J.; Lucas, M.; Jones, H.; Young, N.T.; Klenerman, P. Does Memory Improve with Age? CD85j (ILT-2/LIR-1) Expression on CD8 T Cells Correlates with “memory Inflation” in Human Cytomegalovirus Infection. Immunol. Cell Biol. 2005, 83, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Wagner, C.S.; Walther-Jallow, L.; Buentke, E.; Ljunggren, H.-G.; Achour, A.; Chambers, B.J. Human Cytomegalovirus-Derived Protein UL18 Alters the Phenotype and Function of Monocyte-Derived Dendritic Cells. J. Leukoc. Biol. 2008, 83, 56–63. [Google Scholar] [CrossRef]
- Saverino, D.; Fabbi, M.; Ghiotto, F.; Merlo, A.; Bruno, S.; Zarcone, D.; Tenca, C.; Tiso, M.; Santoro, G.; Anastasi, G.; et al. The CD85/LIR-1/ILT2 Inhibitory Receptor Is Expressed by All Human T Lymphocytes and down-Regulates Their Functions. J. Immunol. 2000, 165, 3742–3755. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Coles, A. Multiple Sclerosis. Lancet 2008, 372, 1502–1517. [Google Scholar] [CrossRef]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between Genetic, Lifestyle and Environmental Risk Factors for Multiple Sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef]
- Lucas, R.M.; Hughes, A.M.; Lay, M.-L.J.; Ponsonby, A.-L.; Dwyer, D.E.; Taylor, B.V.; Pender, M.P. Epstein-Barr Virus and Multiple Sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1142–1148. [Google Scholar] [CrossRef]
- Ascherio, A. Environmental Factors in Multiple Sclerosis. Expert Rev. Neurother. 2013, 13, 3–9. [Google Scholar] [CrossRef]
- Alari-Pahissa, E.; Moreira, A.; Zabalza, A.; Alvarez-Lafuente, R.; Munteis, E.; Vera, A.; Arroyo, R.; Alvarez-Cermeño, J.C.; Villar, L.M.; López-Botet, M.; et al. Low Cytomegalovirus Seroprevalence in Early Multiple Sclerosis: A Case for the ‘Hygiene Hypothesis’? Eur. J. Neurol. 2018, 25, 925–933. [Google Scholar] [CrossRef]
- White, D.W.; Suzanne Beard, R.; Barton, E.S. Immune Modulation during Latent Herpesvirus Infection. Immunol. Rev. 2012, 245, 189–208. [Google Scholar] [CrossRef]
- Sanadgol, N.; Ramroodi, N.; Ahmadi, G.A.; Komijani, M.; Moghtaderi, A.; Bouzari, M.; Rezaei, M.; Kardi, M.T.; Dabiri, S.; Moradi, M.; et al. Prevalence of Cytomegalovirus Infection and Its Role in Total Immunoglobulin Pattern in Iranian Patients with Different Subtypes of Multiple Sclerosis. New Microbiol. 2011, 34, 263–274. [Google Scholar] [PubMed]
- Najafi, S.; Ghane, M.; Poortahmasebi, V.; Jazayeri, S.M.; Yousefzadeh-Chabok, S. Prevalence of Cytomegalovirus in Patients With Multiple Sclerosis: A Case-Control Study in Northern Iran. Jundishapur J. Microbiol. 2016, 9, e36582. [Google Scholar] [CrossRef]
- Vanheusden, M.; Stinissen, P.; ’t Hart, B.A.; Hellings, N. Cytomegalovirus: A Culprit or Protector in Multiple Sclerosis? Trends Mol. Med. 2015, 21, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Clerico, M.; De Mercanti, S.; Artusi, C.A.; Durelli, L.; Naismith, R.T. Active CMV Infection in Two Patients with Multiple Sclerosis Treated with Alemtuzumab. Mult. Scler. 2017, 23, 874–876. [Google Scholar] [CrossRef] [PubMed]
- Costa-Garcia, M.; Vera, A.; Moraru, M.; Vilches, C.; López-Botet, M.; Muntasell, A. Antibody-Mediated Response of NKG2Cbright NK Cells against Human Cytomegalovirus. J. Immunol. 2015, 194, 2715–2724. [Google Scholar] [CrossRef] [PubMed]
- López-Montañés, M.; Alari-Pahissa, E.; Sintes, J.; Martínez-Rodríguez, J.E.; Muntasell, A.; López-Botet, M. Antibody-Dependent NK Cell Activation Differentially Targets EBV-Infected Cells in Lytic Cycle and Bystander B Lymphocytes Bound to Viral Antigen-Containing Particles. J. Immunol. 2017, 199, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Lünemann, J.D.; Tintoré, M.; Messmer, B.; Strowig, T.; Rovira, A.; Perkal, H.; Caballero, E.; Münz, C.; Montalban, X.; Comabella, M. Elevated Epstein-Barr Virus-Encoded Nuclear Antigen-1 Immune Responses Predict Conversion to Multiple Sclerosis. Ann. Neurol. 2010, 67, 159–169. [Google Scholar] [CrossRef]
- Maple, P.A.C.; Tanasescu, R.; Gran, B.; Constantinescu, C.S. A Different Response to Cytomegalovirus (CMV) and Epstein-Barr Virus (EBV) Infection in UK People with Multiple Sclerosis (PwMS) Compared to Controls. J. Infect. 2020, 80, 320–325. [Google Scholar] [CrossRef]
- Baroco, A.L.; Oldfield, E.C. Gastrointestinal Cytomegalovirus Disease in the Immunocompromised Patient. Curr. Gastroenterol. Rep. 2008, 10, 409–416. [Google Scholar] [CrossRef]
- Esclatine, A.; Lemullois, M.; Servin, A.L.; Quero, A.M.; Geniteau-Legendre, M. Human Cytomegalovirus Infects Caco-2 Intestinal Epithelial Cells Basolaterally Regardless of the Differentiation State. J. Virol. 2000, 74, 513–517. [Google Scholar] [CrossRef]
- Jarvis, M.A.; Wang, C.E.; Meyers, H.L.; Smith, P.P.; Corless, C.L.; Henderson, G.J.; Vieira, J.; Britt, W.J.; Nelson, J.A. Human Cytomegalovirus Infection of Caco-2 Cells Occurs at the Basolateral Membrane and Is Differentiation State Dependent. J. Virol. 1999, 73, 4552–4560. [Google Scholar] [CrossRef]
- Nagai, S.; Mangus, R.S.; Anderson, E.; Ekser, B.; Kubal, C.A.; Fridell, J.A.; Tector, A.J. Cytomegalovirus Infection After Intestinal/Multivisceral Transplantation: A Single-Center Experience With 210 Cases. Transplantation 2016, 100, 451–460. [Google Scholar] [CrossRef]
- Chiereghin, A.; Gabrielli, L.; Zanfi, C.; Petrisli, E.; Lauro, A.; Piccirilli, G.; Baccolini, F.; Dazzi, A.; Cescon, M.; Morelli, M.C.; et al. Monitoring Cytomegalovirus T-Cell Immunity in Small Bowel/Multivisceral Transplant Recipients. Transplant. Proc. 2010, 42, 69–73. [Google Scholar] [CrossRef]
- Varani, S.; Landini, M.P. Cytomegalovirus-Induced Immunopathology and Its Clinical Consequences. Herpesviridae 2011, 2, 6. [Google Scholar] [CrossRef]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Jentzer, A.; Veyrard, P.; Roblin, X.; Saint-Sardos, P.; Rochereau, N.; Paul, S.; Bourlet, T.; Pozzetto, B.; Pillet, S. Cytomegalovirus and Inflammatory Bowel Diseases (IBD) with a Special Focus on the Link with Ulcerative Colitis (UC). Microorganisms 2020, 8, 1078. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767. [Google Scholar] [CrossRef]
- Powell, R.D.; Warner, N.E.; Levine, R.S.; Kirsner, J.B. Cytomegalic Inclusion Disease and Ulcerative Colitis: Report of a Case in a Young Adult. Am. J. Med. 1961, 30, 334–340. [Google Scholar] [CrossRef]
- Hendler, S.A.; Barber, G.E.; Okafor, P.N.; Chang, M.S.; Limsui, D.; Limketkai, B.N. Cytomegalovirus Infection Is Associated with Worse Outcomes in Inflammatory Bowel Disease Hospitalizations Nationwide. Int. J. Colorectal. Dis. 2020, 35, 897–903. [Google Scholar] [CrossRef]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J.H. Monocytes Are a Major Site of Persistence of Human Cytomegalovirus in Peripheral Blood Mononuclear Cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef] [PubMed]
- Söderberg-Nauclér, C.; Fish, K.N.; Nelson, J.A. Reactivation of Latent Human Cytomegalovirus by Allogeneic Stimulation of Blood Cells from Healthy Donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Reeves, M.B.; Lehner, P.J.; Sissons, J.G.P.; Sinclair, J.H. An in Vitro Model for the Regulation of Human Cytomegalovirus Latency and Reactivation in Dendritic Cells by Chromatin Remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef] [PubMed]
- Hommes, D.W.; Sterringa, G.; van Deventer, S.J.H.; Tytgat, G.N.J.; Weel, J. The Pathogenicity of Cytomegalovirus in Inflammatory Bowel Disease: A Systematic Review and Evidence-Based Recommendations for Future Research. Inflamm. Bowel. Dis. 2004, 10, 245–250. [Google Scholar] [CrossRef]
- Julka, K.; Surawicz, C.M. Cytomegalovirus in Inflammatory Bowel Disease: Time for Another Look? Gastroenterology 2009, 137, 1163–1166. [Google Scholar] [CrossRef] [PubMed]
- Kuwabara, A.; Okamoto, H.; Suda, T.; Ajioka, Y.; Hatakeyama, K. Clinicopathologic Characteristics of Clinically Relevant Cytomegalovirus Infection in Inflammatory Bowel Disease. J. Gastroenterol. 2007, 42, 823–829. [Google Scholar] [CrossRef]
- Lawlor, G.; Moss, A.C. Cytomegalovirus in Inflammatory Bowel Disease: Pathogen or Innocent Bystander? Inflamm. Bowel. Dis. 2010, 16, 1620–1627. [Google Scholar] [CrossRef]
- Pillet, S.; Pozzetto, B.; Roblin, X. Cytomegalovirus and Ulcerative Colitis: Place of Antiviral Therapy. World J. Gastroenterol. 2016, 22, 2030–2045. [Google Scholar] [CrossRef] [PubMed]
- Mourad, F.H.; Hashash, J.G.; Kariyawasam, V.C.; Leong, R.W. Ulcerative Colitis and Cytomegalovirus Infection: From A to Z. J. Crohns Colitis 2020, 14, 1162–1171. [Google Scholar] [CrossRef]
- Matsuoka, K.; Iwao, Y.; Mori, T.; Sakuraba, A.; Yajima, T.; Hisamatsu, T.; Okamoto, S.; Morohoshi, Y.; Izumiya, M.; Ichikawa, H.; et al. Cytomegalovirus Is Frequently Reactivated and Disappears without Antiviral Agents in Ulcerative Colitis Patients. Am. J. Gastroenterol. 2007, 102, 331–337. [Google Scholar] [CrossRef]
- Domènech, E.; Vega, R.; Ojanguren, I.; Hernández, A.; Garcia-Planella, E.; Bernal, I.; Rosinach, M.; Boix, J.; Cabré, E.; Gassull, M.A. Cytomegalovirus Infection in Ulcerative Colitis: A Prospective, Comparative Study on Prevalence and Diagnostic Strategy. Inflamm. Bowel Dis. 2008, 14, 1373–1379. [Google Scholar] [CrossRef]
- Lv, Y.-L.; Han, F.-F.; Jia, Y.-J.; Wan, Z.-R.; Gong, L.-L.; Liu, H.; Liu, L.-H. Is Cytomegalovirus Infection Related to Inflammatory Bowel Disease, Especially Steroid-Resistant Inflammatory Bowel Disease? A Meta-Analysis. Infect. Drug Resist. 2017, 10, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Hissong, E.; Chen, Z.; Yantiss, R.K. Cytomegalovirus Reactivation in Inflammatory Bowel Disease: An Uncommon Occurrence Related to Corticosteroid Dependence. Mod. Pathol. 2019, 32, 1210–1216. [Google Scholar] [CrossRef] [PubMed]
- Criscuoli, V.; Rizzuto, M.-R.; Cottone, M. Cytomegalovirus and Inflammatory Bowel Disease: Is There a Link? World J. Gastroenterol. 2006, 12, 4813–4818. [Google Scholar] [CrossRef] [PubMed]
- Rahbar, A.; Boström, L.; Lagerstedt, U.; Magnusson, I.; Söderberg-Naucler, C.; Sundqvist, V.-A. Evidence of Active Cytomegalovirus Infection and Increased Production of IL-6 in Tissue Specimens Obtained from Patients with Inflammatory Bowel Diseases. Inflamm. Bowel Dis. 2003, 9, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Simpson, N.; Klipfel, N.; Debose, R.; Barr, N.; Laine, L. Cytomegalovirus Infection in Patients with Active Inflammatory Bowel Disease. Dig. Dis. Sci. 2010, 55, 1059–1065. [Google Scholar] [CrossRef]
- Dimitroulia, E.; Spanakis, N.; Konstantinidou, A.E.; Legakis, N.J.; Tsakris, A. Frequent Detection of Cytomegalovirus in the Intestine of Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2006, 12, 879–884. [Google Scholar] [CrossRef]
- Roblin, X.; Pillet, S.; Berthelot, P.; Del Tedesco, E.; Phelip, J.-M.; Chambonnière, M.-L.; Peyrin-Biroulet, L.; Pozzetto, B. Prevalence of Cytomegalovirus Infection in Steroid-Refractory Crohn’s Disease. Inflamm. Bowel Dis. 2012, 18, E1396–E1397. [Google Scholar] [CrossRef]
- Römkens, T.E.H.; Bulte, G.J.; Nissen, L.H.C.; Drenth, J.P.H. Cytomegalovirus in Inflammatory Bowel Disease: A Systematic Review. World J. Gastroenterol. 2016, 22, 1321–1330. [Google Scholar] [CrossRef]
- Knösel, T.; Schewe, C.; Petersen, N.; Dietel, M.; Petersen, I. Prevalence of Infectious Pathogens in Crohn’s Disease. Pathol. Res. Pract. 2009, 205, 223–230. [Google Scholar] [CrossRef]
- Takahashi, Y.; Tange, T. Prevalence of Cytomegalovirus Infection in Inflammatory Bowel Disease Patients. Dis. Colon Rectum. 2004, 47, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Papadakis, K.A.; Tung, J.K.; Binder, S.W.; Kam, L.Y.; Abreu, M.T.; Targan, S.R.; Vasiliauskas, E.A. Outcome of Cytomegalovirus Infections in Patients with Inflammatory Bowel Disease. Am. J. Gastroenterol. 2001, 96, 2137–2142. [Google Scholar] [CrossRef] [PubMed]
- Kojima, T.; Watanabe, T.; Hata, K.; Shinozaki, M.; Yokoyama, T.; Nagawa, H. Cytomegalovirus Infection in Ulcerative Colitis. Scand. J. Gastroenterol. 2006, 41, 706–711. [Google Scholar] [CrossRef]
- Yoshino, T.; Nakase, H.; Ueno, S.; Uza, N.; Inoue, S.; Mikami, S.; Matsuura, M.; Ohmori, K.; Sakurai, T.; Nagayama, S.; et al. Usefulness of Quantitative Real-Time PCR Assay for Early Detection of Cytomegalovirus Infection in Patients with Ulcerative Colitis Refractory to Immunosuppressive Therapies. Inflamm. Bowel Dis. 2007, 13, 1516–1521. [Google Scholar] [CrossRef]
- Wada, Y.; Matsui, T.; Matake, H.; Sakurai, T.; Yamamoto, J.; Kikuchi, Y.; Yorioka, M.; Tsuda, S.; Yao, T.; Yao, S.; et al. Intractable Ulcerative Colitis Caused by Cytomegalovirus Infection: A Prospective Study on Prevalence, Diagnosis, and Treatment. Dis. Colon. Rectum. 2003, 46, S59-65. [Google Scholar] [CrossRef]
- Minami, M.; Ohta, M.; Ohkura, T.; Ando, T.; Ohmiya, N.; Niwa, Y.; Goto, H. Cytomegalovirus Infection in Severe Ulcerative Colitis Patients Undergoing Continuous Intravenous Cyclosporine Treatment in Japan. World J. Gastroenterol. 2007, 13, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Kambham, N.; Vij, R.; Cartwright, C.A.; Longacre, T. Cytomegalovirus Infection in Steroid-Refractory Ulcerative Colitis: A Case-Control Study. Am. J. Surg. Pathol. 2004, 28, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Cottone, M.; Pietrosi, G.; Martorana, G.; Casà, A.; Pecoraro, G.; Oliva, L.; Orlando, A.; Rosselli, M.; Rizzo, A.; Pagliaro, L. Prevalence of Cytomegalovirus Infection in Severe Refractory Ulcerative and Crohn’s Colitis. Am. J. Gastroenterol. 2001, 96, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Barahona-Garrido, J.; Martínez-Benítez, B.; Espinosa-Cárdenas, E.; Sarti, H.M.; Gutiérrez-Manjarrez, J.I.; Aguirre-Gutiérrez, R.; Tellez-Avila, F.I.; Coss-Adame, E.; García-Juárez, I.; Yamamoto-Furusho, J.K. Cytomegalovirus Infection in Patients Who Required Colectomy for Toxic Megacolon or Severe Steroid-Refractory Ulcerative Colitis. Dig. Dis. Sci. 2010, 55, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Maher, M.M.; Nassar, M.I. Acute Cytomegalovirus Infection Is a Risk Factor in Refractory and Complicated Inflammatory Bowel Disease. Dig. Dis. Sci. 2009, 54, 2456–2462. [Google Scholar] [CrossRef]
- Dignass, A.; Lindsay, J.O.; Sturm, A.; Windsor, A.; Colombel, J.-F.; Allez, M.; D’Haens, G.; D’Hoore, A.; Mantzaris, G.; Novacek, G.; et al. Second European Evidence-Based Consensus on the Diagnosis and Management of Ulcerative Colitis Part 2: Current Management. J. Crohns. Colitis 2012, 6, 991–1030. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.T.; Portela, F.A.; Tomé, L. Cytomegalovirus, Inflammatory Bowel Disease, and Anti-TNFα. Int. J. Colorectal. Dis. 2017, 32, 645–650. [Google Scholar] [CrossRef]
- Stein, J.; Volk, H.D.; Liebenthal, C.; Krüger, D.H.; Prösch, S. Tumour Necrosis Factor Alpha Stimulates the Activity of the Human Cytomegalovirus Major Immediate Early Enhancer/Promoter in Immature Monocytic Cells. J. Gen. Virol. 1993, 74, 2333–2338. [Google Scholar] [CrossRef]
- Nakase, H.; Honzawa, Y.; Toyonaga, T.; Yamada, S.; Minami, N.; Yoshino, T.; Matsuura, M. Diagnosis and Treatment of Ulcerative Colitis with Cytomegalovirus Infection: Importance of Controlling Mucosal Inflammation to Prevent Cytomegalovirus Reactivation. Intest. Res. 2014, 12, 5–11. [Google Scholar] [CrossRef][Green Version]
- Matsumura, K.; Nakase, H.; Kosugi, I.; Honzawa, Y.; Yoshino, T.; Matsuura, M.; Kawasaki, H.; Arai, Y.; Iwashita, T.; Nagasawa, T.; et al. Establishment of a Novel Mouse Model of Ulcerative Colitis with Concomitant Cytomegalovirus Infection: In Vivo Identification of Cytomegalovirus Persistent Infected Cells. Inflamm. Bowel Dis. 2013, 19, 1951–1963. [Google Scholar] [CrossRef]
- Chang, W.L.W.; Barry, P.A.; Szubin, R.; Wang, D.; Baumgarth, N. Human Cytomegalovirus Suppresses Type I Interferon Secretion by Plasmacytoid Dendritic Cells through Its Interleukin 10 Homolog. Virology 2009, 390, 330–337. [Google Scholar] [CrossRef]
- Park, S.C.; Jeen, Y.M.; Jeen, Y.T. Approach to Cytomegalovirus Infections in Patients with Ulcerative Colitis. Korean J. Intern. Med. 2017, 32, 383–392. [Google Scholar] [CrossRef]
- Fakhreddine, A.Y.; Frenette, C.T.; Konijeti, G.G. A Practical Review of Cytomegalovirus in Gastroenterology and Hepatology. Gastroenterol. Res. Pract. 2019, 2019, 6156581. [Google Scholar] [CrossRef]
- Yerushalmy-Feler, A.; Padlipsky, J.; Cohen, S. Diagnosis and Management of CMV Colitis. Curr. Infect. Dis. Rep. 2019, 21, 5. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, Y.; Yamakawa, T.; Hirano, T.; Kazama, T.; Hirayama, D.; Wagatsuma, K.; Nakase, H. Current Diagnostic and Therapeutic Approaches to Cytomegalovirus Infections in Ulcerative Colitis Patients Based on Clinical and Basic Research Data. Int. J. Mol. Sci. 2020, 21, 2438. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Racca, F.; Paolucci, S.; Campanini, G.; Pozzi, L.; Betti, E.; Riboni, R.; Vanoli, A.; Baldanti, F.; Corazza, G.R. Human Cytomegalovirus and Epstein-Barr Virus Infection in Inflammatory Bowel Disease: Need for Mucosal Viral Load Measurement. World J. Gastroenterol. 2015, 21, 1915–1926. [Google Scholar] [CrossRef] [PubMed]
- Ciccocioppo, R.; Racca, F.; Scudeller, L.; Piralla, A.; Formagnana, P.; Pozzi, L.; Betti, E.; Vanoli, A.; Riboni, R.; Kruzliak, P.; et al. Differential Cellular Localization of Epstein-Barr Virus and Human Cytomegalovirus in the Colonic Mucosa of Patients with Active or Quiescent Inflammatory Bowel Disease. Immunol. Res. 2016, 64, 191–203. [Google Scholar] [CrossRef]
- Jones, A.; McCurdy, J.D.; Loftus, E.V.; Bruining, D.H.; Enders, F.T.; Killian, J.M.; Smyrk, T.C. Effects of Antiviral Therapy for Patients with Inflammatory Bowel Disease and a Positive Intestinal Biopsy for Cytomegalovirus. Clin. Gastroenterol. Hepatol. 2015, 13, 949–955. [Google Scholar] [CrossRef]
- Beswick, L.; Ye, B.; van Langenberg, D.R. Toward an Algorithm for the Diagnosis and Management of CMV in Patients with Colitis. Inflamm. Bowel Dis. 2016, 22, 2966–2976. [Google Scholar] [CrossRef]
- Shukla, T.; Singh, S.; Loftus, E.V.; Bruining, D.H.; McCurdy, J.D. Antiviral Therapy in Steroid-Refractory Ulcerative Colitis with Cytomegalovirus: Systematic Review and Meta-Analysis. Inflamm. Bowel Dis. 2015, 21, 2718–2725. [Google Scholar] [CrossRef]
- Roblin, X.; Pillet, S.; Oussalah, A.; Berthelot, P.; Del Tedesco, E.; Phelip, J.-M.; Chambonnière, M.-L.; Garraud, O.; Peyrin-Biroulet, L.; Pozzetto, B. Cytomegalovirus Load in Inflamed Intestinal Tissue Is Predictive of Resistance to Immunosuppressive Therapy in Ulcerative Colitis. Am. J. Gastroenterol. 2011, 106, 2001–2008. [Google Scholar] [CrossRef]
- Lévêque, N.; Brixi-Benmansour, H.; Reig, T.; Renois, F.; Talmud, D.; Brodard, V.; Coste, J.-F.; De Champs, C.; Andréoletti, L.; Diebold, M.-D. Low Frequency of Cytomegalovirus Infection during Exacerbations of Inflammatory Bowel Diseases. J. Med. Virol. 2010, 82, 1694–1700. [Google Scholar] [CrossRef]
- Hommel, C.; Pillet, S.; Rahier, J.-F. Comment on: “Resolution of CMV Infection in the Bowel on Vedolizumab Therapy”. J. Crohns Colitis 2020, 14, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Kopylov, U.; Papamichael, K.; Katsanos, K.; Waterman, M.; Bar-Gil Shitrit, A.; Boysen, T.; Portela, F.; Peixoto, A.; Szilagyi, A.; Silva, M.; et al. Impact of Infliximab and Cyclosporine on the Risk of Colectomy in Hospitalized Patients with Ulcerative Colitis Complicated by Cytomegalovirus-A Multicenter Retrospective Study. Inflamm. Bowel Dis. 2017, 23, 1605–1613. [Google Scholar] [CrossRef] [PubMed]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Henmi, Y.; Kakimoto, K.; Inoue, T.; Nakazawa, K.; Kubota, M.; Hara, A.; Mikami, T.; Naka, Y.; Hirata, Y.; Hirata, Y.; et al. Cytomegalovirus Infection in Ulcerative Colitis Assessed by Quantitative Polymerase Chain Reaction: Risk Factors and Effects of Immunosuppressants. J. Clin. Biochem. Nutr. 2018, 63, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, C.L.; Luo, Y.X.; Isaacs, S.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. The Virome in Early Life and Childhood and Development of Islet Autoimmunity and Type 1 Diabetes: A Systematic Review and Meta-Analysis of Observational Studies. Rev. Med. Virol. 2020, e2209. [Google Scholar] [CrossRef]
- Mine, K.; Yoshikai, Y.; Takahashi, H.; Mori, H.; Anzai, K.; Nagafuchi, S. Genetic Susceptibility of the Host in Virus-Induced Diabetes. Microorganisms 2020, 8, 1133. [Google Scholar] [CrossRef]
- Aarnisalo, J.; Veijola, R.; Vainionpää, R.; Simell, O.; Knip, M.; Ilonen, J. Cytomegalovirus Infection in Early Infancy: Risk of Induction and Progression of Autoimmunity Associated with Type 1 Diabetes. Diabetologia 2008, 51, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Hiltunen, M.; Hyöty, H.; Karjalainen, J.; Leinikki, P.; Knip, M.; Lounamaa, R.; Akerblom, H.K. Serological Evaluation of the Role of Cytomegalovirus in the Pathogenesis of IDDM: A Prospective Study. The Childhood Diabetes in Finland Study Group. Diabetologia 1995, 38, 705–710. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ivarsson, S.A.; Lindberg, B.; Nilsson, K.O.; Ahlfors, K.; Svanberg, L. The Prevalence of Type 1 Diabetes Mellitus at Follow-up of Swedish Infants Congenitally Infected with Cytomegalovirus. Diabet Med. 1993, 10, 521–523. [Google Scholar] [CrossRef]
- Ward, K.P.; Galloway, W.H.; Auchterlonie, I.A. Congenital Cytomegalovirus Infection and Diabetes. Lancet 1979, 1, 497. [Google Scholar] [CrossRef]
- Hyöoty, H.; Räasäanen, L.; Hiltunen, M.; Lehtinen, M.; Huupponen, T.; Leinikki, P. Decreased Antibody Reactivity to Epstein-Barr Virus Capsid Antigen in Type 1 (Insulin-Dependent) Diabetes Mellitus. Apmis 1991, 99, 359–363. [Google Scholar] [CrossRef]
- Saber, A.Z.A.B.I.; Mohammed, A.H. The Roles of Human Cytomegalovirus and Epstein-Barr Virus in Type 1 Diabetes Mellitus. Ann. Trop. Med. Health 2019, 22, 90–99. [Google Scholar] [CrossRef]
- Hjelmesæth, J.; Müller, F.; Jenssen, T.; Rollag, H.; Sagedal, S.; Hartmann, A. Is There a Link between Cytomegalovirus Infection and New-Onset Posttransplantation Diabetes Mellitus? Potential Mechanisms of Virus Induced β-Cell Damage. Nephrol. Dial. Transplant. 2005, 20, 2311–2315. [Google Scholar] [CrossRef]
- Adland, E.; Klenerman, P.; Goulder, P.; Matthews, P.C. Ongoing Burden of Disease and Mortality from HIV/CMV Coinfection in Africa in the Antiretroviral Therapy Era. Front. Microbiol. 2015, 6, 1016. [Google Scholar] [CrossRef]
- Clement, M.; Humphreys, I.R. Cytokine-Mediated Induction and Regulation of Tissue Damage During Cytomegalovirus Infection. Front. Immunol. 2019, 10, 78. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Autoimmune Diseases | References * |
|---|---|
| Rheumatologic diseases | |
| Systemic Lupus Erythematosus | [86,87,89,118,119,120,121,122,123,124,126,128,129,130,131,132,133,134] |
| Systemic sclerosis | [33,83,111,138,139,140,141,143,144,145,146,147,149] |
| Rheumatoid arthritis | [92,95,100,144,151,152,153,154,155,156,179,183,185] |
| Neurological diseases | |
| Multiple sclerosis | [98,200,201,202,203,204,205,209] |
| Enteropathies | |
| Crohn disease & ulcerative colitis | [217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,263,264,265,266,267,268,269,270,271] |
| Metabolic diseases | |
| Type 1 diabetes | [88,274,275,276,277,278,279,280] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gugliesi, F.; Pasquero, S.; Griffante, G.; Scutera, S.; Albano, C.; Pacheco, S.F.C.; Riva, G.; Dell’Oste, V.; Biolatti, M. Human Cytomegalovirus and Autoimmune Diseases: Where Are We? Viruses 2021, 13, 260. https://doi.org/10.3390/v13020260
Gugliesi F, Pasquero S, Griffante G, Scutera S, Albano C, Pacheco SFC, Riva G, Dell’Oste V, Biolatti M. Human Cytomegalovirus and Autoimmune Diseases: Where Are We? Viruses. 2021; 13(2):260. https://doi.org/10.3390/v13020260
Chicago/Turabian StyleGugliesi, Francesca, Selina Pasquero, Gloria Griffante, Sara Scutera, Camilla Albano, Sergio Fernando Castillo Pacheco, Giuseppe Riva, Valentina Dell’Oste, and Matteo Biolatti. 2021. "Human Cytomegalovirus and Autoimmune Diseases: Where Are We?" Viruses 13, no. 2: 260. https://doi.org/10.3390/v13020260
APA StyleGugliesi, F., Pasquero, S., Griffante, G., Scutera, S., Albano, C., Pacheco, S. F. C., Riva, G., Dell’Oste, V., & Biolatti, M. (2021). Human Cytomegalovirus and Autoimmune Diseases: Where Are We? Viruses, 13(2), 260. https://doi.org/10.3390/v13020260