
Imaging Viral Infection by Fluorescence Microscopy: Focus on HIV-1 Early Stage



Abstract
1. Introduction
2. Fluorescent Labeling of HIV-1
3. Viral Entry
3.1. Fusion with the Plasma Membrane
3.2. Endocytosis Mediated Entry
3.3. Factors Enhancing the Viral Entry
4. Journey toward the Nucleus
5. Cytoplasmic Remodeling of the Viral Core
5.1. Cytoplasmic Release of Viral Proteins
5.2. Reverse Transcription and Proviral DNA Imaging
6. Uncoating
6.1. Capsid Opening
6.2. Uncoating and Cytoskeleton
6.3. Uncoating at the Nuclear Pore
6.4. Capsid in the Nucleus
6.5. Uncoating in the Nucleus
7. Nuclear Entry
8. Inside the Nucleus
8.1. Intranuclear Trafficking towards the Site of Integration
8.2. Factors Determining the Choice of the Integration Site
9. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Chen, B. Molecular Mechanism of HIV-1 Entry. Trends Microbiol. 2019, 27, 878–891. [Google Scholar] [CrossRef]
- Hulme, A.E.; Hope, T.J. Live Cell Imaging of Retroviral Entry. Annu. Rev. Virol. 2014, 1, 501–515. [Google Scholar] [CrossRef]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef]
- Dharan, A.; Talley, S.; Tripathi, A.; Mamede, J.I.; Majetschak, M.; Hope, T.; Campbell, E.M. KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 2016, 12, e1005700. [Google Scholar] [CrossRef]
- Malikov, V.; Da, D.S.A.V.; Jovasevic, V.; Bennett, G.; Vieira, D.A.D.S.A.; Schulte, B.; Diaz-Griffero, F.; Walsh, D.; Naghavi, M.H. HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef]
- Lyonnais, S.; Gorelick, R.J.; Heniche-Boukhalfa, F.; Bouaziz, S.; Parissi, V.; Mouscadet, J.-F.; Restle, T.; Gatell, J.M.; Le Cam, E.; Mirambeau, G. A protein ballet around the viral genome orchestrated by HIV-1 reverse transcriptase leads to an architectural switch: From nucleocapsid-condensed RNA to Vpr-bridged DNA. Virus Res. 2013, 171, 287–303. [Google Scholar] [CrossRef]
- Blanco-Rodriguez, G.; Gazi, A.; Monel, B.; Frabetti, S.; Scoca, V.; Mueller, F.; Schwartz, O.; Krijnse-Locker, J.; Charneau, P.; Di Nunzio, F. Remodeling of the Core Leads HIV-1 Preintegration Complex into the Nucleus of Human Lymphocytes. J. Virol. 2020, 94, 00135-20. [Google Scholar] [CrossRef]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Genet. 2015, 13, 471–483. [Google Scholar] [CrossRef]
- Arhel, N.J. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, 96. [Google Scholar] [CrossRef]
- Arhel, N.; Genovesio, A.; Kim, K.-A.; Miko, S.; Perret, E.; Olivo-Marin, J.-C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar] [CrossRef]
- Delaney, M.K.; Malikov, V.; Chai, Q.; Zhao, G.; Naghavi, M.H. Distinct functions of diaphanous-related formins regulate HIV-1 uncoating and transport. Proc. Natl. Acad. Sci. USA 2017, 114, E6932–E6941. [Google Scholar] [CrossRef]
- Burdick, R.C.; Delviks-Frankenberry, K.; Chen, J.; Janaka, S.K.; Sastri, J.; Hu, W.-S.; Pathak, V.K. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017, 13, e1006570. [Google Scholar] [CrossRef]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef]
- Melikyan, G.B. HIV entry: A game of hide-and-fuse? Curr. Opin. Virol. 2014, 4, 1–7. [Google Scholar] [CrossRef]
- Mamede, J.I.; Cianci, G.C.; Anderson, M.R.; Hope, T. Early cytoplasmic uncoating is associated with infectivity of HIV-1. Proc. Natl. Acad. Sci. USA 2017, 114, E7169–E7178. [Google Scholar] [CrossRef]
- Xu, H.; Franks, T.; Gibson, G.A.; Huber, K.; Rahm, N.; Strambio-De-Castillia, C.; Luban, J.; Aiken, C.; Watkins, S.C.; Sluis-Cremer, N.; et al. Evidence for biphasic uncoating during HIV-1 infection from a novel imaging assay. Retrovirology 2013, 10, 70. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Shi, J.; Aiken, C.; Melikyan, G.B. Time-Resolved Imaging of Single HIV-1 Uncoating In Vitro and in Living Cells. PLoS Pathog. 2016, 12, e1005709. [Google Scholar] [CrossRef]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.-S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Zila, V.; Mueller, T.G.; Laketa, V.; Mueller, B.; Kräusslich, H.-G. Analysis of CA Content and CPSF6 Dependence of Early HIV-1 Replication Complexes in SupT1-R5 Cells. mBio 2019, 10. [Google Scholar] [CrossRef]
- Desai, T.M.; Marin, M.; Sood, C.; Shi, J.; Nawaz, F.; Aiken, C.; Melikyan, G.B. Fluorescent protein-tagged Vpr dissociates from HIV-1 core after viral fusion and rapidly enters the cell nucleus. Retrovirology 2015, 12, 88. [Google Scholar] [CrossRef]
- Borrenberghs, D.; Dirix, L.; De Wit, F.; Rocha, S.; Blokken, J.; De Houwer, S.; Gijsbers, R.; Christ, F.; Hofkens, J.; Hendrix, J.; et al. Dynamic Oligomerization of Integrase Orchestrates HIV Nuclear Entry. Sci. Rep. 2016, 6, 36485. [Google Scholar] [CrossRef]
- Zgheib, S.; Lysova, I.; Réal, E.; Dukhno, O.; Vauchelles, R.; Pires, M.; Anton, H.; Mély, Y. Quantitative monitoring of the cytoplasmic release of NCp7 proteins from individual HIV-1 viral cores during the early steps of infection. Sci. Rep. 2019, 9, 945. [Google Scholar] [CrossRef]
- Ferns, R.B.; Tedder, R.S.; Weiss, R.A. Characterization of Monoclonal Antibodies Against the Human Immunodeficiency Virus (HIV) gag Products and Their Use in Monitoring HIV Isolate Variation. J. Gen. Virol. 1987, 68, 1543–1551. [Google Scholar] [CrossRef]
- Niedrig, M.; Rabanus, J.-P.; Stehr, J.L.; Gelderblom, H.R.; Pauli, G. Monoclonal Antibodies Directed against Human Immunodeficiency Virus (HIV) gag Proteins with Specificity for Conserved Epitopes in HIV-1, HIV-2 and Simian Immunodeficiency Virus. J. Gen. Virol. 1988, 69, 2109–2114. [Google Scholar] [CrossRef]
- Apostolski, S.; McAlarney, T.; Quattrini, A.; Levison, S.W.; Rosoklija, G.; Lugaressi, A.; Corbo, M.; Sadiq, S.A.; Lederman, S.; Hays, A.P.; et al. The gp 120 glycoprotein of human immunodeficiency virus type 1 binds to sensory ganglion neurons. Ann. Neurol. 1993, 34, 855–863. [Google Scholar] [CrossRef]
- Stauber, R.H.; Rulong, S.; Palm, G.; Tarasova, N.I. Direct Visualization of HIV-1 Entry: Mechanisms and Role of Cell Surface Receptors. Biochem. Biophys. Res. Commun. 1999, 258, 695–702. [Google Scholar] [CrossRef]
- Ma, Y.; He, Z.; Tan, T.; Li, W.; Zhang, Z.; Song, S.; Zhang, X.; Hu, Q.; Zhou, P.; Wu, Y.; et al. Real-Time Imaging of Single HIV-1 Disassembly with Multicolor Viral Particles. ACS Nano 2016, 10, 6273–6282. [Google Scholar] [CrossRef]
- Bönisch, I.Z.; Dirix, L.; Lemmens, V.; Borrenberghs, D.; De Wit, F.; Vernaillen, F.; Rocha, S.; Christ, F.; Hendrix, J.; Hofkens, J.; et al. Capsid-Labelled HIV to Investigate the Role of Capsid during Nuclear Import and Integration. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Hulme, A.E.; Kelley, Z.L.; Foley, D.; Hope, T.J. Complementary Assays Reveal a Low Level of CA Associated with Viral Complexes in the Nuclei of HIV-1-Infected Cells. J. Virol. 2015, 89, 5350–5361. [Google Scholar] [CrossRef]
- Albanese, A.; Arosio, D.; Terreni, M.; Cereseto, A. HIV-1 Pre-Integration Complexes Selectively Target Decondensed Chromatin in the Nuclear Periphery. PLoS ONE 2008, 3, e2413. [Google Scholar] [CrossRef]
- Francis, A.C.; Di Primio, C.; Quercioli, V.; Valentini, P.; Boll, A.; Girelli, G.; Demichelis, F.; Arosio, D.; Cereseto, A. Second Generation Imaging of Nuclear/Cytoplasmic HIV-1 Complexes. AIDS Res. Hum. Retrovir. 2014, 30, 717–726. [Google Scholar] [CrossRef]
- Sood, C.; Francis, A.C.; Desai, T.M.; Melikyan, G.B. An improved labeling strategy enables automated detection of single-virus fusion and assessment of HIV-1 protease activity in single virions. J. Biol. Chem. 2017, 292, 20196–20207. [Google Scholar] [CrossRef]
- Hubner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of Human Immunodeficiency Virus Type 1 (HIV-1) Gag Localization and Oligomerization Monitored with Live Confocal Imaging of a Replication-Competent, Fluorescently Tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef]
- Padilla-Parra, S.; Marin, M.; Gahlaut, N.; Suter, R.; Kondo, N.; Melikyan, G.B. Fusion of Mature HIV-1 Particles Leads to Complete Release of a Gag-GFP-Based Content Marker and Raises the Intraviral pH. PLoS ONE 2013, 8, e71002. [Google Scholar] [CrossRef]
- Pereira, C.F.; Ellenberg, P.C.; Jones, K.L.; Fernandez, T.L.; Smyth, R.P.; Hawkes, D.J.; Hijnen, M.; Vivet-Boudou, V.; Marquet, R.; Johnson, I.; et al. Labeling of Multiple HIV-1 Proteins with the Biarsenical-Tetracysteine System. PLoS ONE 2011, 6, e17016. [Google Scholar] [CrossRef]
- Lysova, I.; Spiegelhalter, C.; Réal, E.; Zgheib, S.; Anton, H.; Mély, Y. ReAsH/tetracystein-based correlative light-electron microscopy for HIV-1 imaging during the early stages of infection. Methods Appl. Fluoresc. 2018, 6, 045001. [Google Scholar] [CrossRef]
- Burdick, R.C.; Hu, W.-S.; Pathak, V.K. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc. Natl. Acad. Sci. USA 2013, 110, E4780–E4789. [Google Scholar] [CrossRef]
- Chin, C.R.; Perreira, J.M.; Savidis, G.; Portmann, J.M.; Aker, A.M.; Feeley, E.M.; Smith, M.C.; Brass, A.L. Direct Visualization of HIV-1 Replication Intermediates Shows that Capsid and CPSF6 Modulate HIV-1 Intra-nuclear Invasion and Integration. Cell Rep. 2015, 13, 1717–1731. [Google Scholar] [CrossRef]
- Puray-Chavez, M.; Tedbury, P.R.; Huber, A.D.; Ukah, O.B.; Yapo, V.; Liu, D.; Ji, J.; Wolf, J.J.; Engelman, A.N.; Sarafianos, S.G. Multiplex single-cell visualization of nucleic acids and protein during HIV infection. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Stultz, R.D.; Cenker, J.J.; McDonald, D. Imaging HIV-1 Genomic DNA from Entry through Productive Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Müller, B.; Kräusslich, H.-G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. eLife 2014, 3, e04114. [Google Scholar] [CrossRef]
- Oum, Y.H.; Desai, T.M.; Marin, M.; Melikyan, G.B. Click labeling of unnatural sugars metabolically incorporated into viral envelope glycoproteins enables visualization of single particle fusion. J. Virol. Methods 2016, 233, 62–71. [Google Scholar] [CrossRef]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef]
- Li, Q.; Li, W.; Yin, W.; Guo, J.; Zhang, Z.-P.; Zeng, D.; Zhang, X.; Wu, Y.; Zhang, X.-E.; Cui, Z. Single-Particle Tracking of Human Immunodeficiency Virus Type 1 Productive Entry into Human Primary Macrophages. ACS Nano 2017, 11, 3890–3903. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV Enters Cells via Endocytosis and Dynamin-Dependent Fusion with Endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef]
- Di Primio, C.; Quercioli, V.; Allouch, A.; Gijsbers, R.; Christ, F.; Debyser, Z.; Arosio, D.; Cereseto, A. Single-Cell Imaging of HIV-1 Provirus (SCIP). Proc. Natl. Acad. Sci. USA 2013, 110, 5636–5641. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, M.; Li, W.; Zhang, Z.; Zhang, X.; Wu, G.; Tan, T.; Cui, Z.; Zhang, M. Live Visualization of HIV-1 Proviral DNA Using a Dual-Color-Labeled CRISPR System. Anal. Chem. 2017, 89, 12896–12901. [Google Scholar] [CrossRef]
- Campbell, E.M.; Perez, O.; Melar, M.; Hope, T.J. Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell. Virology 2007, 360, 286–293. [Google Scholar] [CrossRef]
- Markosyan, R.M.; Cohen, F.S.; Melikyan, G.B. Time-resolved Imaging of HIV-1 Env-mediated Lipid and Content Mixing between a Single Virion and Cell Membrane. Mol. Biol. Cell 2005, 16, 5502–5513. [Google Scholar] [CrossRef]
- Miyauchi, K.; Marin, M.; Melikyan, G.B. Visualization of retrovirus uptake and delivery into acidic endosomes. Biochem. J. 2011, 434, 559–569. [Google Scholar] [CrossRef]
- Nathan, L.; Daniel, S. Single Virion Tracking Microscopy for the Study of Virus Entry Processes in Live Cells and Biomimetic Platforms. Adv. Exp. Med. Biol. 2019, 1215, 13–43. [Google Scholar] [CrossRef]
- Cavrois, M.; De Noronha, C.; Greene, W.C. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol. 2002, 20, 1151–1154. [Google Scholar] [CrossRef]
- Andres, I.C.; Padilla-Parra, S. Quantitative FRET-FLIM-BlaM to Assess the Extent of HIV-1 Fusion in Live Cells. Viruses 2020, 12, 206. [Google Scholar] [CrossRef]
- Yin, W.; Li, W.; Li, Q.; Liu, Y.; Liu, J.; Ren, M.; Ma, Y.; Zhang, Z.; Zhang, X.; Wu, Y.; et al. Real-time imaging of individual virion-triggered cortical actin dynamics for human immunodeficiency virus entry into resting CD4 T cells. Nanoscale 2019, 12, 115–129. [Google Scholar] [CrossRef]
- Herold, N.; Anders-Osswein, M.; Glass, B.; Eckhardt, M.; Müller, B.; Kräusslich, H.-G.; Anders-Ößwein, M. HIV-1 Entry in SupT1-R5, CEM-ss, and Primary CD4+ T Cells Occurs at the Plasma Membrane and Does Not Require Endocytosis. J. Virol. 2014, 88, 13956–13970. [Google Scholar] [CrossRef]
- Fortin, J.F.; Cantin, R.; Lamontagne, G.; Tremblay, M. Host-derived ICAM-1 glycoproteins incorporated on human immunodeficiency virus type 1 are biologically active and enhance viral infectivity. J. Virol. 1997, 71, 3588–3596. [Google Scholar] [CrossRef]
- Sood, C.; Marin, M.; Mason, C.S.; Melikyan, G.B. Visualization of Content Release from Cell Surface-Attached Single HIV-1 Particles Carrying an Extra-Viral Fluorescent pH-Sensor. PLoS ONE 2016, 11, e0148944. [Google Scholar] [CrossRef]
- Jakobsdottir, G.M.; Iliopoulou, M.; Nolan, R.; Alvarez, L.; Compton, A.A.; Padilla-Parra, S. On the Whereabouts of HIV-1 Cellular Entry and Its Fusion Ports. Trends Mol. Med. 2017, 23, 932–944. [Google Scholar] [CrossRef]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611.e5–626.e5. [Google Scholar] [CrossRef]
- A Coomer, C.; Carlon-Andres, I.; Iliopoulou, M.; Dustin, M.L.; Compeer, E.B.; Compton, A.A.; Padilla-Parra, S. Single-cell glycolytic activity regulates membrane tension and HIV-1 fusion. PLoS Pathog. 2020, 16, e1008359. [Google Scholar] [CrossRef]
- Li, W.; Yu, X.; Xie, F.; Zhang, B.; Shao, S.; Geng, C.; Aziz, A.U.R.; Liao, X.; Liu, B. A Membrane-Bound Biosensor Visualizes Shear Stress-Induced Inhomogeneous Alteration of Cell Membrane Tension. iScience 2018, 7, 180–190. [Google Scholar] [CrossRef]
- Yang, S.-T.; Kreutzberger, A.J.B.; Kiessling, V.; Ganser-Pornillos, B.K.; White, J.M.; Tamm, L.K. HIV virions sense plasma membrane heterogeneity for cell entry. Sci. Adv. 2017, 3, e1700338. [Google Scholar] [CrossRef]
- Mitra, S.; Shanmugapriya, S.; Da Silva, E.S.; Naghavi, M.H. HIV-1 Exploits CLASP2 To Induce Microtubule Stabilization and Facilitate Virus Trafficking to the Nucleus. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef]
- Pereira, C.; Rossy, J.; Owen, D.M.; Mak, J.; Gaus, K. HIV taken by STORM: Super-resolution fluorescence microscopy of a viral infection. Virol. J. 2012, 9, 84. [Google Scholar] [CrossRef]
- Briggs, J.A.G.; Kräusslich, H.-G. The Molecular Architecture of HIV. J. Mol. Biol. 2011, 410, 491–500. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef]
- Panté, N.; Kann, M. Nuclear Pore Complex Is Able to Transport Macromolecules with Diameters of ∼39 nm. Mol. Biol. Cell 2002, 13, 425–434. [Google Scholar] [CrossRef]
- Lelek, M.; Di Nunzio, F.; Henriques, R.; Charneau, P.; Arhel, N.J.; Zimmer, C. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. USA 2012, 109, 8564–8569. [Google Scholar] [CrossRef]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging Intracellular Fluorescent Proteins at Nanometer Resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef]
- Diaz-Griffero, F. The Role of TNPO3 in HIV-1 Replication. Mol. Biol. Int. 2012, 2012, 868597. [Google Scholar] [CrossRef]
- Ciuffi, A.; Llano, M.; Poeschla, E.; Hoffmann, C.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005, 11, 1287–1289. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Prellberg, M.J.; Palermino-Rowland, K.; Melikyan, G.B. HIV-1 Uncoating and Nuclear Import Precede the Completion of Reverse Transcription in Cell Lines and in Primary Macrophages. Viruses 2020, 12, 1234. [Google Scholar] [CrossRef]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1–8. [Google Scholar] [CrossRef]
- Bejarano, D.A.; Peng, K.; Laketa, V.; Boerner, K.; Jost, K.L.; Lucic, B.; Glass, B.; Lusic, M.; Mueller, B.; Kräusslich, H.-G. HIV-1 nuclear import in macrophages is regulated by CPSF6-capsid interactions at the nuclear pore complex. eLife 2019, 8. [Google Scholar] [CrossRef]
- Francis, A.C.; Marin, M.; Singh, P.K.; Achuthan, V.; Prellberg, M.J.; Palermino-Rowland, K.; Lan, S.; Tedbury, P.R.; Sarafianos, S.G.; Engelman, A.N.; et al. HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains. Nat. Commun. 2020, 11, 3505. [Google Scholar] [CrossRef]
- Mattei, S.; Glass, B.; Hagen, W.J.H.; Kräusslich, H.-G.; Briggs, J.A.G. The structure and flexibility of conical HIV-1 capsids determined within intact virions. Science 2016, 354, 1434–1437. [Google Scholar] [CrossRef]
- Ganser-Pornillos, B.K.; Cheng, A.; Yeager, M. Structure of Full-Length HIV-1 CA: A Model for the Mature Capsid Lattice. Cell 2007, 131, 70–79. [Google Scholar] [CrossRef]
- Hulme, A.E.; Perez, O.; Hope, T. Complementary assays reveal a relationship between HIV-1 uncoating and reverse transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 9975–9980. [Google Scholar] [CrossRef]
- Rankovic, S.; Varadarajan, J.; Ramalho, R.; Aiken, C.; Rousso, I. Reverse Transcription Mechanically Initiates HIV-1 Capsid Disassembly. J. Virol. 2017, 91, e00289-17. [Google Scholar] [CrossRef]
- Strunze, S.; Engelke, M.F.; Wang, I.-H.; Puntener, D.; Boucke, K.; Schleich, S.; Way, M.; Schoenenberger, P.; Burckhardt, C.J.; Greber, U.F. Kinesin-1-Mediated Capsid Disassembly and Disruption of the Nuclear Pore Complex Promote Virus Infection. Cell Host Microbe 2011, 10, 210–223. [Google Scholar] [CrossRef]
- Revelo, N.H.; Kamin, D.; Truckenbrodt, S.; Wong, A.B.; Reuter-Jessen, K.; Reisinger, E.; Moser, T.; Rizzoli, S.O. A new probe for super-resolution imaging of membranes elucidates trafficking pathways. J. Cell Biol. 2014, 205, 591–606. [Google Scholar] [CrossRef]
- Jayappa, K.D.; Ao, Z.; Yao, X. The HIV-1 passage from cytoplasm to nucleus: The process involving a complex exchange between the components of HIV-1 and cellular machinery to access nucleus and successful integration. Int. J. Biochem. Mol. Boil. 2012, 3, 70–85. [Google Scholar]
- Di Nunzio, F.; Danckaert, A.; Fricke, T.; Perez, P.; Fernandez, J.; Perret, E.; Roux, P.; Shorte, S.; Charneau, P.; Diaz-Griffero, F.; et al. Human Nucleoporins Promote HIV-1 Docking at the Nuclear Pore, Nuclear Import and Integration. PLoS ONE 2012, 7, e46037. [Google Scholar] [CrossRef]
- Di Nunzio, F.; Fricke, T.; Miccio, A.; Valle-Casuso, J.C.; Perez, P.; Souque, P.; Rizzi, E.; Severgnini, M.; Mavilio, F.; Charneau, P.; et al. Nup153 and Nup98 bind the HIV-1 core and contribute to the early steps of HIV-1 replication. Virology 2013, 440, 8–18. [Google Scholar] [CrossRef]
- Bin Hamid, F.; Kim, J.; Shin, C.-G. Cellular and viral determinants of retroviral nuclear entry. Can. J. Microbiol. 2016, 62, 1–15. [Google Scholar] [CrossRef]
- Matreyek, K.A.; Yücel, S.S.; Li, X.; Engelman, A.N. Nucleoporin NUP153 Phenylalanine-Glycine Motifs Engage a Common Binding Pocket within the HIV-1 Capsid Protein to Mediate Lentiviral Infectivity. PLoS Pathog. 2013, 9, e1003693. [Google Scholar] [CrossRef]
- Buffone, C.; Martinez-Lopez, A.; Fricke, T.; Opp, S.; Severgnini, M.; Cifola, I.; Petiti, L.; Frabetti, S.; Skorupka, K.; Zadrozny, K.K.; et al. Nup153 Unlocks the Nuclear Pore Complex for HIV-1 Nuclear Translocation in Nondividing Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- De Iaco, A.; Santoni, F.; Vannier, A.; Guipponi, M.; Antonarakis, S.E.; Luban, J. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 2013, 10, 20. [Google Scholar] [CrossRef]
- Demeulemeester, J.; Blokken, J.; De Houwer, S.; Dirix, L.; Klaassen, H.; Marchand, A.; Chaltin, P.; Christ, F.; Debyser, Z. Inhibitors of the integrase-transportin-SR2 interaction block HIV nuclear import. Retrovirology 2018, 15, 5. [Google Scholar] [CrossRef]
- Fernandez, J.; Machado, A.K.; Lyonnais, S.; Chamontin, C.; Gärtner, K.; Léger, T.; Henriquet, C.; Garcia, C.; Portilho, D.M.; Pugnière, M.; et al. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 2019, 4, 1840–1850. [Google Scholar] [CrossRef]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F.; et al. Nuclear architecture dictates HIV-1 integration site selection. Nat. Cell Biol. 2015, 521, 227–231. [Google Scholar] [CrossRef]
- Quercioli, V.; Di Primio, C.; Casini, A.; Mulder, L.C.F.; Vranckx, L.S.; Borrenberghs, D.; Gijsbers, R.; Debyser, Z.; Cereseto, A. Comparative Analysis of HIV-1 and Murine Leukemia Virus Three-Dimensional Nuclear Distributions. J. Virol. 2016, 90, 5205–5209. [Google Scholar] [CrossRef]
- Lelek, M.; Casartelli, N.; Pellin, D.; Rizzi, E.; Souque, P.; Severgnini, M.; Di Serio, C.; Fricke, T.; Diaz-Griffero, F.; Zimmer, C.; et al. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015, 6, 6483. [Google Scholar] [CrossRef]
- Fricke, T.; Diaz-Griffero, F. HIV-1 Capsid Stabilization Assay. Adv. Struct. Saf. Stud. 2016, 1354, 39–47. [Google Scholar] [CrossRef]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef]
- Müller, B.; Heilemann, M. Shedding new light on viruses: Super-resolution microscopy for studying human immunodeficiency virus. Trends Microbiol. 2013, 21, 522–533. [Google Scholar] [CrossRef]
- Chojnacki, J.; Eggeling, C. Super-resolution fluorescence microscopy studies of human immunodeficiency virus. Retrovirology 2018, 15, 1–16. [Google Scholar] [CrossRef]
- Juillerat, A.; Heinis, C.; Sielaff, I.; Barnikow, J.; Jaccard, H.; Kunz, B.; Terskikh, A.; Johnsson, K. Engineering Substrate Specificity of O6-Alkylguanine-DNA Alkyltransferase for Specific Protein Labeling in Living Cells. ChemBioChem 2005, 6, 1263–1269. [Google Scholar] [CrossRef]
- Gautier, A.; Juillerat, A.; Heinis, C.; Corrêa, I.R.; Kindermann, M.; Beaufils, F.; Johnsson, K. An Engineered Protein Tag for Multiprotein Labeling in Living Cells. Chem. Biol. 2008, 15, 128–136. [Google Scholar] [CrossRef]
- Vicidomini, G.; Bianchini, P.; Diaspro, A. STED super-resolved microscopy. Nat. Methods 2018, 15, 173–182. [Google Scholar] [CrossRef]
- Sahl, S.J.; Hell, S.W.; Bille, J.F. High-Resolution 3D Light Microscopy with STED and RESOLFT. High Resolut. Imaging Microsc. Ophthalmol. 2019, 3–32. [Google Scholar]
- Ries, J. SMAP: A modular super-resolution microscopy analysis platform for SMLM data. Nat. Methods 2020, 17, 1–2. [Google Scholar] [CrossRef]
- Sage, D.; Pham, T.-A.; Babcock, H.; Lukes, T.; Pengo, T.; Chao, J.; Velmurugan, R.; Herbert, A.; Agrawal, A.; Colabrese, S.; et al. Super-resolution fight club: Assessment of 2D and 3D single-molecule localization microscopy software. Nat. Methods 2019, 16, 387–395. [Google Scholar] [CrossRef]
- Blanc, T.; El Beheiry, M.; Caporal, C.; Masson, J.-B.; Hajj, B. Genuage: Visualize and analyze multidimensional single-molecule point cloud data in virtual reality. Nat. Methods 2020, 17, 1100–1102. [Google Scholar] [CrossRef]
- Schnitzbauer, J.; Strauss, M.T.; Schlichthaerle, T.; Schueder, F.; Jungmann, R. Super-resolution microscopy with DNA-PAINT. Nat. Protoc. 2017, 12, 1198–1228. [Google Scholar] [CrossRef]
- Khater, I.M.; Nabi, I.R.; Hamarneh, G. A Review of Super-Resolution Single-Molecule Localization Microscopy Cluster Analysis and Quantification Methods. Gene Expr. Patterns 2020, 1, 100038. [Google Scholar] [CrossRef]
- Manley, S.; Gillette, J.M.; Patterson, G.H.; Shroff, H.; Hess, H.F.; Betzig, E.; Lippincott-Schwartz, J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat. Methods 2008, 5, 155–157. [Google Scholar] [CrossRef]
- Jun, J.V.; Chenoweth, D.M.; Petersson, E.J. Rational design of small molecule fluorescent probes for biological applications. Org. Biomol. Chem. 2020, 18, 5747–5763. [Google Scholar] [CrossRef]
- AshokKumar, P.; Ashoka, A.H.; Collot, M.; Das, A.; Klymchenko, A.S. A fluorogenic BODIPY molecular rotor as an apoptosis marker. Chem. Commun. 2019, 55, 6902–6905. [Google Scholar] [CrossRef]
- Collot, M.; AshokKumar, P.; Anton, H.; Boutant, E.; Faklaris, O.; Galli, T.; Mély, Y.; Danglot, L.; Klymchenko, A.S. MemBright: A Family of Fluorescent Membrane Probes for Advanced Cellular Imaging and Neuroscience. Cell Chem. Biol. 2019, 26, 600.e7–614.e7. [Google Scholar] [CrossRef]
- Danylchuk, D.I.; Moon, S.; Xu, K.; Klymchenko, A.S. Switchable Solvatochromic Probes for Live-Cell Super-resolution Imaging of Plasma Membrane Organization. Angew. Chem. Int. Ed. 2019, 58, 14920–14924. [Google Scholar] [CrossRef]
- Despras, G.; Zamaleeva, A.I.; Dardevet, L.; Tisseyre, C.; Magalhaes, J.G.; Garner, C.; Waard, M.; Amigorena, S.; Feltz, A.; Mallet, J.-M.; et al. H-Rubies, a new family of red emitting fluorescent pH sensors for living cells. Chem. Sci. 2015, 6, 5928–5937. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
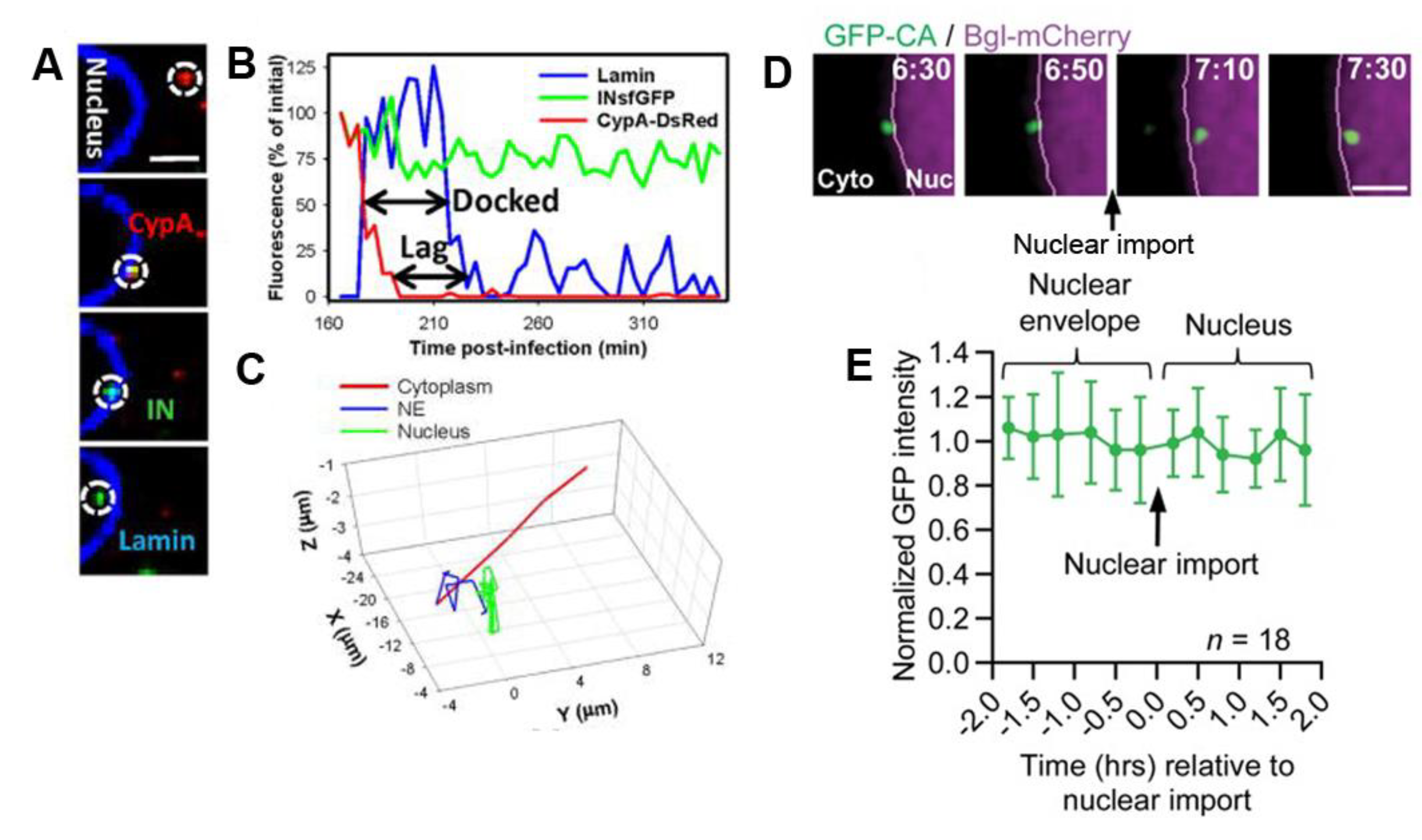{kind=link}
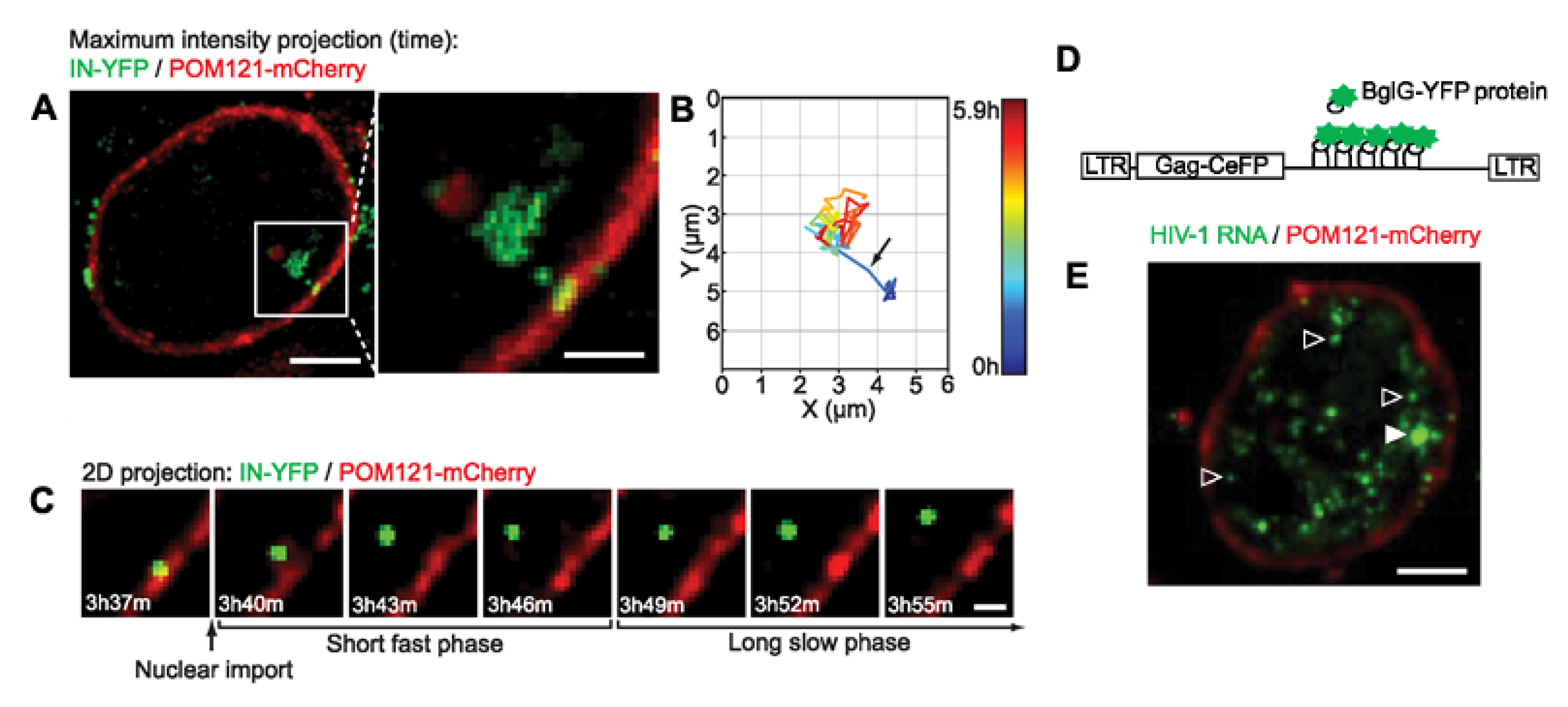{kind=link}
| Viral Component | Labeling Strategy | References |
|---|---|---|
| Capsid | CA-TC/FlAsH, ReAsH | [27] |
| Cyclophilin A-DsRed—CypA-DsRed | [17] | |
| CA-eGFP | [28] | |
| GFP-CA | [18] | |
| Matrix | MA-GFP | [27] |
| Vpr | Vpr-FP | [3,26] |
| mCherry-2CL-YFP-Vpr (bifunctional marker for the content release and viral core tracking; 2 protease cleavage sites are inserted between mCherry and YFP-Vpr leading to their release upon maturation) | [32] | |
| Vpr-QDs | [44] | |
| Integrase | IN-TC/FlAsH | [10] |
| Vpr-IN-FP (protease cleavage site inserted between Vpr and IN) | [30,31] | |
| Gag-IN-eGFP (protease cleavage site inserted between Gag and IN leading to release of IN-GFP upon maturation) | [29] | |
| Nucleocapsid | NC-GFP (GFP is inserted in place of Pol in Gag-Pol, which eliminates a viral protease cleavage site downstream of NC) | [45] |
| NCp7-TC/FlAsH, ReAsH | [22,36] | |
| vRNA | APOBEC3F-FP (A3F-FP) (cytidine deaminase that is incorporated into virions during production) | [37] |
| MICDDRP (multiplex immunofluorescent-cell-based detection of DNA, RNA and proteins; bDNA FISH based vDNA and vRNA labeling combined with immunostaining) | [39] | |
| [Ru(phen)2(dppz)]2+ | [27] | |
| BglG and MS2 technologies (insertion of specific RNA sequences into viral genome in combination with expression of BglG or MS2 proteins fused to FP in infected cells | [12,43] | |
| Incorporation of 5-ethynyl uridine (EU) into vRNA during viral production followed by click labeling | [16] | |
| vDNA | ANCHOR technology: specific DNA sequence –ANCH3 is inserted into vDNA and recognized by OR protein (derived from bacterial parB protein) fused to GFP. | [7] |
| ViewHIV (bDNA-FISH combined with immunostaining of capsid) | [38] | |
| MICDDRP | [39] | |
| Incorporation of EdU into vDNA during reverse transcription followed by click labeling | [40,41] | |
| Transcription sites | Insertion of 18 Bgl binding stem loops into the viral genome in combination with expression of BglG-mCherry in infected cells | [12,18] |
| Integration sites | I-Sce1 reporter system (insertion of ISce1 target site into the viral genome results in induction of I-Sce1 specific double-strand break repair activity at the site of viral integration that leads to H2AX phosphorylation. The site of integration is then detected via immunoabeling of phosphorylated H2AX histone) | [46] |
| Integrated DNA | CRISPR Cas9-QD: U3 region of HIV-1 proviral DNA is targeted by guide RNA and labeled by QD-conjugated Cas9 mutants lacking the endonuclease activity | [47] |
| Membrane/Envelope | S15-mCherry (S15 corresponds to the 15 N-terminal amino acids of p60c-SRC protein that specifically targets the cell plasma membrane. Expression of this truncated form fused to mCherry (S15-mCherry) in producer cells leads to its incorporation into the membrane of newly formed viral particles) | [48] |
| DiD lipid dye (Dioctadecyl-3,3,3 3 Tetramethylindodicarbocyanine) | [49] | |
| EcpH-ICAM1 (GFP related pH sensor fused to intercellular adhesion molecule 1, that is incorporated into HIV-1 virions during viral assembly) | [50] | |
| Incorporation of modified sugars (peracylated azidomannosamine (Ac4ManNAz)) in viral envelope glycoproteins followed by click labeling | [42] | |
| Viral Content markers | Gag-iGFP, Gag-imCherry (FP flanked by a protease cleavage site inserted between CA and MA domains of Gag) | [33,34] |
| Capsid Labeling Strategy | Cells | Timing | Conclusions | Ref. |
|---|---|---|---|---|
| IF | CHO A745 HeLa | 6 h.p.i. 7.5 h.p.i. | Late uncoating CA present in the nucleus | [29] |
| MDMs | 48 h.p.i. | No capsid loss between cytoplasm and nucleus | [75] | |
| SupT1-R5 (CD+T cell line) | 0.5 h.p.i | Cytoplasmic loss of ~50% of CA molecules | [19] | |
| SupT1-R5 (CD+T cell line) | 5.5 h.p.i. | Uncoating at NPC | ||
| MDMs | 48 h.p.i. | No capsid loss between cytoplasm and nucleus | ||
| Cyp-DsRed | HeLa derived and MDMs | <1 h.p.i. | Early cytoplasmic uncoating results in proteolytic degradation of the viral complex | [13,17,73] |
| HeLa derived MDMs | ~4 h.p.i. ~11.5 h.p.i. | Uncoating at the NPC with a fraction of capsid molecules entering into the nucleus with PICs | ||
| CA-eGFP | HeLa | 6 h.p.i | Uncoating at the NE CA nuclear entry independent of PICs | [28] |
| GFP-CA | HeLa | 10.5 h.p.i. | Intact capsid enters the nucleus | [18] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Boutant, E.; Réal, E.; Mély, Y.; Anton, H. Imaging Viral Infection by Fluorescence Microscopy: Focus on HIV-1 Early Stage. Viruses 2021, 13, 213. https://doi.org/10.3390/v13020213
Mukherjee S, Boutant E, Réal E, Mély Y, Anton H. Imaging Viral Infection by Fluorescence Microscopy: Focus on HIV-1 Early Stage. Viruses. 2021; 13(2):213. https://doi.org/10.3390/v13020213
Chicago/Turabian StyleMukherjee, Soumajit, Emmanuel Boutant, Eleonore Réal, Yves Mély, and Halina Anton. 2021. "Imaging Viral Infection by Fluorescence Microscopy: Focus on HIV-1 Early Stage" Viruses 13, no. 2: 213. https://doi.org/10.3390/v13020213
APA StyleMukherjee, S., Boutant, E., Réal, E., Mély, Y., & Anton, H. (2021). Imaging Viral Infection by Fluorescence Microscopy: Focus on HIV-1 Early Stage. Viruses, 13(2), 213. https://doi.org/10.3390/v13020213