
Prediction of SARS-CoV-2 Variant Lineages Using the S1-Encoding Region Sequence Obtained by PacBio Single-Molecule Real-Time Sequencing

 ,
,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. SARS-CoV-2 RNA Extraction
2.3. PacBio SMRT Sequencing
2.4. Illumina COVIDSeq Sequencing
3. Results
3.1. Determination of the Clades and Lineages Based on Full-Length Genomes
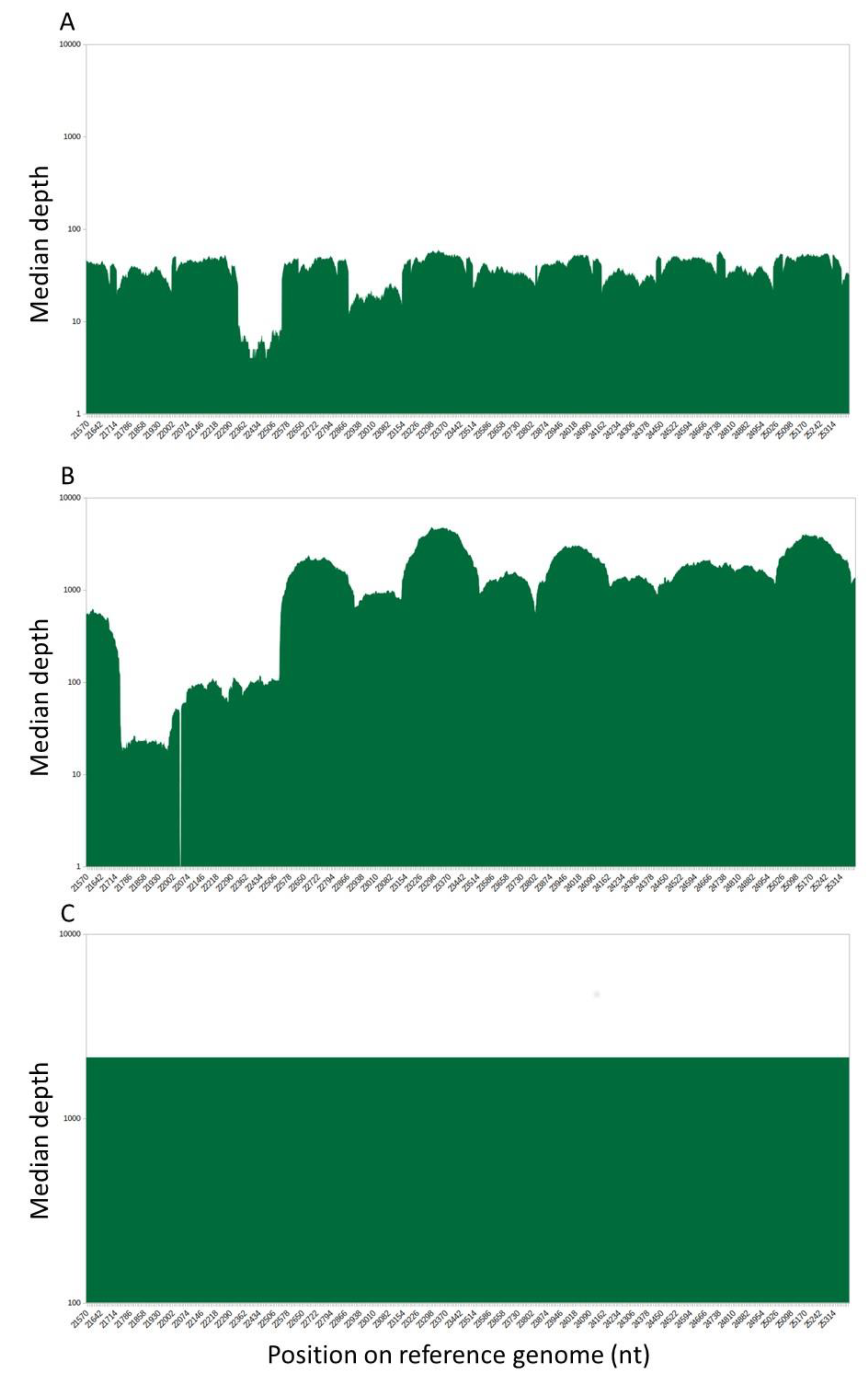
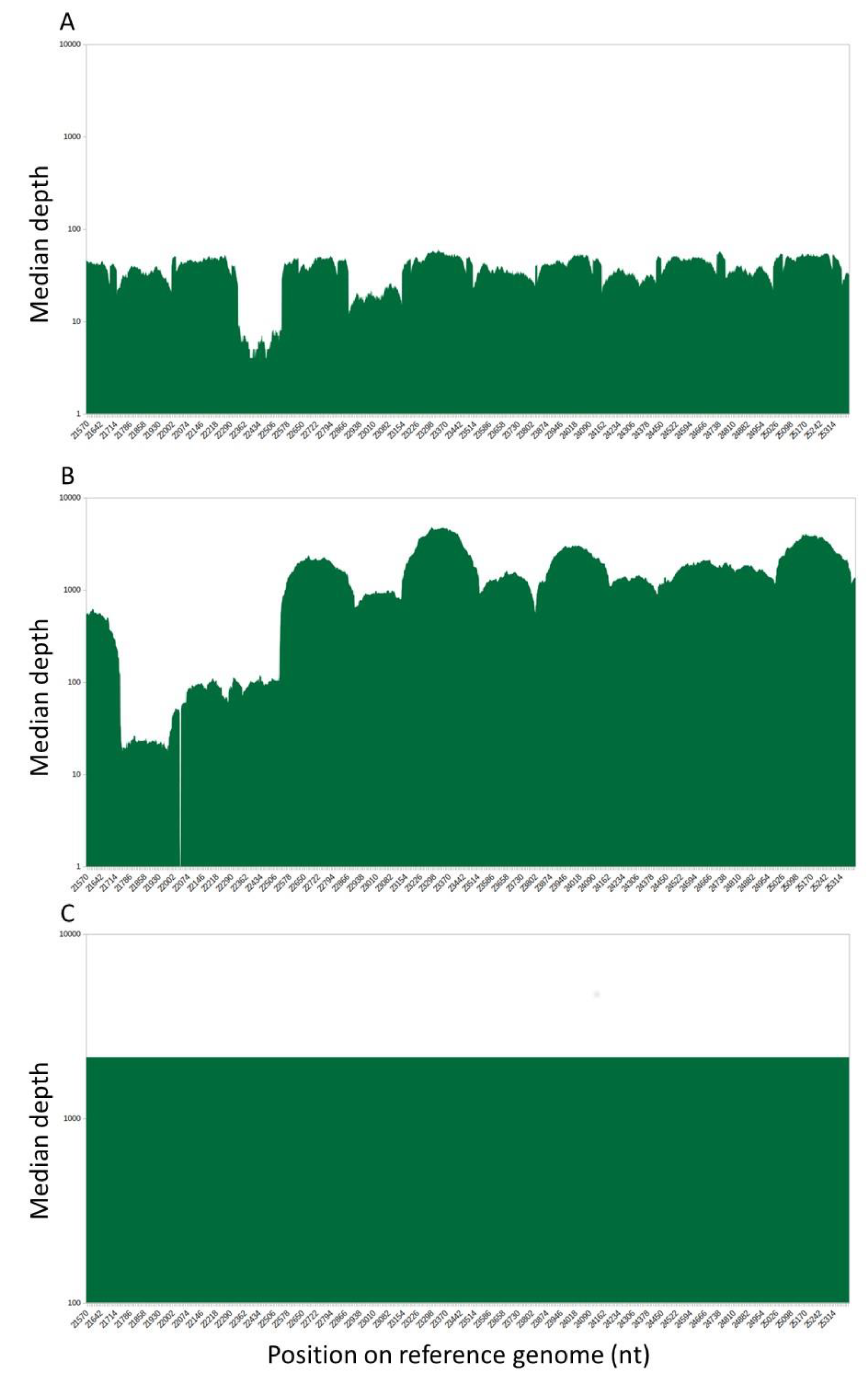
3.2. Comparison of the Spike Sequences Obtained with the Illumina and Pacbio SMRT Systems
3.3. Determination of SARS-CoV-2 Lineage by S1 Domain Key Position Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Zaldivar-Lopez, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e278. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.J.; Chiba, S.; Halfmann, P.; Ehre, C.; Kuroda, M.; Dinnon, K.H., III; Leist, S.R.; Schäfer, A.; Nakajima, N.; Takahashi, K.; et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science 2020, 370, 1464–1468. [Google Scholar] [CrossRef]
- Rotondo, J.C.; Martini, F.; Maritati, M.; Mazziotta, C.; Di Mauro, G.; Lanzillotti, C.; Barp, N.; Gallerani, A.; Tognon, M.; Contini, C. SARS-CoV-2 Infection: New Molecular, Phylogenetic, and Pathogenetic Insights. Efficacy of Current Vaccines and the Potential Risk of Variants. Viruses 2021, 13, 1687. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Vaughan, T.G.; Crawford, K.H.; Althaus, C.L.; Reichmuth, M.L.; Bowen, J.E.; Walls, A.C.; Corti, D.; et al. Spread of a SARS-CoV-2 variant through Europe in the summer of 2020. Nature 2021, 595, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Lessells, R.J.; Giandhari, J.; Pillay, S.; Msomi, N.; Mlisana, K.; Bhiman, J.N.; von Gottberg, A.; Walaza, S.; et al. Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat. Med. 2021, 27, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; St Denis, K.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e2379. [Google Scholar] [CrossRef]
- Voloch, C.M.; da Silva Francisco, R., Jr.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C., Jr.; et al. Genomic characterization of a novel SARS-CoV-2 lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95. [Google Scholar] [CrossRef]
- Peng, J.; Liu, J.; Mann, S.A.; Mitchell, A.M.; Laurie, M.T.; Sunshine, S.; Pilarowski, G.; Ayscue, P.; Kistler, A.; Vanaerschot, M.; et al. Estimation of secondary household attack rates for emergent spike L452R SARS-CoV-2 variants detected by genomic surveillance at a community-based testing site in San Francisco. Clin. Infect. Dis 2021, 31, ciab283. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef]
- Corti, D.; Purcell, L.A.; Snell, G.; Veesler, D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell 2021, 184, 3086–3108. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Kruger, N.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Arora, P.; Sidarovich, A.; Moldenhauer, A.S.; Winkler, M.S.; et al. SARS-CoV-2 variant B.1.617 is resistant to bamlanivimab and evades antibodies induced by infection and vaccination. Cell Rep. 2021, 36, 109415. [Google Scholar] [CrossRef]
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S.; et al. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVIDSeq next-generation sequencing. PLoS ONE 2021, 16, e0247115. [Google Scholar] [CrossRef] [PubMed]
- Harilal, D.; Ramaswamy, S.; Loney, T.; Suwaidi, H.A.; Khansaheb, H.; Alkhaja, A.; Varghese, R.; Deesi, Z.; Nowotny, N.; Alsheikh-Ali, A.; et al. SARS-CoV-2 Whole Genome Amplification and Sequencing for Effective Population-Based Surveillance and Control of Viral Transmission. Clin. Chem. 2020, 66, 1450–1458. [Google Scholar] [CrossRef] [PubMed]
- Bal, A.; Destras, G.; Gaymard, A.; Bouscambert-Duchamp, M.; Valette, M.; Escuret, V.; Frobert, E.; Billaud, G.; Trouillet-Assant, S.; Cheynet, V.; et al. Molecular characterization of SARS-CoV-2 in the first COVID-19 cluster in France reveals an amino acid deletion in nsp2 (Asp268del). Clin. Microbiol. Infect. 2020, 26, 960–962. [Google Scholar] [CrossRef] [PubMed]
- Eid, J.; Fehr, A.; Gray, J.; Luong, K.; Lyle, J.; Otto, G.; Peluso, P.; Rank, D.; Baybayan, P.; Bettman, B.; et al. Real-time DNA sequencing from single polymerase molecules. Science 2009, 323, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Vellas, C.; Del Bello, A.; Debard, A.; Steinmeyer, Z.; Tribaudeau, L.; Ranger, N.; Jeanne, N.; Martin-Blondel, G.; Delobel, P.; Kamar, N.; et al. Influence of treatment with neutralizing monoclonal antibodies on the SARS-CoV-2 nasopharyngeal load and quasispecies. Clin. Microbiol. Infect. 2021. [Google Scholar] [CrossRef]
- Ko, S.H.; Bayat Mokhtari, E.; Mudvari, P.; Stein, S.; Stringham, C.D.; Wagner, D.; Ramelli, S.; Ramos-Benitez, M.J.; Strich, J.R.; Davey, R.T., Jr.; et al. High-throughput, single-copy sequencing reveals SARS-CoV-2 spike variants coincident with mounting humoral immunity during acute COVID-19. PLoS Pathog. 2021, 17, e1009431. [Google Scholar] [CrossRef]
- Sun, F.; Wang, X.; Tan, S.; Dan, Y.; Lu, Y.; Zhang, J.; Xu, J.; Tan, Z.; Xiang, X.; Zhou, Y.; et al. SARS-CoV-2 Quasispecies Provides an Advantage Mutation Pool for the Epidemic Variants. Microbiol. Spectr. 2021, 9, e0026121. [Google Scholar] [CrossRef] [PubMed]
- Eden, J.S.; Rockett, R.; Carter, I.; Rahman, H.; de Ligt, J.; Hadfield, J.; Storey, M.; Ren, X.; Tulloch, R.; Basile, K.; et al. An emergent clade of SARS-CoV-2 linked to returned travellers from Iran. Virus Evol. 2020, 6, veaa027. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acid. Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [Green Version]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e49. [Google Scholar] [CrossRef] [PubMed]
- Jangra, S.; Ye, C.; Rathnasinghe, R.; Stadlbauer, D.; Krammer, F.; Simon, V.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Schotsaert, M. SARS-CoV-2 spike E484K mutation reduces antibody neutralisation. Lancet Microbe 2021, 2, e283–e284. [Google Scholar] [CrossRef]
- Paiva, M.H.S.; Guedes, D.R.D.; Docena, C.; Bezerra, M.F.; Dezordi, F.Z.; Machado, L.C.; Krokovsky, L.; Helvecio, E.; da Silva, A.F.; Vasconcelos, L.R.S.; et al. Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil. Viruses 2020, 12, 1414. [Google Scholar] [CrossRef]
- Planas, D.; Bruel, T.; Grzelak, L.; Guivel-Benhassine, F.; Staropoli, I.; Porrot, F.; Planchais, C.; Buchrieser, J.; Rajah, M.M.; Bishop, E.; et al. Sensitivity of infectious SARS-CoV-2 B.1.1.7 and B.1.351 variants to neutralizing antibodies. Nat. Med. 2021, 27, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Gaymard, A.; Bosetti, P.; Feri, A.; Destras, G.; Enouf, V.; Andronico, A.; Burrel, S.; Behillil, S.; Sauvage, C.; Bal, A.; et al. Early assessment of diffusion and possible expansion of SARS-CoV-2 Lineage 20I/501Y.V1 (B.1.1.7, variant of concern 202012/01) in France, January to March 2021. Euro Surveill 2021, 26, 2100133. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; Haim-Boukobza, S.; Foulongne, V.; Verdurme, L.; Trombert-Paolantoni, S.; Lecorche, E.; Roquebert, B.; Sofonea, M.T. Rapid spread of the SARS-CoV-2 Delta variant in some French regions, June 2021. Eurosurveillance 2021, 26, 2100573. [Google Scholar] [CrossRef]
- Ramesh, S.; Govindarajulu, M.; Parise, R.S.; Neel, L.; Shankar, T.; Patel, S.; Lowery, P.; Smith, F.; Dhanasekaran, M.; Moore, T. Emerging SARS-CoV-2 Variants: A Review of Its Mutations, Its Implications and Vaccine Efficacy. Vaccines 2021, 9, 1195. [Google Scholar] [CrossRef]
- Wang, R.; Chen, J.; Gao, K.; Wei, G.W. Vaccine-escape and fast-growing mutations in the United Kingdom, the United States, Singapore, Spain, India, and other COVID-19-devastated countries. Genomics 2021, 113, 2158–2170. [Google Scholar] [CrossRef]

{kind=link}
| Clade | 20I/ B.1.1.7 | 20I/ B.1.1.7 | 20I/ B.1.1.7 | 20H/ B.1.351 | 21A/ B.1.617.2 | 21A/ B.1.617.2 | 20J/ P.1 | 21H | 20D/ C.36.3 | 20B/ B.1.1.318 | 20A/ B.1.620 | 19B/ A.27 | 19B/ A.28 | 20I/ B.1.1.7 | 20B/P.3 | 20A/ B.1.160 | 20E/ B.1.177 | 20A/ B.1.214.2 | 20A/ B.1.221 | 21C/ B.1.427 | 21D/ B.1.525 | 21F/ B.1.526 | 20C/ B.1.526.1 | 20C/ B.1.616 | 21B/ B.1.617.1 | 20A/ B.1.617.3 | 20A/ B.1.619 | 21G/ C.37 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lineage | B.1.1.7 | B.1.1.7+ E484K | B.1.1.7+ E484Q | B.1.351 | B.1.617.2 | AY.1/AY.2 | P.1 | B.1.621 | C.36.3 | B.1.1.318 | B.1.620 | A.27 | A.28 | B.1.1.7+ L452R | B.1.1.28.3 /P.3 | B.1.160 | B.1.177 | B.1.214.2 | B.1.221 | B.1.427/ B.1.429 | B.1.525 | B.1.526 | B.1.526.1 | B.1.616 | B.1.617.1 | B.1.617.3 | B.1.619 | C.37 |
| WHO | Alpha | Alpha | Alpha | Beta | Delta | Delta | Gamma | Mu | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Epsilon | Eta | Iota | N/A | N/A | Kappa | N/A | N/A | Lambda |
| L5F | F | |||||||||||||||||||||||||||
| P9L | ||||||||||||||||||||||||||||
| S12F | F | |||||||||||||||||||||||||||
| S13I | I | |||||||||||||||||||||||||||
| L18F | (F) | F | F | |||||||||||||||||||||||||
| T19R | (R) | (R) | (R) | |||||||||||||||||||||||||
| T20N | N | |||||||||||||||||||||||||||
| P26S | S | S | ||||||||||||||||||||||||||
| Q52R | R | |||||||||||||||||||||||||||
| H66D | D | |||||||||||||||||||||||||||
| A67V | V | |||||||||||||||||||||||||||
| 69del | del | del | del | del | del | del | del | del | ||||||||||||||||||||
| 70del | del | del | del | del | del | del | del | del | ||||||||||||||||||||
| G75V | V | V | ||||||||||||||||||||||||||
| T76I | I | |||||||||||||||||||||||||||
| D80A/G | A | G | ||||||||||||||||||||||||||
| T95I | (I) | (I) | I | I | I | (I) | ||||||||||||||||||||||
| S98F | F | |||||||||||||||||||||||||||
| V126A | A | |||||||||||||||||||||||||||
| C136F | ||||||||||||||||||||||||||||
| D138Y | Y | |||||||||||||||||||||||||||
| 141del | del | |||||||||||||||||||||||||||
| G142D/del/V | (D) | (D) | del | V | (D) | |||||||||||||||||||||||
| 143del | del | |||||||||||||||||||||||||||
| 144del/T | del | del | del | (T) | (del) | del | del | del | del | del | ||||||||||||||||||
| Y145S/insN | (S) | (insN) | ||||||||||||||||||||||||||
| W152R/C | R | C | ||||||||||||||||||||||||||
| E154K | K * | |||||||||||||||||||||||||||
| E156G | (G) | (G) | ||||||||||||||||||||||||||
| 157del/S | (del) | (del) | S | (del) | ||||||||||||||||||||||||
| 158del | (del) | (del) | (del) | |||||||||||||||||||||||||
| R190S | S | |||||||||||||||||||||||||||
| I210T | T | |||||||||||||||||||||||||||
| 214insTDR | insTDR * | |||||||||||||||||||||||||||
| D215G | (G) | G | ||||||||||||||||||||||||||
| A222V | V | (V) | ||||||||||||||||||||||||||
| 242del | (del) | (del) | ||||||||||||||||||||||||||
| 243del | (del) | del | del | |||||||||||||||||||||||||
| 244del | (del) | del | del | |||||||||||||||||||||||||
| H245Y | Y | |||||||||||||||||||||||||||
| 247del | del | |||||||||||||||||||||||||||
| 248del | del | |||||||||||||||||||||||||||
| 249del | del | |||||||||||||||||||||||||||
| 250del | del | |||||||||||||||||||||||||||
| 251del | del | |||||||||||||||||||||||||||
| 252del | del | |||||||||||||||||||||||||||
| D253G/del | G | del | ||||||||||||||||||||||||||
| R346K/S | K | S | ||||||||||||||||||||||||||
| Q414K | K | |||||||||||||||||||||||||||
| K417N/T | (N) | N * | T | |||||||||||||||||||||||||
| N440K | K * | |||||||||||||||||||||||||||
| Y449H | ||||||||||||||||||||||||||||
| N450K | K | |||||||||||||||||||||||||||
| L452R/Q | R * | (R) | R * | R * | R * | R * | R * | R * | R * | Q * | ||||||||||||||||||
| S477N | N * | N | N^ | |||||||||||||||||||||||||
| T478K | K * | (K) | ||||||||||||||||||||||||||
| V483A | A | |||||||||||||||||||||||||||
| E484K/Q | K * | Q * | K * | K * | (K) | K * | K * | K * | K * | K^ | Q * | Q * | K * | |||||||||||||||
| F490S | S | |||||||||||||||||||||||||||
| N501Y/T | Y * | Y * | Y * | Y * | Y * | Y | Y * | T * | Y * | Y * | ||||||||||||||||||
| A570D | D | D | D | D | ||||||||||||||||||||||||
| D614G | G | G | G | G | G | (G) | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | G | |||
| A653V | V | |||||||||||||||||||||||||||
| H655Y | Y | Y | Y | Y * | ||||||||||||||||||||||||
| G669S | S | |||||||||||||||||||||||||||
| Q677H | H | (H) | H | |||||||||||||||||||||||||
| N679K | ||||||||||||||||||||||||||||
| P681H/R | H | H | H | R | (R) | H | H * | H | H | H | R | R | ||||||||||||||||
| A701V | V | V | ||||||||||||||||||||||||||
| T716I | I | I | I | I | I |
| Clade | Lineage | Month of Collection | Total | |
|---|---|---|---|---|
| January | July | |||
| 20I | B.1.1.7 | 48 | 3 | 51 |
| 20A | B.1.160 | 37 | 37 | |
| 20E(EU1) | B.1.177 | 37 | 37 | |
| 21A | B.1.617.2 | 20 | 20 | |
| 21A | AY.4 | 19 | 19 | |
| 20A | B.1.221 | 8 | 8 | |
| 20A | B.1 | 4 | 4 | |
| 21A | AY.34 | 6 | 6 | |
| 21A | AY.9 | 4 | 4 | |
| 20B | B.1.1.241 | 3 | 3 | |
| 20B | B.1.1.317 | 2 | 2 | |
| 20H | B.1.351 | 2 | 2 | |
| 20C | B.1 | 1 | 1 | |
| 20B | B.1.1.186 | 1 | 1 | |
| 20B | B.1.1.28 | 1 | 1 | |
| 20G | B.1.2 | 1 | 1 | |
| 20A | B.1.214 | 1 | 1 | |
| 21A | AY.23 | 1 | 1 | |
| 21A | AY.7.1 | 1 | 1 | |
| Total | 146 | 54 | 200 | |
| Sample | Clade | Lineage | Mutations Detected with Illumina | Mutations Detected with Pacbio |
|---|---|---|---|---|
| 20 | 20A | B.1.214 | T95I, D614G, T716I | T95I, T478K, D614G, T716I |
| 33 | 20A | B.1.160 | D614G | S477N, D614G |
| 62 | 20A | B.1.160 | D614G | S477N, D614G |
| 79 | 20A | B.1.160 | D614G | S477N, A522S, D614G |
| 110 | 20A | B.1.160 | D614G | S477N, D614G |
| 111 | 20A | B.1.160 | D614G | S477N, D614G |
| 136 | 20B | B.1.1.186 | L18F, D614G | L18F, N440K, D614G |
| 30 | 20H (Beta) | B.1.351 | L18F, D80A, D215G, 241del, 242del, 243del, K417N, N501Y, D614G, A701V | L18F, D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V |
| 134 | 20H (Beta) | B.1.351 | D80A, D215G, 241del, 242del, 243del, K417N, D614G, A701V | D80A, D215G, 241del, 242del, 243del, K417N, E484K, N501Y, D614G, A701V |
| 15 | 20I (Alpha) | B.1.1.7 | 69del, 70del, 144del, A570D, D614G, P681H, T716I | 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I |
| 27 | 20I (Alpha) | B.1.1.7 | 69del, 70del, 144del, A570D, D614G, P681H, T716I | 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I |
| 56 | 20I (Alpha) | B.1.1.7 | 69del, 70del, 144del, A570D, D614G, P681H, T716I | 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I |
| 73 | 20I (Alpha) | B.1.1.7 | 69del, 70del, 144del, A570D, D614G, P681H, T716I | 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I |
| 116 | 20I (Alpha) | B.1.1.7 | 69del, 70del, 144del, A570D, D614G, P681H, T716I | 69del, 70del, 144del, N501Y, A570D, D614G, P681H, T716I |
| 143 | 21A (Delta) | AY.4 | T19R, E156G, 157del, 158del, L452R, T478K, D614G, P681R | T19R, T95I, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 153 | 21A (Delta) | AY.4 | T19R, E156G, 157del, 158del, L452R, T478K, D614G, P681R, D950N | T19R, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 163 | 21A (Delta) | AY.4 | T19R, L452R, T478K, D614G, P681R, D950N | T19R, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 167 | 21A (Delta) | AY.4 | T19R, L452R, T478K, D614G, P681R, D950N | T19R, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 169 | 21A (Delta) | AY.4 | T19R, E156G, 157del, 158del, L452R, T478K, D614G, P681R | T19R, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 170 | 21A (Delta) | AY.4 | T19R, L452R, T478K, D614G, P681R | T19R, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 189 | 21A (Delta) | AY.4 | T19R, E156G, 157del, 158del, L452R, T478K, D614G, P681R | T19R, T95I, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 186 | 21A (Delta) | AY.7.1 | T19R, L452R, T478K, D614G, P681R | T19R, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 142 | 21A (Delta) | AY.9 | T19R, E156G, 157del, 158del, A222V, L452R, T478K, D614G, P681R | T19R, V130F, E156G, 157del, 158del, A222V, L452R, T478K, D614G, P681R |
| 183 | 21A (Delta) | AY.9 | L452R, T478K, D614G, P681R, D950N | T19R, V130F, G142D, E156G, 157del, 158del, A222V, L452R, T478K, D614G, P681R |
| 185 | 21A (Delta) | AY.9 | T19R, E156G, 157del, 158del, A222V, L452R, T478K, D614G, P681R | T19R, G142D, E156G, 157del, 158del, A222V, L452R, T478K, D614G, P681R |
| 157 | 21A (Delta) | AY.23 | T19R, L452R, T478K, D614G, P681R, D950N, V1264L | T19R, G142D, E156G, 157del, 158del, L452R, T478K, D614G, P681R |
| 154 | 21A (Delta) | AY.34 | T19R, E156G, 157del, 158del, L452R, T478K, D614G, Q677H, P681R, D950N | T19R, T95I, G142D, E156G, 157del, 158del, L452R, T478K, D614G, Q677H, P681R |
| Full Length Genome (Illumina) | Spike S1 Region (PacBio) | |||
|---|---|---|---|---|
| Clade | Lineage | n | Lineage | n |
| 20A | B.1.221 | 8 | B.1.221 | 8 |
| 20A | B.1 | 4 | NA | 4 |
| 20A | B.1.214 | 1 | NA | 1 |
| 20A | B.1.160 | 37 | B.1.160 | 37 |
| 20B | B.1.1.186 | 1 | NA | 1 |
| 20B | B.1.1.241 | 3 | NA | 3 |
| 20B | B.1.1.28 | 1 | NA | 1 |
| 20B | B.1.1.317 | 2 | NA | 2 |
| 20C | B.1 | 1 | NA | 1 |
| 20E (EU1) | B.1.177 | 37 | B.1.177 | 37 |
| 20G | B.1.2 | 1 | NA | 1 |
| 20H | B.1.351 | 2 | B.1.351 | 2 |
| 20I | B.1.1.7 | 45 | B.1.1.7 | 45 |
| 21A | B.1.617.2 | 17 | B.1.617.2 | 43 |
| 21A | AY.4 | 16 | ||
| 21A | AY.34 | 4 | ||
| 21A | AY.9 | 4 | ||
| 21A | AY.23 | 1 | ||
| 21A | AY.7.1 | 1 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhomme, S.; Latour, J.; Jeanne, N.; Trémeaux, P.; Ranger, N.; Migueres, M.; Salin, G.; Donnadieu, C.; Izopet, J. Prediction of SARS-CoV-2 Variant Lineages Using the S1-Encoding Region Sequence Obtained by PacBio Single-Molecule Real-Time Sequencing. Viruses 2021, 13, 2544. https://doi.org/10.3390/v13122544
Lhomme S, Latour J, Jeanne N, Trémeaux P, Ranger N, Migueres M, Salin G, Donnadieu C, Izopet J. Prediction of SARS-CoV-2 Variant Lineages Using the S1-Encoding Region Sequence Obtained by PacBio Single-Molecule Real-Time Sequencing. Viruses. 2021; 13(12):2544. https://doi.org/10.3390/v13122544
Chicago/Turabian StyleLhomme, Sébastien, Justine Latour, Nicolas Jeanne, Pauline Trémeaux, Noémie Ranger, Marion Migueres, Gérald Salin, Cécile Donnadieu, and Jacques Izopet. 2021. "Prediction of SARS-CoV-2 Variant Lineages Using the S1-Encoding Region Sequence Obtained by PacBio Single-Molecule Real-Time Sequencing" Viruses 13, no. 12: 2544. https://doi.org/10.3390/v13122544
APA StyleLhomme, S., Latour, J., Jeanne, N., Trémeaux, P., Ranger, N., Migueres, M., Salin, G., Donnadieu, C., & Izopet, J. (2021). Prediction of SARS-CoV-2 Variant Lineages Using the S1-Encoding Region Sequence Obtained by PacBio Single-Molecule Real-Time Sequencing. Viruses, 13(12), 2544. https://doi.org/10.3390/v13122544