
Production- and Purification-Relevant Properties of Human and Murine Cytomegalovirus


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Virus and Exosomes Production
2.3. Infective Virus Quantification
2.4. Total Particle Quantification and Size Determination
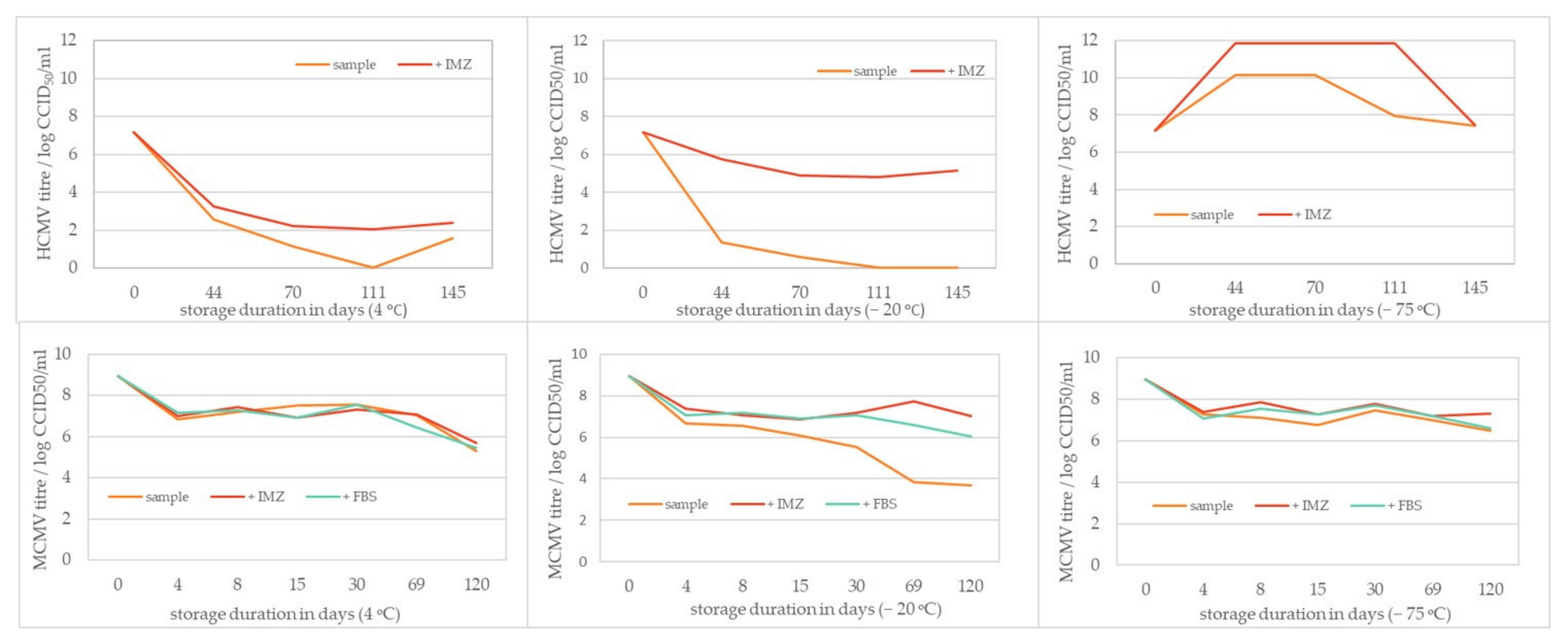
2.5. Storage Stability Study
2.6. Ultracentrifugation of Viruses
2.7. Chromatography
2.8. ELISA Quantification of MRC-5 Host Cell Proteins
2.9. PCR Detection of Genomic DNA
3. Results
3.1. Viral Growth Kinetics In Vitro
3.2. Impact of Clarification on Viral Samples
3.3. HCMV and MCMV Storage Stability
3.4. Ultracentrifugation
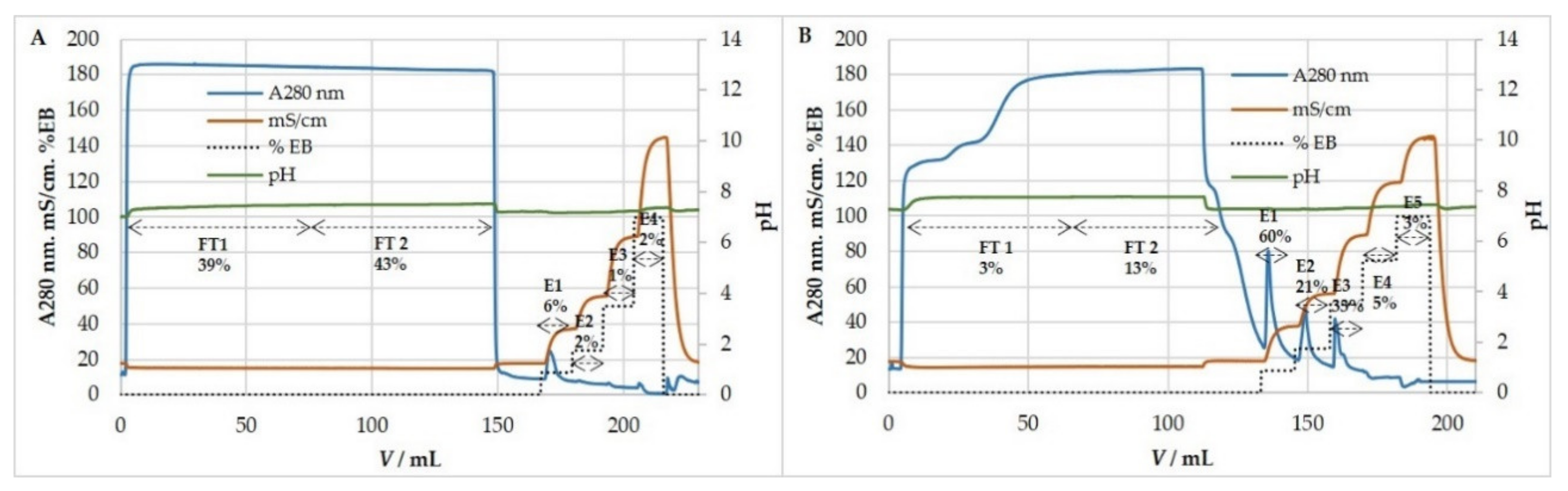
3.5. Ion-Exchange Chromatography
3.5.1. Anion-Exchange Chromatography for Purification and Concentration of HCMV
3.5.2. Anion-Exchange Chromatography for Purification and Concentration of MCMV
3.6. Exosomes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Stern-Ginossar, N.; Weisburd, B.; Michalski, A.; Le, V.T.K.; Hein, M.Y.; Huang, S.-X.; Ma, M.; Shen, B.; Qian, S.-B.; Hengel, H.; et al. Decoding human cytomegalovirus. Science 2012, 338, 1088–1093. [Google Scholar] [CrossRef] [PubMed]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef]
- Boppana, S.B.; Britt, W.J. Synopsis of Clinical Aspects of Human Cytomegalovirus Disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Poole, UK, 2013. [Google Scholar]
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine Transmission of Cytomegalovirus to Infants of Women with Preconceptional Immunity. N. Engl. J. Med. 2001, 344, 1366–1371. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.; Cannon, S.D.G.; Fowler, K.B. The Epidemiology and Public Health Impact of Congenital Cytomegalovirus Infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Poole, UK, 2013. [Google Scholar]
- Tania, C.; Rajiv, K. Immunobiology of Human Cytomegalovirus: From Bench to Bedside. Clin. Microbiol. Rev. 2009, 22, 76–98. [Google Scholar] [CrossRef]
- Sheetal, M.; Emeny, V.C.; Tiziana, L.; Boppana, S.B.; Cupta, R.G. The “Silent” Global Burden of Congenital Cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef]
- Karrer, U.; Sierro, S.; Wagner, M.; Oxenius, A.; Hengel, H.; Koszinowski, U.H.; Phillips, R.E.; Klenerman, P. Memory Inflation: Continuous Accumulation of Antiviral CD8+ T Cells Over Time. J. Immunol. 2003, 170, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Wagner, W.M.; Voehringer, D.; Wikby, A.; Klatt, T.; Walter, S.; Müller, C.A.; Pircher, H.; Pawelec, G. Age-associated accumulation of CMV-specific CD8+ T cells expressing the inhibitory killer cell lectin-like receptor G1 (KLRG1). Exp. Gerontol. 2003, 38, 911–920. [Google Scholar] [CrossRef]
- Sierro, S.; Rothkopf, R.; Klenerman, P. Evolution of diverse antiviral CD8+ T cell populations after murine cytomegalovirus infection. Eur. J. Immunol. 2005, 35, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; van Dommelen, S.; Shellam, G.R.; Hill, A.B. Memory Inflation during Chronic Viral Infection Is Maintained by Continuous Production of Short-Lived, Functional T Cells. Immunity 2008, 29, 650–659. [Google Scholar] [CrossRef]
- Smith, C.J.; Turula, H.; Snyder, C.M. Systemic Hematogenous Maintenance of Memory Inflation by MCMV Infection. PLoS Pathog. 2014, 10, e1004233. [Google Scholar] [CrossRef]
- Rafaela, H.; Doris, T.; Jürgen, P.; Matthias, J.R. Two Antigenic Peptides from Genes m123 and m164 of Murine Cytomegalovirus Quantitatively Dominate CD8 T-Cell Memory in the H-2d Haplotype. J. Virol. 2002, 76, 151–164. [Google Scholar] [CrossRef]
- Méndez, A.C.; Rodríguez-Rojas, C.; Del Val, M. Vaccine vectors: The bright side of cytomegalovirus. Med. Microbiol. Immunol. 2019, 208, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Früh, K. CD8+ T cell programming by cytomegalovirus vectors: Applications in prophylactic and therapeutic vaccination. Physiol. Behav. 2017, 47, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Piatik, M., Jr.; Ventura, A.B.; Hughes, C.M.; Gilbride, R.M.; Ford, J.C.; Oswald, K.; Shoemaker, R.; Li, Y.; Lewis, M.S.; et al. Immune clearance of highly pathogenic SIV infection. Nature 2013, 502, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Zak, D.E.; Xu, G.; Ford, J.C.; Marshall, E.E.; Malouli, D.; Gilbride, R.M.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Prevention of tuberculosis in rhesus macaques by a cytomegalovirus-based vaccine. Nat. Med. 2018, 24, 130–143. [Google Scholar] [CrossRef]
- Hansen, S.G.; Womack, J.; Scholz, I.; Renner, A.; Edgel, K.A.; Xu, G.; Ford, J.C.; Grey, M.; St Laurent, B.; Turner, J.M.; et al. Cytomegalovirus vectors expressing Plasmodium knowlesi antigens induce immune responses that delay parasitemia upon sporozoite challenge. PLoS ONE 2019, 14, e0210252. [Google Scholar] [CrossRef] [PubMed]
- Okoye, A.A.; Hansen, S.G.; Vaidya, M.; Fukazawa, Y.; Park, H.; Duell, D.M.; Lum, R.; Hughes, C.M.; Ventura, A.B.; Ainslie, E.; et al. Early antiretroviral therapy limits SIV reservoir establishment to delay or prevent post-treatment viral rebound. Nat. Med. 2018, 24, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Andreas, M.C.; Jurica, A.; Hermine, M.; Marc, P.; Annelies, W.; Lars, D.; Astrid, K.; David, V.; Zsolt, R.; Ulrich, K.; et al. A Spread-Deficient Cytomegalovirus for Assessment of First-Target Cells in Vaccination. J. Virol. 2010, 84, 7730–7742. [Google Scholar] [CrossRef][Green Version]
- Dekhtiarenko, I.; Čičin-Šain, L.; Messerle, M. Use of Recombinant Approaches to Construct Human Cytomegalovirus Mutants BT–Human Cytomegaloviruses: Methods and Protocols; Yurochko, A.D., Miller, W.E., Eds.; Humana Press: Totowa, NJ, USA, 2014; pp. 59–79. ISBN 978-1-62703-788-4. [Google Scholar]
- Hansen, S.G.; Powers, C.J.; Richards, R.; Ventura, A.B.; Ford, J.C.; Siess, D.; Axthelm, M.K.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J.; et al. Evasion of CD8+ T cells is critical for superinfection by cytomegalovirus. Science 2010, 328, 102–106. [Google Scholar] [CrossRef]
- Qiu, Z.; Huang, H.; Grenier, J.M.; Perez, O.A.; Smilowitz, H.M.; Adler, B.; Khanna, K.M. Cytomegalovirus-Based Vaccine Expressing a Modified Tumor Antigen Induces Potent Tumor-Specific CD8+ T-cell Response and Protects Mice from Melanoma. Cancer Immunol. Res. 2015, 3, 536–546. [Google Scholar] [CrossRef]
- Beverley, P.C.L.; Ruzsics, Z.; Hey, A.; Hutchings, C.; Boos, S.; Bolinger, B.; Marchi, E.; O’Hara, G.; Klenerman, P.; Koszinowski, U.H.; et al. A novel murine cytomegalovirus vaccine vector protects against Mycobacterium tuberculosis. J. Immunol. 2014, 193, 2306–2316. [Google Scholar] [CrossRef] [PubMed]
- Klyushnenkova, E.N.; Kouiavskaia, D.V.; Parkins, C.J.; Caposio, P.; Botto, S.; Alexander, R.B.; Jarvis, M.A. A cytomegalovirus-based vaccine expressing a single tumor-specific CD8+ T-cell epitope delays tumor growth in a murine model of prostate cancer. J. Immunother. 2012, 35, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Parkins, C.J.; Caposio, P.; Feldmann, F.; Botto, S.; Ball, S.; Messaoudi, I.; Cicin-Sain, L.; Feldmann, H.; Jarvis, M.A. A cytomegalovirus-based vaccine provides long-lasting protection against lethal Ebola virus challenge after a single dose. Vaccine 2015, 33, 2261–2266. [Google Scholar] [CrossRef] [PubMed]
- Morabito, K.M.; Ruckwardt, T.R.; Redwood, A.J.; Moin, S.M.; Price, D.A.; Graham, B.S. Intranasal administration of RSV antigen-expressing MCMV elicits robust tissue-resident effector and effector memory CD8+ T cells in the lung. Mucosal Immunol. 2017, 10, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Smith, T.; Grey, F.; Hill, A.B. Cytomegalovirus-based cancer vaccines expressing TRP2 induce rejection of melanoma in mice. Biochem. Biophys. Res. Commun. 2013, 437, 287–291. [Google Scholar] [CrossRef] [PubMed]
- McVoy, M.A. Cytomegalovirus Vaccines. Clin. Infect. Dis. 2013, 57, S196–S199. [Google Scholar] [CrossRef] [PubMed]
- Anderholm, K.M.; Bierle, C.J.; Schleiss, M.R. Cytomegalovirus Vaccines: Current Status and Future Prospects. Drugs 2016, 76, 1625–1645. [Google Scholar] [CrossRef] [PubMed]
- Modlin, J.F.; Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R. Vaccine Development to Prevent Cytomegalovirus Disease: Report from the National Vaccine Advisory Committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar] [CrossRef] [PubMed]
- McTaggart, S.; Al-Rubeai, M. Effects of culture parameters on the production of retroviral vectors by a human packaging cell line. Biotechnol. Prog. 2000, 16, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Genzel, Y.; Fischer, M.; Reichl, U. Serum-free influenza virus production avoiding washing steps and medium exchange in large-scale microcarrier culture. Vaccine 2006, 24, 3261–3272. [Google Scholar] [CrossRef]
- Butler, M.; Burgener, A.; Patrick, M.; Berry, M.; Moffatt, D.; Huzel, N.; Barnabé, N.; Coombs, K. Application of a serum-free medium for the growth of Vero cells and the production of reovirus. Biotechnol. Prog. 2000, 16, 854–858. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.; Zachertowska, A.; Wan, S.; Li, L.; Klimaszewski, D.; Euloth, M.; Hatchette, T.F. Yield increases in intact influenza vaccine virus from chicken allantoic fluid through isolation from insoluble allantoic debris. Vaccine 2007, 25, 4456–4463. [Google Scholar] [CrossRef] [PubMed]
- Iammarino, M.; Nti-Gyabaah, J.; Chandler, M.; Roush, D.; Goklen, K. Impact of cell density and viability on primary clarification of mammalian cell broth. Bioprocess Int. 2007, 5, 38–50. [Google Scholar]
- Pattnaik, P.; Louis, I.; Mahadevan, M.S. Use of membrane technology in bioprocessing therapeutic proteins from inclusion bodies of E. coli. Bioprocess Int. 2009, 7, 54–62. [Google Scholar]
- Raghunath, B.; Bin, W.; Pattnaik, P.; Janssens, J. Best Practices for Optimization and Scale-Up of Microfiltration TFF Processes. Bioprocess. J. 2012, 11, 30–40. [Google Scholar] [CrossRef]
- De las Mercedes Segura, M.; Kamen, A.; Garnier, A. Downstream processing of oncoretroviral and lentiviral gene therapy vectors. Biotechnol. Adv. 2006, 24, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Kumru, O.S.; Joshi, S.B.; Smith, D.E.; Middaugh, C.R.; Prusik, T.; Volkin, D.B. Vaccine instability in the cold chain: Mechanisms, analysis and formulation strategies. Biologicals 2014, 42, 237–259. [Google Scholar] [CrossRef] [PubMed]
- Sviben, D.; Forčić, D.; Kurtović, T.; Halassy, B.; Brgles, M. Stability, biophysical properties and effect of ultracentrifugation and diafiltration on measles virus and mumps virus. Arch. Virol. 2016, 161, 1455–1467. [Google Scholar] [CrossRef]
- Podgornik, A.; Yamamoto, S.; Peterka, M.; Krajnc, N.L. Fast separation of large biomolecules using short monolithic columns. J. Chromatogr. B 2013, 927, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Brgles, M.; Sviben, D.; Forčić, D.; Halassy, B. Nonspecific native elution of proteins and mumps virus in immunoaffinity chromatography. J. Chromatogr. A 2016, 1447, 107–114. [Google Scholar] [CrossRef]
- Sviben, D.; Forcic, D.; Ivancic-Jelecki, J.; Halassy, B.; Brgles, M. Recovery of infective virus particles in ion-exchange and hydrophobic interaction monolith chromatography is influenced by particle charge and total-to-infective particle ratio. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1054, 10–19. [Google Scholar] [CrossRef]
- Flickinger, M.C.; Gagnon, P. Chromatographic Purification of Virus Particles. Encycl. Ind. Biotechnol. 2009, m, 1591. [Google Scholar] [CrossRef]
- Whitfield, R.J.; Battom, S.E.; Barut, M.; Gilham, D.E.; Ball, P.D. Rapid high-performance liquid chromatographic analysis of adenovirus type 5 particles with a prototype anion-exchange analytical monolith column. J. Chromatogr. A 2009, 1216, 2725–2729. [Google Scholar] [CrossRef]
- Rupar, M.; Ravnikar, M.; Tušek-Žnidarič, M.; Kramberger, P.; Glais, L.; Gutiérrez-Aguirre, I. Fast purification of the filamentous Potato virus Y using monolithic chromatographic supports. J. Chromatogr. A 2013, 1272, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Burden, C.S.; Jin, J.; Podgornik, A.; Bracewell, D.G. A monolith purification process for virus-like particles from yeast homogenate. J. Chromatogr. B 2012, 880, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Vandersluis, M.; Stout, J.; Haupts, U.; Sanders, M.; Jacquemart, R. Affinity chromatography for vaccines manufacturing: Finally ready for prime time? Vaccine 2019, 37, 5491–5503. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.W.; Reichl, U. Downstream processing of cell culture-derived virus particles. Expert Rev. Vaccines 2011, 10, 1451–1475. [Google Scholar] [CrossRef]
- Hoen, E.N.; Cremer, T.; Gallo, R.C.; Margolis, L.B. Extracellular vesicles and viruses: Are they close relatives? Proc. Natl. Acad. Sci. USA 2016, 113, 9155–9161. [Google Scholar] [CrossRef] [PubMed]
- Craighead, J.E.; Kanich, R.E.; Almeida, J.D. Nonviral microbodies with viral antigenicity produced in cytomegalovirus-infected cells. J. Virol. 1972, 10, 766–775. [Google Scholar] [CrossRef]
- Irmiere, A.; Gibson, W. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 1983, 130, 118–133. [Google Scholar] [CrossRef]
- Forcic, D.; Košutić-Gulija, T.; Šantak, M.; Jug, R.; Ivancic-Jelecki, J.; Markusic, M.; Mažuran, R. Comparisons of mumps virus potency estimates obtained by 50% cell culture infective dose assay and plaque assay. Vaccine 2010, 28, 1887–1892. [Google Scholar] [CrossRef]
- Barker, K. Phenol-Chloroform Isoamyl Alcohol (PCI) DNA Extraction. Bench 1998. Available online: http://hosted.usf.edu/ecoimmunology/wpcontent/uploads/2014/07/PCI-extraction.pdf (accessed on 1 February 2020).
- Jayme, D.W.; Smith, S.R. Media formulation options and manufacturing process controls to safeguard against introduction of animal origin contaminants in animal cell culture. Cytotechnology 2000, 33, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.F.; de Mucha, J. Replication of poliovirus (LSc 2 ab) in perfused cultures of MRC-5 diploid cells. Dev. Biol. Stand. 1976, 37, 255–258. [Google Scholar] [PubMed]
- Pellegrini, V.; Fineschi, N.; Matteucci, G.; Marsili, I.; Nencioni, L.; Puddu, M.; Garelick, H.; Zuckerman, A.J. Preparation and immunogenicity of an inactivated hepatitis A vaccine. Vaccine 1993, 11, 383–387. [Google Scholar] [CrossRef]
- Fletcher, M.A.; Hessel, L.; Plotkin, S.A. Human diploid cell strains (HDCS) viral vaccines. Dev. Biol. Stand. 1998, 93, 97–107. [Google Scholar] [PubMed]
- Jacobs, J.P. The status of human diploid cell strain MRC-5 as an approved substrate for the production of viral vaccines. J. Biol. Stand. 1976, 4, 97–99. [Google Scholar] [CrossRef]
- Sinzger, C.; Schmidt, K.; Knapp, J.; Kahl, M.; Beck, R.; Waldman, J.; Hebart, H.; Einsele, H.; Jahn, G. Modification of Human Cytomegalovirus Tropism through Propagation In Vitro Is Associated with Changes in the Viral Genome. J. Gen. Virol. 1999, 80, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Hahn, G.; Digel, M.; Katona, R.; Sampaio, K.L.; Messerle, M.; Hengel, H.; Koszinowski, U.; Brune, W.; Adler, B. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 2008, 89, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.W.; Rose, J.A.; Rabson, A.S.; Bond, H.E.; Hall, W.T. Propagation and purification of high-titer human cytomegalovirus. Appl. Microbiol. 1971, 22, 914–918. [Google Scholar] [CrossRef] [PubMed]
- Zurbach, K.A.; Moghbeli, T.; Snyder, C.M. Resolving the titer of murine cytomegalovirus by plaque assay using the M2-10B4 cell line and a low viscosity overlay. Virol. J. 2014, 11, 71. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Carrondo, M.J.T.; Alves, P.M.; Cruz, P.E. Purification of retroviral vectors for clinical application: Biological implications and technological challenges. J. Biotechnol. 2007, 127, 520–541. [Google Scholar] [CrossRef]
- Besnard, L.; Fabre, V.; Fettig, M.; Gousseinov, E.; Kawakami, Y.; Laroudie, N.; Scanlan, C.; Pattnaik, P. Clarification of vaccines: An overview of filter based technology trends and best practices. Biotechnol. Adv. 2016, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Jih, J.; Jiang, J.; Zhou, Z.H. Atomic structure of the human cytomegalovirus capsid with its securing tegument layer of pp150. Science 2017, 356, eaam6892. [Google Scholar] [CrossRef] [PubMed]
- Gibson, W. Structure and assembly of the virion. Intervirology 1996, 39, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Kumru, O.S.; Saleh-Birdjandi, S.; Antunez, L.R.; Sayeed, E.; Robinson, D.; van den Worm, S.; Diemer, G.S.; Perez, W.; Caposio, P.; Früh, K.; et al. Stabilization and formulation of a recombinant Human Cytomegalovirus vector for use as a candidate HIV-1 vaccine. Vaccine 2019, 37, 6696–6706. [Google Scholar] [CrossRef] [PubMed]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., II; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Jairath, S.; Penfold, M.E.; St Jeor, S.; Bohlman, M.C.; Pari, G.S. Identification of persistent RNA-DNA hybrid structures within the origin of replication of human cytomegalovirus. J. Virol. 1998, 72, 6997–7004. [Google Scholar] [CrossRef] [PubMed]
- Sviben, D.; Forcic, D.; Halassy, B.; Allmaier, G.; Marchetti-Deschmann, M.; Brgles, M. Mass spectrometry-based investigation of measles and mumps virus proteome. Virol. J. 2018, 15, 160. [Google Scholar] [CrossRef]
- Brgles, M.; Bonta, M.; Šantak, M.; Jagušić, M.; Forčić, D.; Halassy, B.; Allmaier, G.; Marchetti-Deschmann, M. Identification of mumps virus protein and lipid composition by mass spectrometry. Virol. J. 2016, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Nestola, P.; Peixoto, C.; Silva, R.R.J.S.; Alves, P.M.; Mota, J.P.B.; Carrondo, M.J.T. Improved virus purification processes for vaccines and gene therapy. Biotechnol. Bioeng. 2015, 112, 843–857. [Google Scholar] [CrossRef]
- Wolff, M.W.; Reichl, U. Downstream Processing: From Egg to Cell Culture-Derived Influenza Virus Particles. Chem. Eng. Technol. 2008, 31, 846–857. [Google Scholar] [CrossRef] [PubMed]
- Gomez, P.L.; Robinson, J.M. Vaccine Manufacturing. In Plotkin’s Vaccines, 7th ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 51–60. [Google Scholar] [CrossRef]
- Van Dongen, H.M.; Masoumi, N.; Witwer, K.W.; Pegtel, D.M. Extracellular vesicles exploit viral entry routes for cargo delivery. Microbiol. Mol. Biol. Rev. 2016, 80, 369–386. [Google Scholar] [CrossRef] [PubMed]
- Zicari, S.; Arakelyan, A.; Palomino, R.A.Ñ.; Fitzgerald, W.; Vanpouille, C.; Lebedeva, A.; Schmitt, A.; Bomsel, M.; Britt, W.; Margolis, L. Human cytomegalovirus-infected cells release extracellular vesicles that carry viral surface proteins. Virology 2018, 524, 97–105. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean/nm | Mode/nm | D90/nm | Size Ratio (%) | Particle Recovery (%) | Infectivety Recovery (%) | |||
|---|---|---|---|---|---|---|---|---|
| HCMV (n = 24) | microfiltration 0.45 µm | prior | 224.5 ± 27.20 | 200.7 ± 31.85 | 322.7 ± 52.60 | 100 | 100 ± 22.2 | 76 ± 54.5 |
| post | 226.1 ± 23.84 | 197.0 ± 26.44 | 322 ± 52.32 | |||||
| MRC-5 exosomes | microfiltration 0.45 µm | prior | 216 ± 12.42 | 163.5 ± 15.56 | 321,7 ± 17.72 | 100 | 116 ± 13.7 | |
| post | 218.6 ± 6.13 | 163.0 ± 20.21 | 315.5 ± 9.51 | |||||
| MCMV (n = 10) | centrifugation 3220× g; 7 min | prior | 196.51 ± 10.8 | 155.8 ± 33.9 | 289.55 ± 15.1 | 100 | 95.72 ± 1.35 | 85 ± 17.3 |
| post | 207.62 ± 2.2 | 138.6 ± 10.5 | 307.2 ± 7.1 | |||||
| microfiltration 0.45 µm | prior | 207.62 ± 2.2 | 138.6 ± 10.5 | 307.2 ± 7.1 | 94 | 156 ± 2 | 94 ± 28.3 | |
| post | 195.9 ± 20.2 | 131.7 ± 15.9 | 288.8 ± 26.1 | |||||
| M2-10B4 exosomes | microfiltration 0.45 µm | prior | 189.3 | 140.4 | 266.9 | 101 | 90.71 | |
| post | 191.6 | 133 | 292.3 |
| Infectivity Recovery (%) | Particle Recovery (%) | Size Ratio (%) | Infectivity Recovery (%) | Particle Recovery (%) | Size Ratio (%) | Infectivity Recovery (%) | Particle Recovery (%) | Size Ratio (%) | |
|---|---|---|---|---|---|---|---|---|---|
| HCMV | 1 h | 2 h | 4 h | ||||||
| supernatant | 0.2 ± 0.27 | 9 ± 3.6 | 63 ± 1.9 | 0 | 7 ± 4.4 | 77 ± 12.1 | 0 | 8 ± 8.9 | 74 ± 18.3 |
| pellet | 0.8 ± 1.13 | 59 ± 16.3 | 85 ± 8.8 | 7,6 ± 11.83 | 100 ± 81.6 | 99 ± 15.8 | 0.1 ± 0.23 | 63 ± 24.6 | 99 ± 7.1 |
| MCMV | 1 h | 2 h | 4 h | ||||||
| supernatant | 8.1 ± 3.1 | 9.682 ± 2.01 | 76.13 ± 1.53 | ND | 0 | 1.62 ± 0.81 | 75.52 ± 4.7 | ||
| pellet | 220.87 ± 121.44 | 95.21 ± 18.68 | 96.52 ± 4.41 | 28.22 ± 4.6 | 71.35 ± 1.72 | 105.41 ± 3.4 | |||
| Binding Buffer | Elution Buffer (EB) | Chromatographic Fractions | EB (%) | Elution Molarity (M) | Particle Recovery (%) | Size Ratio (%) | Infectivity Recovery (%) |
|---|---|---|---|---|---|---|---|
| 50 mM MES 0.15M NaCl pH 7.3 | 50 mM MES 1M NaCl pH 7.3 | FT | 2 ± 2.62 | 59 ± 15.2 | 0 ± 0.4 | ||
| E1 | 50 | 0.57 M | 36 ± 4.32 | 87 ± 9.8 | 59 ± 36.8 | ||
| E2 | 100 | 1 M | 41 ± 6.9 | 80 ± 7 | 43 ± 22.2 |
| Binding Buffer | Elution Buffer (EB) | Chromatographic Fractions | EB (%) | Elution Molarity (M) | Particle Recovery (%) | Size Ratio (%) | Infectivity Recovery (%) | Host Cell Proteins Percentage of Starting Sample (%) | Host Cell DNA Percentage of Starting Sample (%) |
|---|---|---|---|---|---|---|---|---|---|
| 50mM MES pH 7.3 | 50mM MES 2M NaCl pH 7.3 | FT | 15.5 ± 11.5 | 79.3 ± 14.5 | 4.8 ± 3.5 | 54.5 ± 9.5 | 0.45 ± 0.35 | ||
| E1 | 12.5 | 0.25 M | 21.7 ± 8.55 | 91 ± 10.4 | 128 ± 101 | 10.5 ± 8.5 | 1.45 ± 0.05 | ||
| E2 | 25 | 0.5 M | 16.3 ± 13.5 | 99.3 ± 9.5 | 8.4 ± 9.8 | 2.5 ± 0.5 | 152 ± 39.4 | ||
| E3 | 50 | 1 M | 11.4 ± 3.5 | 98 ± 6.7 | 15.5 ± 21.6 | 2.5 ± 0.5 | |||
| E4 | 100 | 2 M | 1 ± 0.3 | 111.3 ± 3.4 | 0.03 ± 0.05 | 0 |
| Binding Buffer | Elution Buffer (EB) | Chromatographic Fractions | EB (%) | Elution Molarity (M) | Particle Recovery (%) | Size Ratio (%) | Infectivity Recovery (%) |
|---|---|---|---|---|---|---|---|
| 50 mM MES 0.15 M NaCl pH 7.3 | 50 mM MES 2M NaCl pH7.3 | FT | 0.15 ± 0.2 | 99.4 ± 42 | 0.02 ± 0.01 | ||
| E1 | 12.5 | 0.32 M | 36.7 ± 7.9 | 104.9 ± 6.2 | 47 ± 27.8 | ||
| E2 | 25 | 0.61 M | 10.8 ± 1.8 | 100.3 ± 1.8 | 7.4 ± 3.3 | ||
| E3 | 50 | 1.075 M | 13.9 ± 4.8 | 106.4 ± 6.1 | 5.8 ± 4 | ||
| E4 | 100 | 2 M | 1.9 ± 0.7 | 134.9 ± 19.3 | 1.4 ± 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravlić, S.; Brgles, M.; Hiršl, L.; Jonjić, S.; Halassy, B. Production- and Purification-Relevant Properties of Human and Murine Cytomegalovirus. Viruses 2021, 13, 2481. https://doi.org/10.3390/v13122481
Ravlić S, Brgles M, Hiršl L, Jonjić S, Halassy B. Production- and Purification-Relevant Properties of Human and Murine Cytomegalovirus. Viruses. 2021; 13(12):2481. https://doi.org/10.3390/v13122481
Chicago/Turabian StyleRavlić, Sanda, Marija Brgles, Lea Hiršl, Stipan Jonjić, and Beata Halassy. 2021. "Production- and Purification-Relevant Properties of Human and Murine Cytomegalovirus" Viruses 13, no. 12: 2481. https://doi.org/10.3390/v13122481
APA StyleRavlić, S., Brgles, M., Hiršl, L., Jonjić, S., & Halassy, B. (2021). Production- and Purification-Relevant Properties of Human and Murine Cytomegalovirus. Viruses, 13(12), 2481. https://doi.org/10.3390/v13122481