
Time-Resolved Analysis of N-RNA Interactions during RVFV Infection Shows Qualitative and Quantitative Shifts in RNA Encapsidation and Packaging


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Flow Cytometry
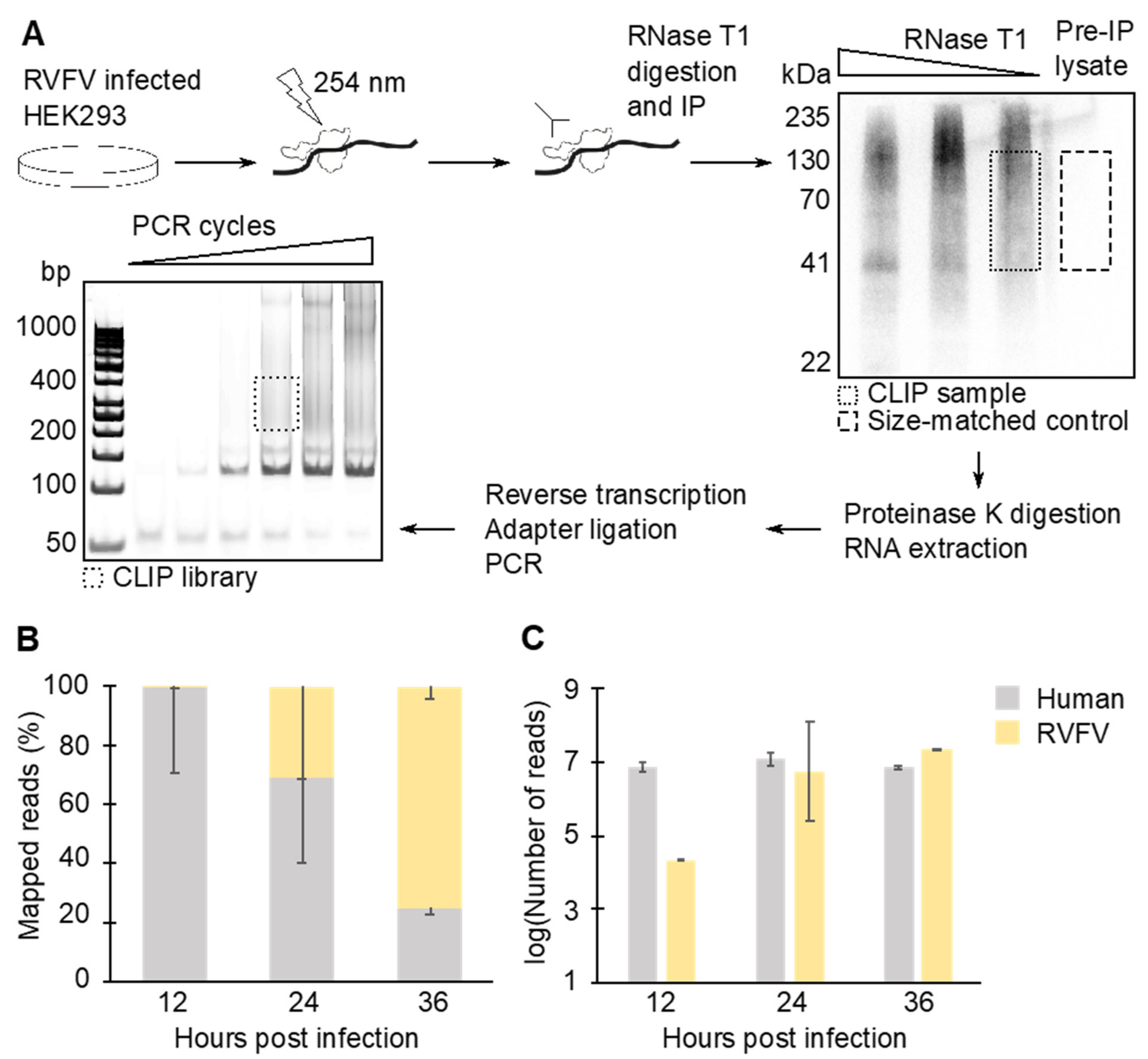
2.3. CLIP-Seq Lysate Preparation
2.4. Immunoprecipitation
2.5. Size Selection of Immunoprecipitated N–RNA Complexes
2.6. Sequencing Library Preparation and Deep Sequencing
2.7. Bioinformatics Analysis
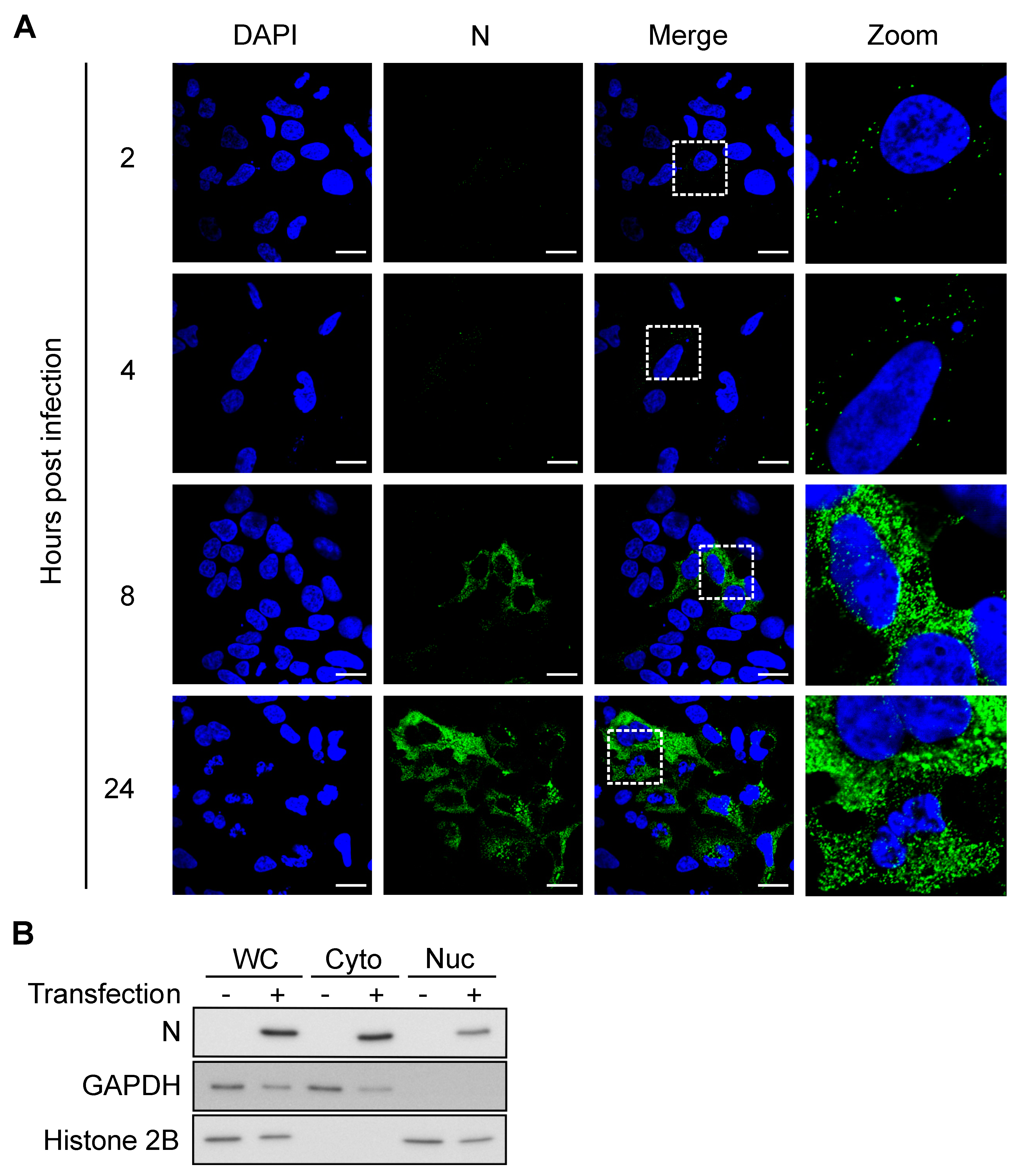
2.8. Immunofluorescence and Confocal Microscopy
2.9. Western Blot
2.10. Strand-Specific RT-qPCR
2.11. Purification of RNAs from Cell Culture and Cell Culture Supernatant
2.12. Preparation of 15N-Labeled N
2.13. Sample Preparation for MRM-MS
2.14. MRM-MS for RVFV N
3. Results
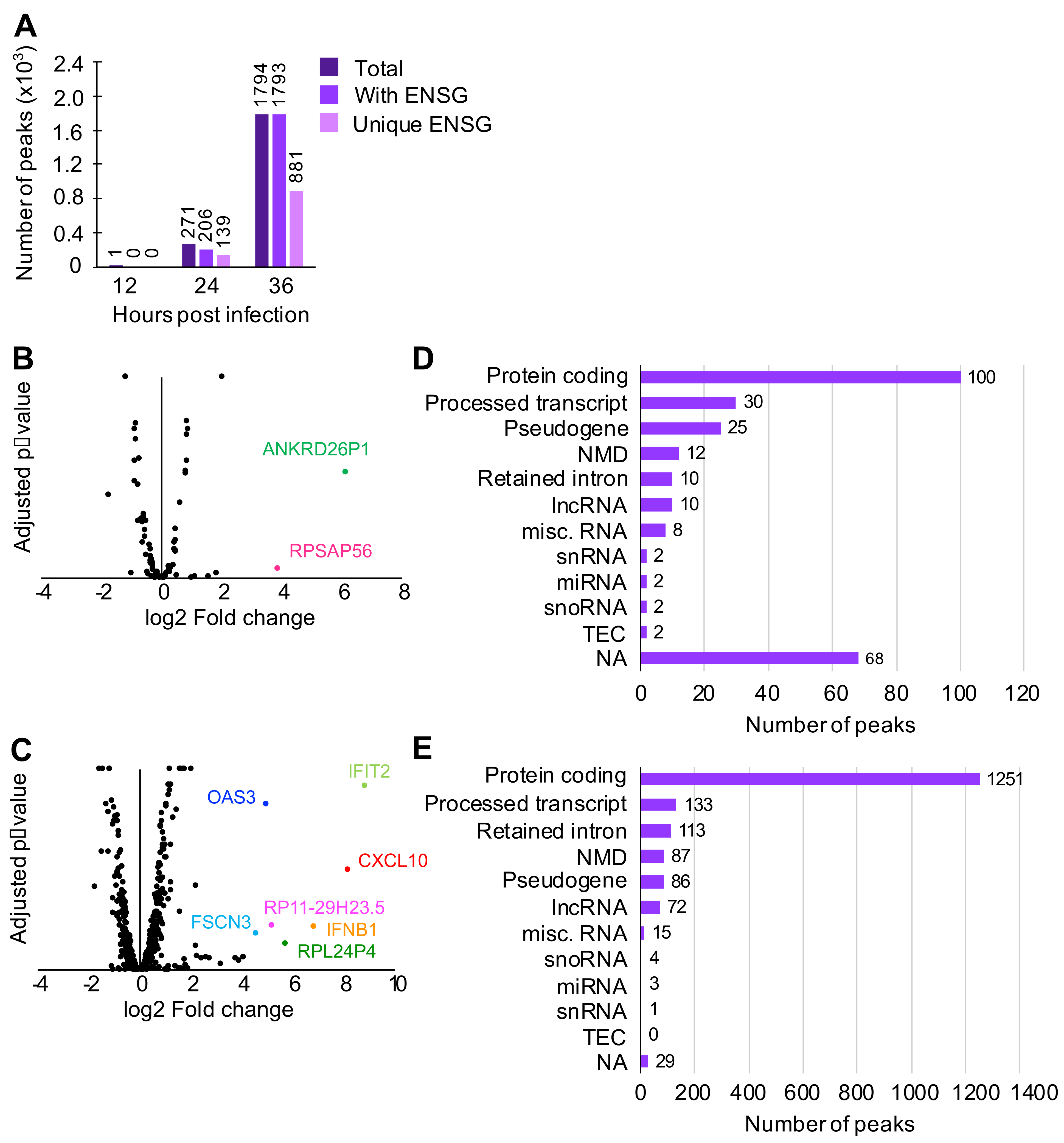
3.1. N Binds Viral and Cellular RNA during Infection
3.2. N Binds Host Cell Protein-Coding Transcripts
3.3. N Exhibits High- and Low-Density Binding Regions on Viral Sense and Antisense RNAs
3.4. N Concentration Increases Exponentially Early during Infection
3.5. Cells Produce More Infectious Virus Later during Infection
4. Discussion
4.1. Consequences of N Binding to Host RNAs
4.2. N Binding Bias on vRNAs
4.3. Dynamics of N Expression and N-vRNA Interactions during RVFV Infection
4.4. Formation and Roles of Incomplete Viral Particles
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jupp, P.G.; Kemp, A.; Grobbelaar, A.; Leman, P.; Burt, F.J.; Alahmed, A.M.; Al Mujalli, D.; Al Khamees, M.; Swanepoel, R. The 2000 epidemic of Rift Valley fever in Saudi Arabia: Mosquito vector studies. Med. Vet. Entomol. 2002, 16, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Bouloy, M.; Weber, F. Molecular biology of rift valley Fever virus. Open Virol. J. 2010, 4, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Linthicum, K.J.; Britch, S.C.; Anyamba, A. Rift Valley Fever: An Emerging Mosquito-Borne Disease. Annu. Rev. Entomol. 2016, 61, 395–415. [Google Scholar] [CrossRef]
- Wilson, M.L.; Chapman, L.E.; Hall, D.B.; Dykstra, E.A.; Ba, K.; Zeller, H.G.; Traore-Lamizana, M.; Hervy, J.P.; Linthicum, K.J.; Peters, C.J. Rift Valley fever in rural northern Senegal: Human risk factors and potential vectors. Am. J. Trop. Med. Hyg. 1994, 50, 663–675. [Google Scholar] [CrossRef]
- McMillen, C.M.; Arora, N.; Boyles, D.A.; Albe, J.R.; Kujawa, M.R.; Bonadio, J.F.; Coyne, C.B.; Hartman, A.L. Rift Valley fever virus induces fetal demise in Sprague-Dawley rats through direct placental infection. Sci. Adv. 2018, 4, 1–13. [Google Scholar] [CrossRef]
- Baudin, M.; Jumaa, A.M.; Jomma, H.J.E.; Karsany, M.S.; Bucht, G.; Näslund, J.; Ahlm, C.; Evander, M.; Mohamed, N. Association of Rift Valley fever virus infection with miscarriage in Sudanese women: A cross-sectional study. Lancet Glob. Health 2016, 4, e864–e871. [Google Scholar] [CrossRef]
- Ikegami, T.; Makino, S. The pathogenesis of rift valley fever. Viruses 2011, 3, 493–519. [Google Scholar] [CrossRef]
- Billecocq, A.; Spiegel, M.; Vialat, P.; Kohl, A.; Weber, F.; Bouloy, M.; Haller, O. NSs Protein of Rift Valley Fever Virus Blocks Interferon Production by Inhibiting Host Gene Transcription. J. Virol. 2004, 78, 9798–9806. [Google Scholar] [CrossRef]
- Bouloy, M.; Janzen, C.; Vialat, P.; Khun, H.; Pavlovic, J.; Huerre, M.; Haller, O. Genetic Evidence for an Interferon-Antagonistic Function of Rift Valley Fever Virus Nonstructural Protein NSs. J. Virol. 2001, 75, 1371–1377. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Pettersson, R.F.; Neve, E.P.A. The Glycoprotein Cytoplasmic Tail of Uukuniemi Virus (Bunyaviridae) Interacts with Ribonucleoproteins and Is Critical for Genome Packaging. J. Virol. 2007, 81, 3198–3205. [Google Scholar] [CrossRef]
- Piper, M.E.; Sorenson, D.R.; Gerrard, S.R. Efficient cellular release of Rift Valley fever virus requires genomic RNA. PLoS ONE 2011, 6, e18070. [Google Scholar] [CrossRef]
- Tercero, B.; Narayanan, K.; Terasaki, K.; Makino, S. Characterization of the Molecular Interactions That Govern the Packaging of Viral RNA Segments into Rift Valley Fever Phlebovirus Particles. J. Virol. 2021, 95, e0042921. [Google Scholar] [CrossRef] [PubMed]
- Albarino, C.G.; Bird, B.H.; Nichol, S.T. A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever Sicilian, and Toscana viruses. J. Virol. 2007, 81, 5246–5256. [Google Scholar] [CrossRef]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Characterization of Rift Valley fever virus transcriptional terminations. J. Virol. 2007, 81, 8421–8438. [Google Scholar] [CrossRef] [PubMed]
- Obijeski, J.F.; Bishop, D.H.; Palmer, E.L.; Murphy, F.A. Segmented genome and nucleocapsid of La Crosse virus. J. Virol. 1976, 20, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, R.F.; Von Bonsdorff, C.H. Ribonucleoproteins of Uukuniemi virus are circular. J. Virol. 1975, 15, 386–392. [Google Scholar] [CrossRef]
- Brennan, B.; Welch, S.R.; McLees, A.; Elliott, R.M. Creation of a recombinant Rift Valley fever virus with a two-segmented genome. J. Virol. 2011, 85, 10310–10318. [Google Scholar] [CrossRef]
- Wichgers Schreur, P.J.; Oreshkova, N.; Moormann, R.J.; Kortekaas, J. Creation of Rift Valley fever viruses with four-segmented genomes reveals flexibility in bunyavirus genome packaging. J. Virol. 2014, 88, 10883–10893. [Google Scholar] [CrossRef]
- Wichgers Schreur, P.J.; Kortekaas, J. Single-Molecule FISH Reveals Non-selective Packaging of Rift Valley Fever Virus Genome Segments. PLoS Pathog. 2016, 12, e1005800. [Google Scholar] [CrossRef]
- Terasaki, K.; Murakami, S.; Lokugamage, K.G.; Makino, S. Mechanism of tripartite RNA genome packaging in Rift Valley fever virus. Proc. Natl. Acad. Sci. USA 2011, 108, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Mendez, E.; Katrukha, E.A.; Spruit, C.M.; Kortekaas, J.; Wichgers Schreur, P.J. Visualizing the ribonucleoprotein content of single bunyavirus virions reveals more efficient genome packaging in the arthropod host. Commun. Biol. 2021, 4, 345. [Google Scholar] [CrossRef]
- Ferron, F.; Li, Z.; Danek, E.I.; Luo, D.; Wong, Y.; Coutard, B.; Lantez, V.; Charrel, R.; Canard, B.; Walz, T.; et al. The hexamer structure of the Rift Valley fever virus nucleoprotein suggests a mechanism for its assembly into ribonucleoprotein complexes. PLoS Pathog. 2011, 7, e1002030. [Google Scholar] [CrossRef]
- Raymond, D.D.; Piper, M.E.; Gerrard, S.R.; Smith, J.L. Structure of the Rift Valley fever virus nucleocapsid protein reveals another architecture for RNA encapsidation. Proc. Natl. Acad. Sci. USA 2010, 107, 11769–11774. [Google Scholar] [CrossRef] [PubMed]
- Raymond, D.D.; Piper, M.E.; Gerrard, S.R.; Skiniotis, G.; Smith, J.L. Phleboviruses encapsidate their genomes by sequestering RNA bases. Proc. Natl. Acad. Sci. USA 2012, 109, 19208–19213. [Google Scholar] [CrossRef]
- Hornak, K.E.; Lanchy, J.-m.; Lodmell, J.S. RNA Encapsidation and Packaging in the Phleboviruses. Viruses 2016, 8, 194. [Google Scholar] [CrossRef] [PubMed]
- Overby, A.K.; Popov, V.L.; Pettersson, R.F.; Neve, E.P. The cytoplasmic tails of Uukuniemi Virus (Bunyaviridae) G(N) and G(C) glycoproteins are important for intracellular targeting and the budding of virus-like particles. J. Virol. 2007, 81, 11381–11391. [Google Scholar] [CrossRef] [PubMed]
- Ellenbecker, M.; Sears, L.; Li, P.; Lanchy, J.M.; Stephen Lodmell, J. Characterization of RNA aptamers directed against the nucleocapsid protein of Rift Valley fever virus. Antivir. Res. 2012, 93, 330–339. [Google Scholar] [CrossRef]
- Ellenbecker, M.; Lanchy, J.M.; Lodmell, J.S. Inhibition of Rift Valley fever virus replication and perturbation of nucleocapsid-RNA interactions by suramin. Antimicrob Agents Chemother 2014, 58, 7405–7415. [Google Scholar] [CrossRef]
- Jiao, L.; Ouyang, S.; Liang, M.; Niu, F.; Shaw, N.; Wu, W.; Ding, W.; Jin, C.; Peng, Y.; Zhu, Y.; et al. Structure of Severe Fever with Thrombocytopenia Syndrome Virus Nucleocapsid Protein in Complex with Suramin Reveals Therapeutic Potential. J. Virol. 2013, 87, 6829–6839. [Google Scholar] [CrossRef] [PubMed]
- Carnec, X.; Ermonval, M.; Kreher, F.; Flamand, M.; Bouloy, M. Role of the cytosolic tails of Rift Valley fever virus envelope glycoproteins in viral morphogenesis. Virology 2014, 448, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Thoulouze, M.I. Why viruses sometimes disperse in groups? Virus Evol. 2019, 5, vez014. [Google Scholar]
- Martin, M. Cutadapt removes adaptor sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kondili, M.; Fust, A.; Preussner, J.; Kuenne, C.; Braun, T.; Looso, M. UROPA: A tool for Universal Robust Peak Annotation. Sci. Rep. 2017, 7, 2593. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic. Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Shihavuddin, A.; Basu, S.; Rexhepaj, E.; Delestro, F.; Menezes, N.; Sigoillot, S.M.; Del Nery, E.; Selimi, F.; Spassky, N.; Genovesio, A. Smooth 2D manifold extraction from 3D image stack. Nat. Commun. 2017, 8, 15554. [Google Scholar] [CrossRef]
- Holden, P.; Horton, W.A. Crude subcellular fractionation of cultured mammalian cell lines. BMC Res. Notes 2009, 2, 243. [Google Scholar] [CrossRef]
- Vashist, S.; Urena, L.; Goodfellow, I. Development of a strand specific real-time RT-qPCR assay for the detection and quantitation of murine norovirus RNA. J. Virol. Methods 2012, 184, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic. Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef]
- Wenger, C.D.; Coon, J.J. A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J. Proteome Res. 2013, 12, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Zhang, C.; Gantman, E.C.; Mele, A.; Darnell, J.C.; Darnell, R.B. Erratum: Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc. 2016, 11, 616. [Google Scholar] [CrossRef]
- Wheeler, E.C.; Van Nostrand, E.L.; Yeo, G.W. Advances and challenges in the detection of transcriptome-wide protein-RNA interactions. Wiley Interdiscip Rev. RNA 2018, 9, e1436. [Google Scholar] [CrossRef] [PubMed]
- Havranek, K.E.; White, L.A.; Lanchy, J.M.; Lodmell, J.S. Transcriptome profiling in Rift Valley fever virus infected cells reveals modified transcriptional and alternative splicing programs. PLoS ONE 2019, 14, e0217497. [Google Scholar] [CrossRef]
- Copeland, A.M.; Altamura, L.A.; Van Deusen, N.M.; Schmaljohn, C.S. Nuclear relocalization of polyadenylate binding protein during rift valley fever virus infection involves expression of the NSs gene. J. Virol. 2013, 87, 11659–11669. [Google Scholar] [CrossRef][Green Version]
- Hopkins, K.C.; Tartell, M.A.; Herrmann, C.; Hackett, B.A.; Taschuk, F.; Panda, D.; Menghani, S.V.; Sabin, L.R.; Cherry, S. Virus-induced translational arrest through 4EBP1/2-dependent decay of 5’-TOP mRNAs restricts viral infection. Proc. Natl. Acad. Sci. USA 2015, 112, E2920–E2929. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.; Crepin, T.; Kolakofsky, D. Nucleoproteins and nucleocapsids of negative-strand RNA viruses. Curr. Opin. Microbiol. 2011, 14, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rescue of infectious rift valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J. Virol. 2006, 80, 2933–2940. [Google Scholar] [CrossRef]
- Murakami, S.; Terasaki, K.; Ramirez, S.I.; Morrill, J.C.; Makino, S. Development of a novel, single-cycle replicable rift valley Fever vaccine. PLoS Negl. Trop. Dis. 2014, 8, e2746. [Google Scholar] [CrossRef]
- Ahsan, N.A.; Sampey, G.C.; Lepene, B.; Akpamagbo, Y.; Barclay, R.A.; Iordanskiy, S.; Hakami, R.M.; Kashanchi, F. Presence of Viral RNA and Proteins in Exosomes from Cellular Clones Resistant to Rift Valley Fever Virus Infection. Front. Microbiol. 2016, 7, 139. [Google Scholar] [CrossRef] [PubMed]
- Timani, K.A.; Liao, Q.; Ye, L.; Zeng, Y.; Liu, J.; Zheng, Y.; Yang, X.; Lingbao, K.; Gao, J.; Zhu, Y. Nuclear/nucleolar localization properties of C-terminal nucleocapsid protein of SARS coronavirus. Virus Res. 2005, 114, 23–34. [Google Scholar] [CrossRef]
- Wulan, W.N.; Heydet, D.; Walker, E.J.; Gahan, M.E.; Ghildyal, R. Nucleocytoplasmic transport of nucleocapsid proteins of enveloped RNA viruses. Front. Microbiol. 2015, 6, 553. [Google Scholar] [CrossRef]
- Wang, R.; Brattain, M.G. The maximal size of protein to diffuse through the nuclear pore is larger than 60 kDa. FEBS Lett. 2007, 581, 3164–3170. [Google Scholar] [CrossRef]
- Han, J.; Xiong, J.; Wang, D.; Fu, X.D. Pre-mRNA splicing: Where and when in the nucleus. Trends Cell Biol. 2011, 21, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.; Lubyova, B.; Pitha, P.M. On the role of IRF in host defense. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2002, 22, 59–71. [Google Scholar] [CrossRef]
- Zhou, A.; Dong, X.; Liu, M.; Tang, B. Comprehensive Transcriptomic Analysis Identifies Novel Antiviral Factors Against Influenza A Virus Infection. Front. Immunol. 2021, 12, 632798. [Google Scholar] [CrossRef]
- Perwitasari, O.; Cho, H.; Diamond, M.S.; Gale, M. Inhibitor of κB kinase epsilon (IKK(epsilon)), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J. Biol. Chem. 2011, 286, 44412–44423. [Google Scholar] [CrossRef]
- Lin, R.J.; Yu, H.P.; Chang, B.L.; Tang, W.C.; Liao, C.L.; Lin, Y.L. Distinct antiviral roles for human 2′,5′;-oligoadenylate synthetase family members against dengue virus infection. J. Immunol. 2009, 183, 8035–8043. [Google Scholar] [CrossRef]
- Bréhin, A.C.; Casadémont, I.; Frenkiel, M.P.; Julier, C.; Sakuntabhai, A.; Desprès, P. The large form of human 2′,5′-Oligoadenylate Synthetase (OAS3) exerts antiviral effect against Chikungunya virus. Virology 2009, 384, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, Q.Z.; Lu, W.; Yang, Y.L.; Chen, R.; Huang, Y.W.; Wang, B. A Comparative Analysis of Coronavirus Nucleocapsid (N) Proteins Reveals the SADS-CoV N Protein Antagonizes IFN-β Production by Inducing Ubiquitination of RIG-I. Front. Immunol. 2021, 12, 688758. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar] [CrossRef] [PubMed]
- Ontiveros, S.J.; Li, Q.; Jonsson, C.B. Modulation of apoptosis and immune signaling pathways by the Hantaan virus nucleocapsid protein. Virology 2010, 401, 165–178. [Google Scholar] [CrossRef]
- Brennan, B.; Welch, S.R.; Elliott, R.M. The consequences of reconfiguring the ambisense S genome segment of Rift Valley fever virus on viral replication in mammalian and mosquito cells and for genome packaging. PLoS Pathog. 2014, 10, e1003922. [Google Scholar] [CrossRef]
- Murakami, S.; Terasaki, K.; Narayanan, K.; Makino, S. Roles of the coding and noncoding regions of rift valley Fever virus RNA genome segments in viral RNA packaging. J. Virol. 2012, 86, 4034–4039. [Google Scholar] [CrossRef] [PubMed]
- Jeeva, S.; Mir, S.; Velasquez, A.; Ragan, J.; Leka, A.; Wu, S.; Sevarany, A.T.; Royster, A.D.; Almeida, N.A.; Chan, F.; et al. Crimean-Congo hemorrhagic fever virus nucleocapsid protein harbors distinct RNA-binding sites in the stalk and head domains. J. Biol. Chem. 2019, 294, 5023–5037. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1936. [Google Scholar] [CrossRef] [PubMed]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.R.; Milin, A.N.; Moosa, M.M.; Onuchic, P.L.; Deniz, A.A. Reentrant Phase Transition Drives Dynamic Substructure Formation in Ribonucleoprotein Droplets. Angew. Chem. Int. Ed. Engl. 2017, 56, 11354–11359. [Google Scholar] [CrossRef] [PubMed]
- Oymans, J.; Wichgers Schreur, P.J.; van Keulen, L.; Kant, J.; Kortekaas, J. Rift Valley fever virus targets the maternal-foetal interface in ovine and human placentas. PLoS Negl. Trop. Dis. 2020, 14, e0007898. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, M.; Schultz, E.P.; Lanchy, J.-M.; Lodmell, J.S. Time-Resolved Analysis of N-RNA Interactions during RVFV Infection Shows Qualitative and Quantitative Shifts in RNA Encapsidation and Packaging. Viruses 2021, 13, 2417. https://doi.org/10.3390/v13122417
Hayashi M, Schultz EP, Lanchy J-M, Lodmell JS. Time-Resolved Analysis of N-RNA Interactions during RVFV Infection Shows Qualitative and Quantitative Shifts in RNA Encapsidation and Packaging. Viruses. 2021; 13(12):2417. https://doi.org/10.3390/v13122417
Chicago/Turabian StyleHayashi, Miyuki, Eric P. Schultz, Jean-Marc Lanchy, and J. Stephen Lodmell. 2021. "Time-Resolved Analysis of N-RNA Interactions during RVFV Infection Shows Qualitative and Quantitative Shifts in RNA Encapsidation and Packaging" Viruses 13, no. 12: 2417. https://doi.org/10.3390/v13122417
APA StyleHayashi, M., Schultz, E. P., Lanchy, J.-M., & Lodmell, J. S. (2021). Time-Resolved Analysis of N-RNA Interactions during RVFV Infection Shows Qualitative and Quantitative Shifts in RNA Encapsidation and Packaging. Viruses, 13(12), 2417. https://doi.org/10.3390/v13122417