
Killing Two Birds with One Stone by Administration of Soluble ACE2: A Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection

 , ,
, , 
Abstract
:1. Introduction
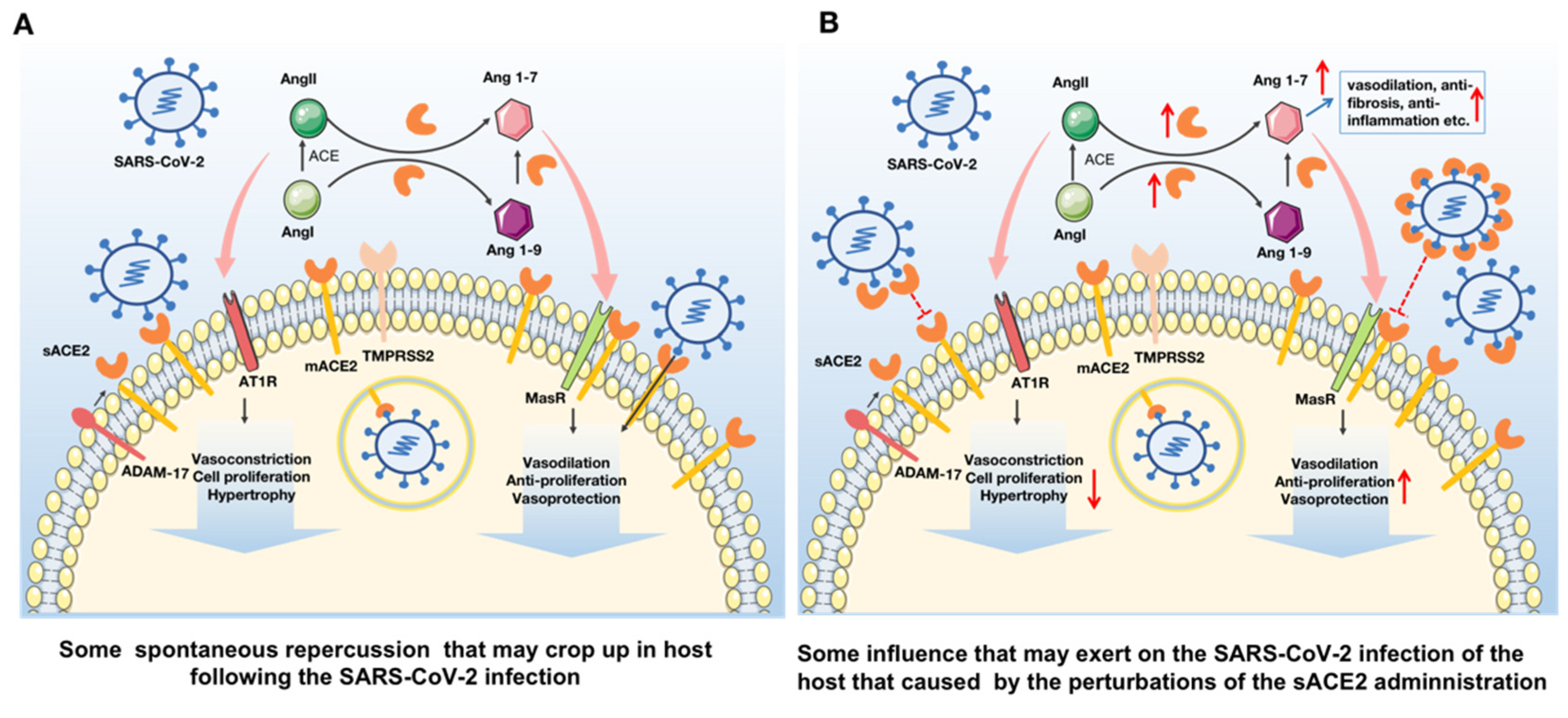
2. Administration of sACE2 in COVID-19 Patients as a Promising Therapeutic Treatment for Both Cardiovascular Diseases and SARS-CoV-2 Infection
2.1. ACE2 Perform a Crucial Role in the Cardiovascular System through Modulating the Renin Angiotensin System
2.2. SARS-CoV-2 Enters into the Host Cell via the ACE2 Receptor and Causes a Series of Deleterious Response
2.3. sACE2 Could Be Developed as a Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection
3. Several Optimized Strategies to Improve the Therapeutic Effect of sACE2 against SARS-CoV-2 Infection
3.1. Prolonging the Half-Life of sACE2 by Fusion with a Varied Fc Fragment
3.2. Increasing the Affinity between sACE2 and RBD by Gene Engineering to Create High-Affinity Decoys for SARS-CoV-2
3.3. Optimizing Truncated sACE2 Peptides to Enhance Its In Vivo Safety
3.4. Other Strategies for sACE2-Based Therapeutic Agents against SARS-CoV-2 Infection
4. Conclusions and Perspective
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Monteil, V.; Dyczynski, M.; Lauschke, V.M.; Kwon, H.; Wirnsberger, G.; Youhanna, S.; Zhang, H.; Slutsky, A.S.; Hurtado Del Pozo, C.; Horn, M.; et al. Human soluble ACE2 improves the effect of remdesivir in SARS-CoV-2 infection. EMBO Mol. Med. 2021, 13, e13426. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Song, K.; Huang, Y. Coronavirus Disease-2019 (COVID-19) and Cardiovascular Complications. J. Cardiothorac. Vasc. Anesth. 2021, 35, 1860–1865. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Malla, A.; Shanmugaraj, B.; Ramalingam, S. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Emerging Zoonotic Respiratory Pathogen in Humans. J. Pure Appl. Microbiol. 2020, 14, 931–936. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Scialo, F.; Daniele, A.; Amato, F.; Pastore, L.; Matera, M.G.; Cazzola, M.; Castaldo, G.; Bianco, A. ACE2: The Major Cell Entry Receptor for SARS-CoV-2. Lung 2020, 198, 867–877. [Google Scholar] [CrossRef]
- Li, H.; Zhou, Y.; Zhang, M.; Wang, H.; Zhao, Q.; Liu, J. Updated Approaches against SARS-CoV-2. Antimicrob. Agents Chemother. 2020, 64, e00483-20. [Google Scholar] [CrossRef] [Green Version]
- Lei, C.; Qian, K.; Li, T.; Zhang, S.; Fu, W.; Ding, M.; Hu, S. Neutralization of SARS-CoV-2 spike pseudotyped virus by recombinant ACE2-Ig. Nat. Commun. 2020, 11, 2070. [Google Scholar] [CrossRef] [Green Version]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkruys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [Green Version]
- Lew, R.A.; Warner, F.J.; Hanchapola, I.; Yarski, M.A.; Ramchand, J.; Burrell, L.M.; Smith, A.I. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp. Physiol. 2008, 93, 685–693. [Google Scholar] [CrossRef]
- Pang, J.; Liu, M.; Ling, W.; Jin, T. Friend or foe? ACE2 inhibitors and GLP-1R agonists in COVID-19 treatment. Obes. Med. 2021, 22, 100312. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Zhong, J.; Guo, D.; Chen, C.B.; Wang, W.; Schuster, M.; Loibner, H.; Penninger, J.M.; Scholey, J.W.; Kassiri, Z.; Oudit, G.Y. Prevention of angiotensin II-mediated renal oxidative stress, inflammation, and fibrosis by angiotensin-converting enzyme 2. Hypertension 2011, 57, 314–322. [Google Scholar] [CrossRef]
- Trask, A.J.; Averill, D.B.; Ganten, D.; Chappell, M.C.; Ferrario, C.M. Primary role of angiotensin-converting enzyme-2 in cardiac production of angiotensin-(1-7) in transgenic Ren-2 hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H3019–H3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.H. Soluble angiotensin-converting enzyme 2 in human heart failure: Relation with myocardial function and clinical outcomes. J. Card Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [Green Version]
- Flores-Munoz, M.; Smith, N.J.; Haggerty, C.; Milligan, G.; Nicklin, S.A. Angiotensin1-9 antagonises pro-hypertrophic signalling in cardiomyocytes via the angiotensin type 2 receptor. J. Physiol. 2011, 589 Pt 4, 939–951. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Lavandero, S.; Jalil, J.E.; Moya, J.; Pinto, M.; Novoa, U.; Apablaza, F.; Gonzalez, L.; Hernandez, C.; Varas, M.; et al. Angiotensin-(1-9) regulates cardiac hypertrophy in vivo and in vitro. J. Hypertens. 2010, 28, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Basu, R.; Guo, D.; Chow, F.L.; Byrns, S.; Schuster, M.; Loibner, H.; Wang, X.H.; Penninger, J.M.; Kassiri, Z.; et al. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation 2010, 122, 717–728. [Google Scholar] [CrossRef]
- Uri, K.; Fagyas, M.; Manyine Siket, I.; Kertesz, A.; Csanadi, Z.; Sandorfi, G.; Clemens, M.; Fedor, R.; Papp, Z.; Edes, I.; et al. New perspectives in the renin-angiotensin-aldosterone system (RAAS) IV: Circulating ACE2 as a biomarker of systolic dysfunction in human hypertension and heart failure. PLoS ONE 2014, 9, e87845. [Google Scholar] [CrossRef]
- Patel, V.B.; Zhong, J.C.; Grant, M.B.; Oudit, G.Y. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ. Res. 2016, 118, 1313–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, J.; Ye, M.; Rodriguez, E.; Gonzalez-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: Prevention of angiotensin II-dependent hypertension. Hypertension 2010, 55, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka-Tojo, M. Vascular Endothelial Glycocalyx Damage in COVID-19. Int. J. Mol. Sci. 2020, 21, 9712. [Google Scholar] [CrossRef]
- Evans, P.C.; Rainger, G.E.; Mason, J.C.; Guzik, T.J.; Osto, E.; Stamataki, Z.; Neil, D.; Hoefer, I.E.; Fragiadaki, M.; Waltenberger, J.; et al. Endothelial dysfunction in COVID-19: A position paper of the ESC Working Group for Atherosclerosis and Vascular Biology, and the ESC Council of Basic Cardiovascular Science. Cardiovasc. Res. 2020, 116, 2177–2184. [Google Scholar] [CrossRef]
- Dong, B.; Zhang, C.; Feng, J.B.; Zhao, Y.X.; Li, S.Y.; Yang, Y.P.; Dong, Q.L.; Deng, B.P.; Zhu, L.; Yu, Q.T.; et al. Overexpression of ACE2 enhances plaque stability in a rabbit model of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1270–1276. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J.; et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Ohishi, M.; Katsuya, T.; Ito, N.; Ikushima, M.; Kaibe, M.; Tatara, Y.; Shiota, A.; Sugano, S.; Takeda, S.; et al. Deletion of angiotensin-converting enzyme 2 accelerates pressure overload-induced cardiac dysfunction by increasing local angiotensin II. Hypertension 2006, 47, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Koibuchi, N.; Nishimatsu, H.; Higashikuni, Y.; Hirata, Y.; Kugiyama, K.; Nagai, R.; Sata, M. Candesartan ameliorates cardiac dysfunction observed in angiotensin-converting enzyme 2-deficient mice. Hypertens. Res. 2008, 31, 1953–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuba, K.; Imai, Y.; Penninger, J.M. Multiple functions of angiotensin-converting enzyme 2 and its relevance in cardiovascular diseases. Circ. J. 2013, 77, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Santos, R.A.; Ferreira, A.J.; Nadu, A.P.; Braga, A.N.; de Almeida, A.P.; Campagnole-Santos, M.J.; Baltatu, O.; Iliescu, R.; Reudelhuber, T.L.; Bader, M. Expression of an angiotensin-(1-7)-producing fusion protein produces cardioprotective effects in rats. Physiol. Genomics 2004, 17, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, P.; Desikan, R.; Dixit, N.M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS Comput. Biol. 2020, 16, e1008461. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.X.; Gnirss, K.; et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, J.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef] [Green Version]
- Kawase, M.; Shirato, K.; van der Hoek, L.; Taguchi, F.; Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012, 86, 6537–6545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.A.; Sacco, O.; Mancino, E.; Cristiani, L.; Midulla, F. Differences and similarities between SARS-CoV and SARS-CoV-2: Spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection 2020, 48, 665–669. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Khan, N.; Chen, X.; Geiger, J.D. Role of Endolysosomes in Severe Acute Respiratory Syndrome Coronavirus-2 Infection and Coronavirus Disease 2019 Pathogenesis: Implications for Potential Treatments. Front. Pharmacol. 2020, 11, 595888. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.Y.; Xu, M.Y.; Wang, X.; Zhang, H.Y.; Hu, H.R.; Li, Y.F.; Hu, Z.H.; Zhong, W.; Wang, M.L. Hydroxychloroquine, a less toxic derivative of chloroquine, is effective in inhibiting SARS-CoV-2 infection in vitro. Cell Discov. 2020, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, N.; Halcrow, P.W.; Lakpa, K.L.; Afghah, Z.; Miller, N.M.; Dowdy, S.F.; Geiger, J.D.; Chen, X. Two-pore channels regulate Tat endolysosome escape and Tat-mediated HIV-1 LTR transactivation. FASEB J. 2020, 34, 4147–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Zhang, Y.; Alharbi, A.; Hanbashi, A.; Alhoshani, A.; Parrington, J. Targeting Two-Pore Channels: Current Progress and Future Challenges. Trends Pharmacol. Sci. 2020, 41, 582–594. [Google Scholar] [CrossRef]
- Khan, N.; Chen, X.; Geiger, J.D. Possible Therapeutic Use of Natural Compounds Against COVID-19. J. Cell Signal. 2021, 2, 63–79. [Google Scholar] [PubMed]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Santaolalla, A.; Beckmann, K.; Kibaru, J.; Josephs, D.; Van Hemelrijck, M.; Irshad, S. Association Between Vitamin D and Novel SARS-CoV-2 Respiratory Dysfunction—A Scoping Review of Current Evidence and Its Implication for COVID-19 Pandemic. Front. Physiol. 2020, 11, 564387. [Google Scholar] [CrossRef]
- Breithaupt-Faloppa, A.C.; Correia, C.J.; Prado, C.M.; Stilhano, R.S.; Ureshino, R.P.; Moreira, L.F.P. 17beta-Estradiol, a potential ally to alleviate SARS-CoV-2 infection. Clinics 2020, 75, e1980. [Google Scholar] [CrossRef]
- Tirupathi Pichiah, P.B.; Suriyakalaa, U.; Kamalakkannan, S.; Kokilavani, P.; Kalaiselvi, S.; SankarGanesh, D.; Gowri, J.; Archunan, G.; Cha, Y.S.; Achiraman, S. Spermidine may decrease ER stress in pancreatic beta cells and may reduce apoptosis via activating AMPK dependent autophagy pathway. Med. Hypotheses 2011, 77, 677–679. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.B.; Clarke, N.; Wang, Z.; Fan, D.; Parajuli, N.; Basu, R.; Putko, B.; Kassiri, Z.; Turner, A.J.; Oudit, G.Y. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM-17: A positive feedback mechanism in the RAS. J. Mol. Cell Cardiol. 2014, 66, 167–176. [Google Scholar] [CrossRef]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B., Jr. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef] [Green Version]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-alpha-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-alpha production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Hasan, M.; Ahmed, A. Potential detrimental role of soluble ACE2 in severe COVID-19 comorbid patients. Rev. Med. Virol. 2021, 31, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Man, L.Y.; Teng, J.; Jia, L.; Zhang, C.; Yuen, K.Y. Soluble ACE2-mediated cell entry of SARS-CoV-2 via interaction with proteins related to the renin-angiotensin system. Cell 2021, 184, 2212–2228.e2212. [Google Scholar]
- Cocozza, F.; Nevo, N.; Piovesana, E.; Lahaye, X.; Buchrieser, J.; Schwartz, O.; Manel, N.; Tkach, M.; Thery, C.; Martin-Jaular, L. Extracellular vesicles containing ACE2 efficiently prevent infection by SARS-CoV-2 Spike protein-containing virus. J. Extracell. Vesicles 2020, 10, e12050. [Google Scholar] [CrossRef]
- Lu, J.; Sun, P.D. High affinity binding of SARS-CoV-2 spike protein enhances ACE2 carboxypeptidase activity. J. Biol. Chem. 2020, 295, 18579–18588. [Google Scholar] [CrossRef] [PubMed]
- Case, J.B.; Rothlauf, P.W.; Chen, R.E.; Liu, Z.; Zhao, H.; Kim, A.S.; Bloyet, L.M.; Zeng, Q.; Tahan, S.; Droit, L.; et al. Neutralizing Antibody and Soluble ACE2 Inhibition of a Replication-Competent VSV-SARS-CoV-2 and a Clinical Isolate of SARS-CoV-2. Cell Host Microbe 2020, 28, 475–485.e475. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Critical Care 2017, 21, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamai, L. Upregulation of the Renin-Angiotensin System Pathways and SARS-CoV-2 Infection: The Rationale for the Administration of Zinc-Chelating Agents in COVID-19 Patients. Cells 2021, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.; Krahenbuhl, S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharm. 2013, 52, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.; Cheng, Y.; Ling, R.; Dai, Y.; Huang, B.; Huang, W.; Zhang, S.; Jiang, Y. Antibody-dependent enhancement of coronavirus. Int. J. Infect. Dis. 2020, 100, 483–489. [Google Scholar] [CrossRef]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef]
- Iwanaga, N.; Cooper, L.; Rong, L.; Beddingfield, B.; Crabtree, J.; Tripp, R.A.; Qin, X.; Kolls, J.K. Novel ACE2-IgG1 fusions with improved in vitro and in vivo activity against SARS-CoV2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, E.; Zhang, L.; Wang, W.; Jin, Y.; Sun, J.; Huang, S.; Yin, W.; Dai, J.; Zhuang, Z.; et al. Potent prophylactic and therapeutic efficacy of recombinant human ACE2-Fc against SARS-CoV-2 infection in vivo. Cell Discov. 2021, 7, 65. [Google Scholar] [CrossRef]
- Tada, T.; Fan, C.; Chen, J.S.; Kaur, R.; Stapleford, K.A.; Gristick, H.; Dcosta, B.M.; Wilen, C.B.; Nimigean, C.M.; Landau, N.R. An ACE2 Microbody Containing a Single Immunoglobulin Fc Domain Is a Potent Inhibitor of SARS-CoV-2. Cell Rep. 2020, 33, 108528. [Google Scholar] [CrossRef]
- Sheikhi, A.; Hojjat-Farsangi, M. An immunotherapeutic method for COVID-19 patients: A soluble ACE2-Anti-CD16 VHH to block SARS-CoV-2 Spike protein. Hum. Vaccin. Immunother. 2021, 17, 92–97. [Google Scholar] [CrossRef]
- Frenken, L.; Linden, R.; Hermans, P.J.; Bos, J.W.; Ruuls, R.C.; Geus, B.D.; Verrips, C.T. Isolation of antigen specific Llama VHH antibody fragments and their high level secretion by Saccharomyces cerevisiae. J. Biotechnol. 2000, 78, 11–21. [Google Scholar] [CrossRef]
- Bazl, M.R.; Rasaee, M.J.; Foruzandeh, M.; Rahimpour, A.; Kiani, J.; Rahbarizadeh, F.; Alirezapour, B.; Mohammadi, M. Production of chimeric recombinant single domain antibody-green fluorescent fusion protein in Chinese hamster ovary cells. Hybridoma 2007, 26, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 28046–28055. [Google Scholar] [CrossRef]
- Devaux, C.A.; Pinault, L.; Osman, I.O.; Raoult, D. Can ACE2 Receptor Polymorphism Predict Species Susceptibility to SARS-CoV-2? Front. Public Health 2020, 8, 608765. [Google Scholar] [CrossRef]
- Calcagnile, M.; Forgez, P.; Iannelli, A.; Bucci, C.; Alifano, M.; Alifano, P. Molecular docking simulation reveals ACE2 polymorphisms that may increase the affinity of ACE2 with the SARS-CoV-2 Spike protein. Biochimie 2021, 180, 143–148. [Google Scholar] [CrossRef]
- Chan, K.K.; Dorosky, D.; Sharma, P.; Abbasi, S.A.; Dye, J.M.; Kranz, D.M.; Herbert, A.S.; Procko, E. Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2. Science 2020, 369, 1261–1265. [Google Scholar] [CrossRef]
- Chan, K.K.; Tan, T.J.C.; Narayanan, K.K.; Procko, E. An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants. Sci. Adv. 2021, 7, eabf1738. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J.; Dorfman, T.; Li, W.; Wong, S.K.; Li, Y.; Kuhn, J.H.; Coderre, J.; Vasilieva, N.; Han, Z.; Greenough, T.C.; et al. Retroviruses pseudotyped with the severe acute respiratory syndrome coronavirus spike protein efficiently infect cells expressing angiotensin-converting enzyme 2. J. Virol. 2004, 78, 10628–10635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basit, A.; Ali, T.; Rehman, S.U. Truncated human angiotensin converting enzyme 2; a potential inhibitor of SARS-CoV-2 spike glycoprotein and potent COVID-19 therapeutic agent. J. Biomol. Struct. Dyn. 2021, 39, 3605–3614. [Google Scholar] [CrossRef]
- Zhang, G.; Pomplun, S.; Loftis, A.R.; Tan, X.; Loas, A.; Pentelute, B.L. Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Mukherjee, A.; Nelson, D.; Jozic, A.; Sahay, G. Rapid generation of circulating and mucosal decoy ACE2 using mRNA nanotherapeutics for the potential treatment of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schafer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef] [PubMed]
- Inal, J.M. Decoy ACE2-expressing extracellular vesicles that competitively bind SARS-CoV-2 as a possible COVID-19 therapy. Clin. Sci. 2020, 134, 1301–1304. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Lu, X.; Xu, C.; Wang, Y.; Sun, W.; Xi, J. CD-sACE2 inclusion compounds: An effective treatment for coronavirus disease 2019 (COVID-19). J. Med. Virol. 2020, 92, 1721–1723. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Li, D.; Wei, D.Q.; Zhao, J.; Wang, J. Human Intestinal Defensin 5 Inhibits SARS-CoV-2 Invasion by Cloaking ACE2. Gastroenterology 2020, 159, 1145–1147.e1144. [Google Scholar] [CrossRef]
- Watson, A.; Ferreira, L.; Hwang, P.; Xu, J.; Stroud, R. Peptide Antidotes to SARS-CoV-2 (COVID-19). bioRxiv 2020. [Google Scholar] [CrossRef]
- Beddingfield, B.J.; Iwanaga, N.; Chapagain, P.P.; Zheng, W.; Roy, C.J.; Hu, T.Y.; Kolls, J.K.; Bix, G.J. The Integrin Binding Peptide, ATN-161, as a Novel Therapy for SARS-CoV-2 Infection. JACC Basic Transl. Sci. 2021, 6, 1–8. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Hansen, J.; Baum, A.; Pascal, K.E.; Russo, V.; Giordano, S.; Wloga, E.; Fulton, B.O.; Yan, Y.; Koon, K.; Patel, K.; et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020, 369, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.A.; van Haperen, R.; Osterhaus, A.; van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251. [Google Scholar] [CrossRef]
- Sun, S.H.; Chen, Q.; Gu, H.J.; Yang, G.; Wang, Y.X.; Huang, X.Y.; Liu, S.S.; Zhang, N.N.; Li, X.F.; Xiong, R.; et al. A Mouse Model of SARS-CoV-2 Infection and Pathogenesis. Cell Host Microbe 2020, 28, 124–133.e4. [Google Scholar] [CrossRef]
- Imai, M.; Iwatsuki-Horimoto, K.; Hatta, M.; Loeber, S.; Halfmann, P.J.; Nakajima, N.; Watanabe, T.; Ujie, M.; Takahashi, K.; Ito, M.; et al. Syrian hamsters as a small animal model for SARS-CoV-2 infection and countermeasure development. Proc. Natl. Acad. Sci. USA 2020, 117, 16587–16595. [Google Scholar] [CrossRef]
- Everett, H.E.; Lean, F.Z.X.; Byrne, A.M.P.; van Diemen, P.M.; Rhodes, S.; James, J.; Mollett, B.; Coward, V.J.; Skinner, P.; Warren, C.J.; et al. Intranasal Infection of Ferrets with SARS-CoV-2 as a Model for Asymptomatic Human Infection. Viruses 2021, 13, 113. [Google Scholar] [CrossRef]
- Cleary, S.J.; Pitchford, S.C.; Amison, R.T.; Carrington, R.; Robaina Cabrera, C.L.; Magnen, M.; Looney, M.R.; Gray, E.; Page, C.P. Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. Br. J. Pharmacol. 2020, 177, 4851–4865. [Google Scholar] [CrossRef] [PubMed]
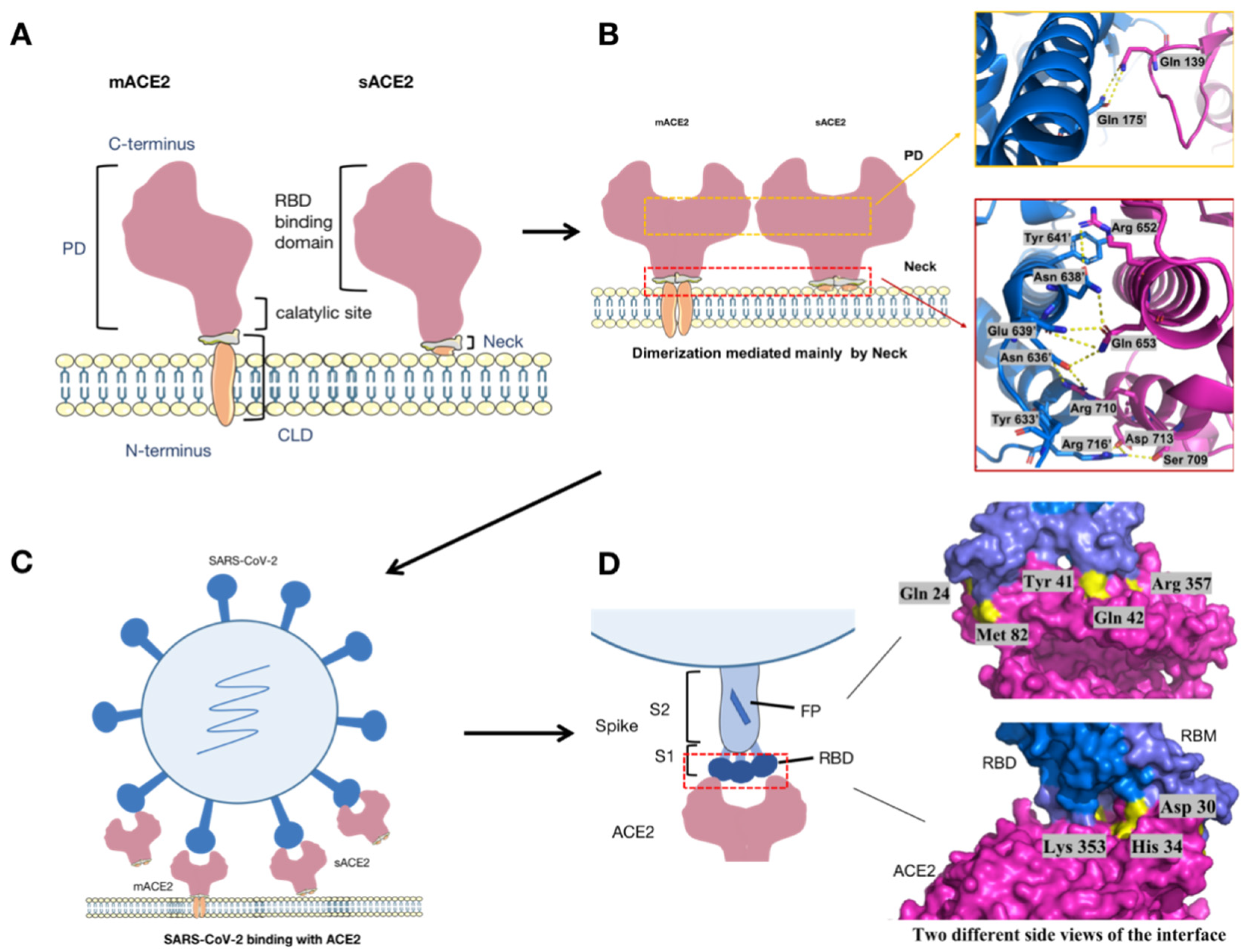

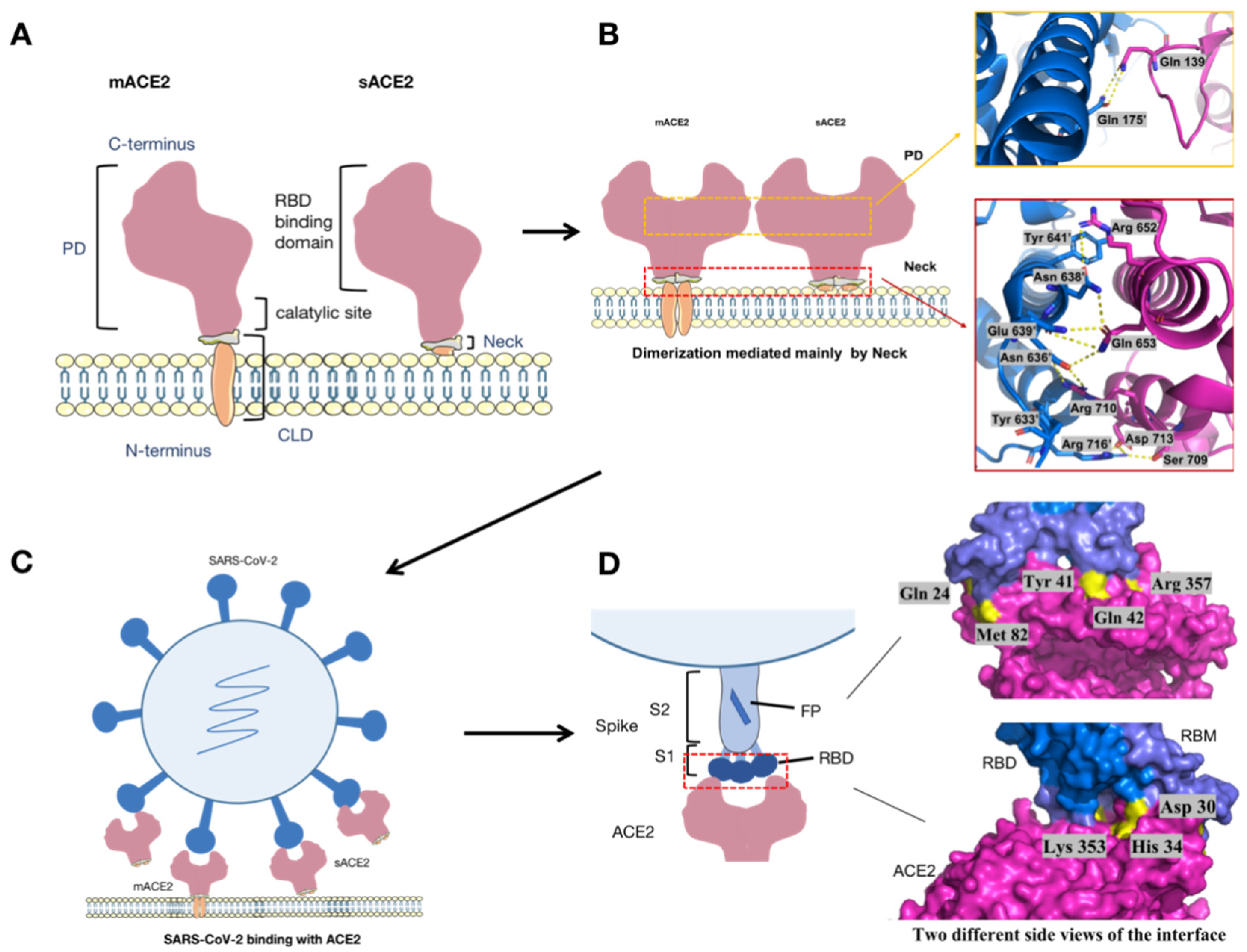{kind=link}
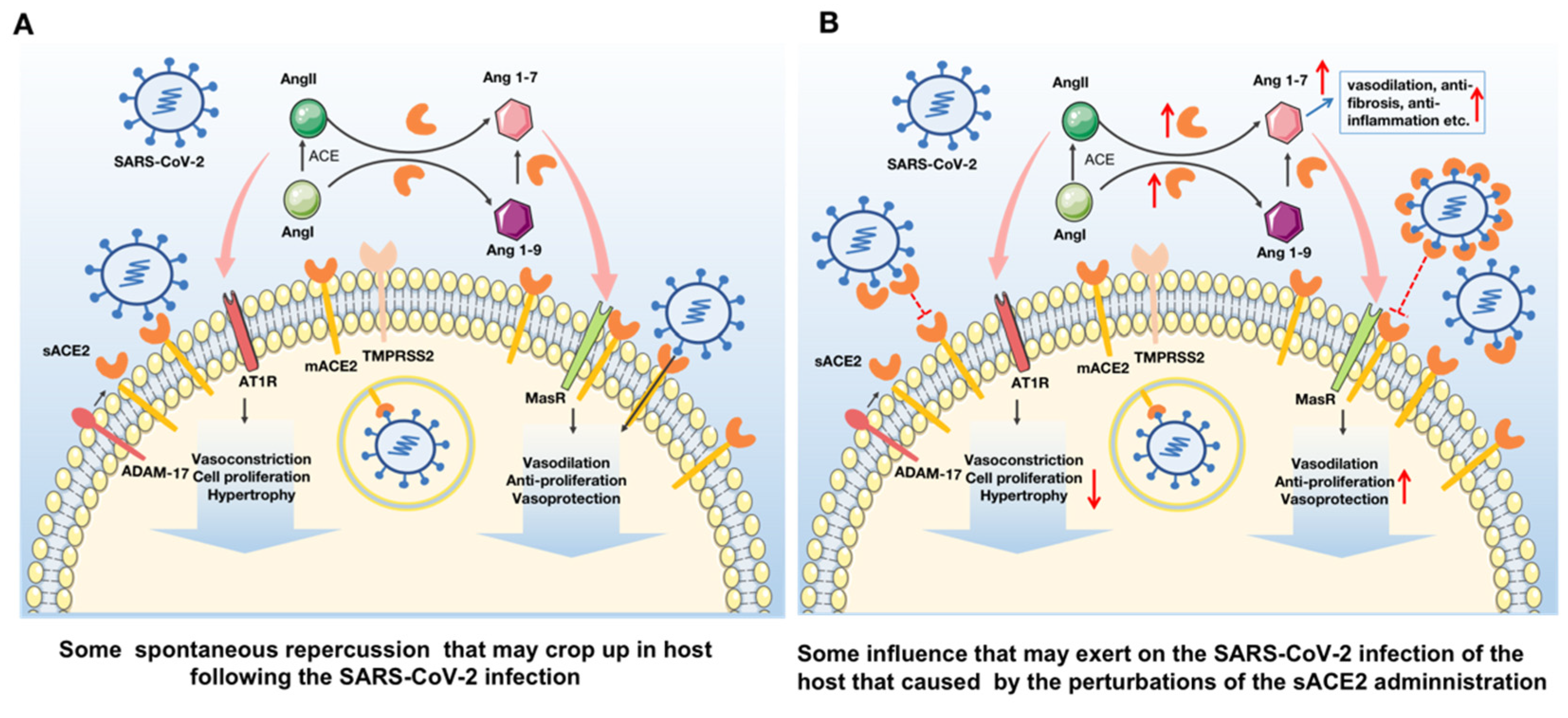
{kind=link}
| sACE2 Optimization Strategy | Name | Animal or Human Trials | Advantages | Disadvantages | References |
|---|---|---|---|---|---|
| Fused with varied Fc | |||||
| Fused with the Fc fragment of human immunoglobulin IgG1 | ACE2-Ig | BALB/c mice exhibit desirable pharmacological properties after a single intravenous dose of the fusion proteins | Enhanced half-life; immunoreactive functions; cross-reactivity against both SARS-CoV and SARS-CoV-2. | Possibility to compromise serum stability or activate FcRγ in myeloid cells | C. Lei. et al. Nat Commun, 11 (2020). |
| Fused with the Fc domain 3 of immunoglobulin (Ig) heavy chain | ACE2 “microbody” | K18-hACE2 mice treated with the ACE2 microbody protein is able to prevent lethal SARS-CoV-2 disease. | Enhanced half-life; smaller than ACE2-Ig. | Possibility to cause antibody-dependent enhancement | T. Tada. et al. Cell Rep, 33 (2020). |
| Linked to a chimeric molecule named VHH or nanobodies | sACE2-anti-CD16 VHH | More evidence in vivo is needed. | Rapid permeation into different tissues; produced in large quantities in prokaryotic and eukaryotic cell lines. | Possibility to cause antibody-dependent enhancement | A. Sheikhi. et al. Hum Vaccin Immunother, 17 (2021). |
| Fused with the N terminal of human IgG-Fc region | hACE2-Fc | Both prophylactic and therapeutic hACE2-Fc treatments effectively protected Ad5-hACE2-transduced BALB/c mice from SARS-CoV-2 infection. | Enhanced half-life. | Possibility to cause antibody-dependent enhancement | Zhang, Z.et al. Cell Discov 7 (2021). |
| Gene engineering | |||||
| A two-stage flexible protein backbone design process | ACE2 variants | More evidence in vivo is needed | Increased affinity | More anti-viral evidence is needed; preparation is cumbersome and expensive | A. Glasgow. et al. Proc Natl Acad Sci U S A, (2020). |
| Deep mutagenesis | sACE22.v2.4 | More evidence in vivo is needed | High affinity; cross-reactivity against diverse SARS-associated beta coronaviruses | More anti-viral evidence isneeded; preparation is cumbersome and expensive | K.K. Chan. et al. Science, 369 (2020). |
| Truncated ACE2 and polypeptide | |||||
| Truncated ACE2 | tACE2 (21-119aa) | More evidence in vivo is needed | Enhanced binding affinity for S protein; improved safety | Antiviral activity remains to be analyzed | Basit, A. et al. J Biomol Struct Dyn, (2020). |
| 23-mer peptide derived from α1-helix (SBP1) | ACE2(21-43aa) | More evidence in vivo is needed | Association with SARS-CoV-2 RBD with low nanomolar affinity | Antiviral activity remains to be analyzed | G. Zhang, S. et al. bioRxiv, (2020). |
| Screened from five antibacterial peptide databases and a chimeric peptide design approach | AC20, AC23, DBP6, and cnCoVP-1- cnCoVP-7 | More evidence in vivo is needed | Ease of synthesis and modifications; low toxicity; high target specificity and selectivity | Antiviral activity remains to be analyzed | Barh, D. et al. F1000 Research 9 (2020). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, F.; Chen, J.; Zhao, J.; Li, Y.; Li, M.; Sun, C. Killing Two Birds with One Stone by Administration of Soluble ACE2: A Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection. Viruses 2021, 13, 2243. https://doi.org/10.3390/v13112243
Feng F, Chen J, Zhao J, Li Y, Li M, Sun C. Killing Two Birds with One Stone by Administration of Soluble ACE2: A Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection. Viruses. 2021; 13(11):2243. https://doi.org/10.3390/v13112243
Chicago/Turabian StyleFeng, Fengling, Jiaoshan Chen, Jin Zhao, Yanjun Li, Minchao Li, and Caijun Sun. 2021. "Killing Two Birds with One Stone by Administration of Soluble ACE2: A Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection" Viruses 13, no. 11: 2243. https://doi.org/10.3390/v13112243
APA StyleFeng, F., Chen, J., Zhao, J., Li, Y., Li, M., & Sun, C. (2021). Killing Two Birds with One Stone by Administration of Soluble ACE2: A Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection. Viruses, 13(11), 2243. https://doi.org/10.3390/v13112243