
Molecular Docking and Virtual Screening of an Influenza Virus Inhibitor That Disrupts Protein–Protein Interactions

Abstract
1. Introduction
2. Materials and Methods
2.1. Pretreatment of the PB1 and PB2 Proteins of the Influenza Virus
2.2. Ligand Preparation
2.3. Molecular Docking
2.4. Simulation of Molecular Dynamics
2.5. Calculation of the Binding Free Energy
2.6. Binding Force Analysis
2.7. ADMET Prediction
3. Results
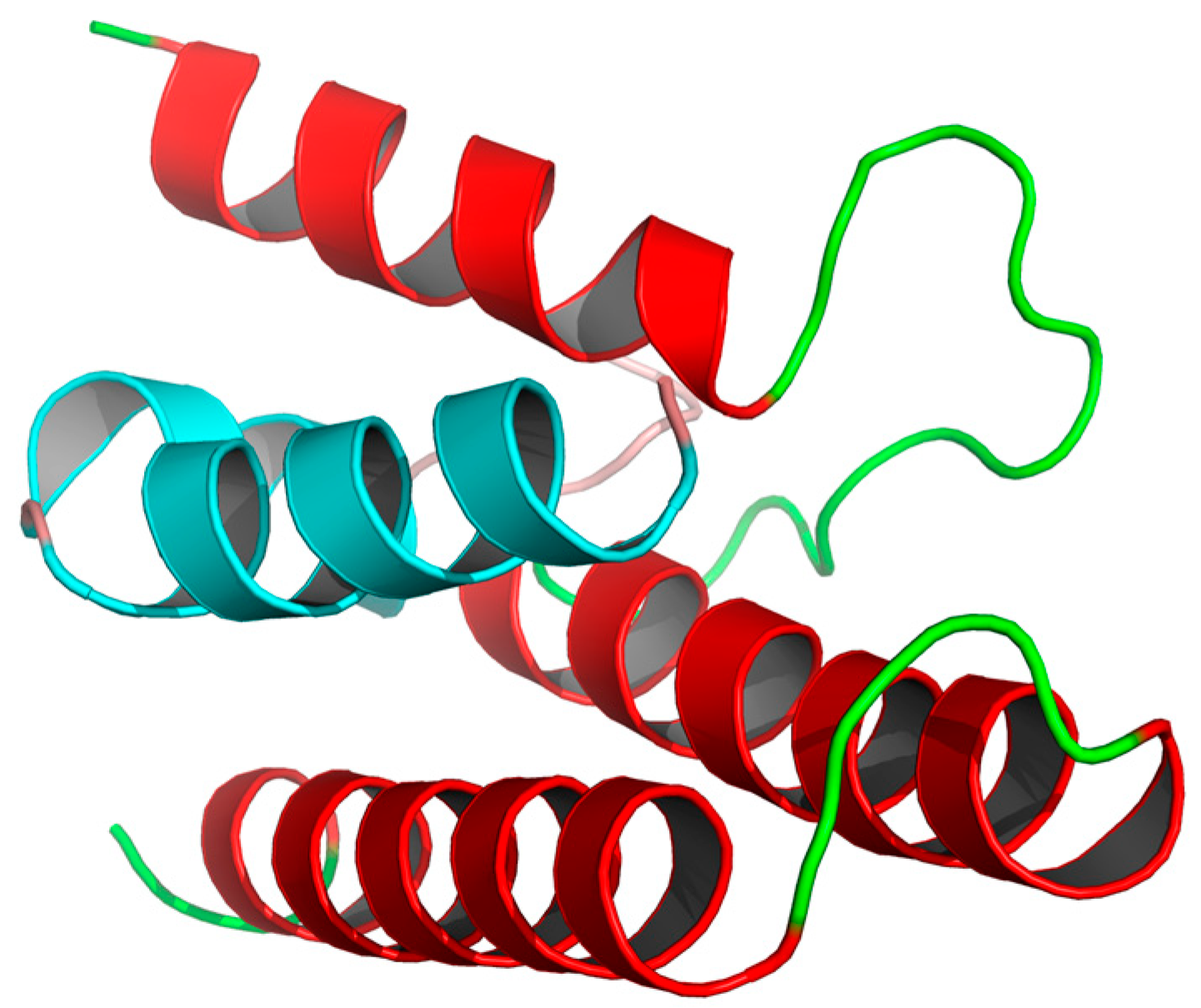
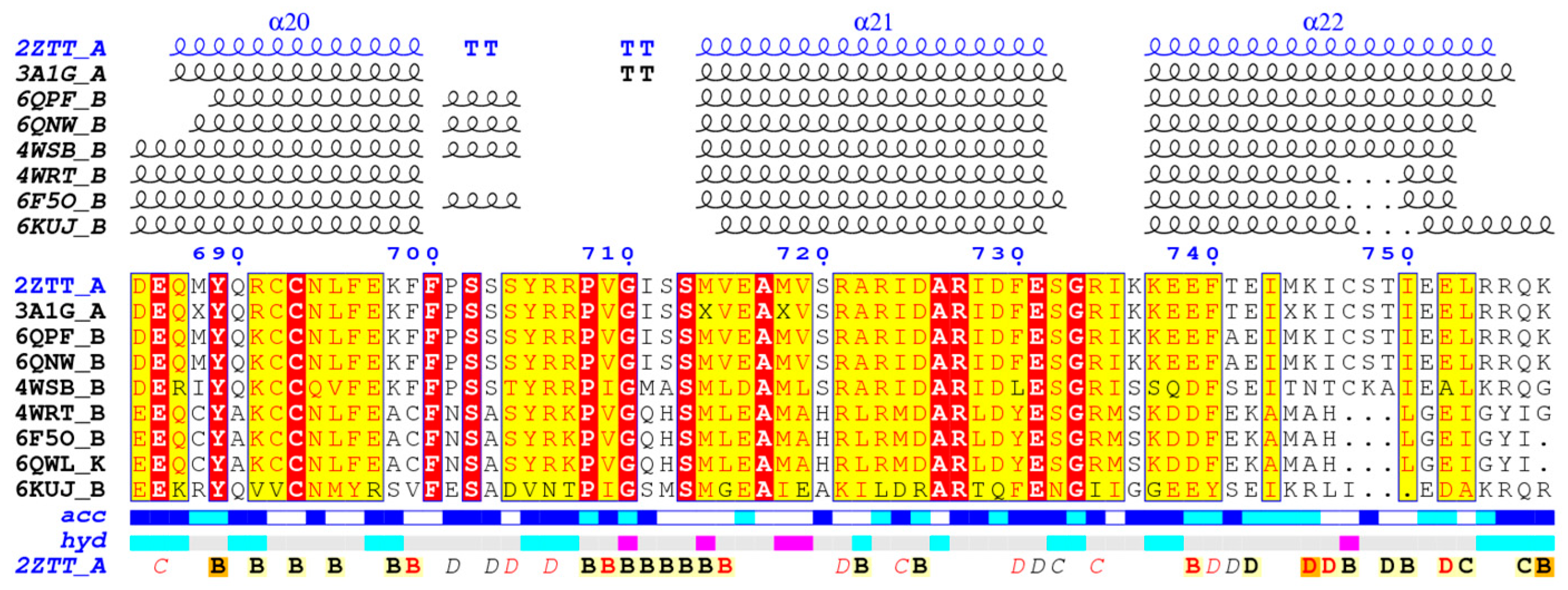
3.1. Protein Preparation
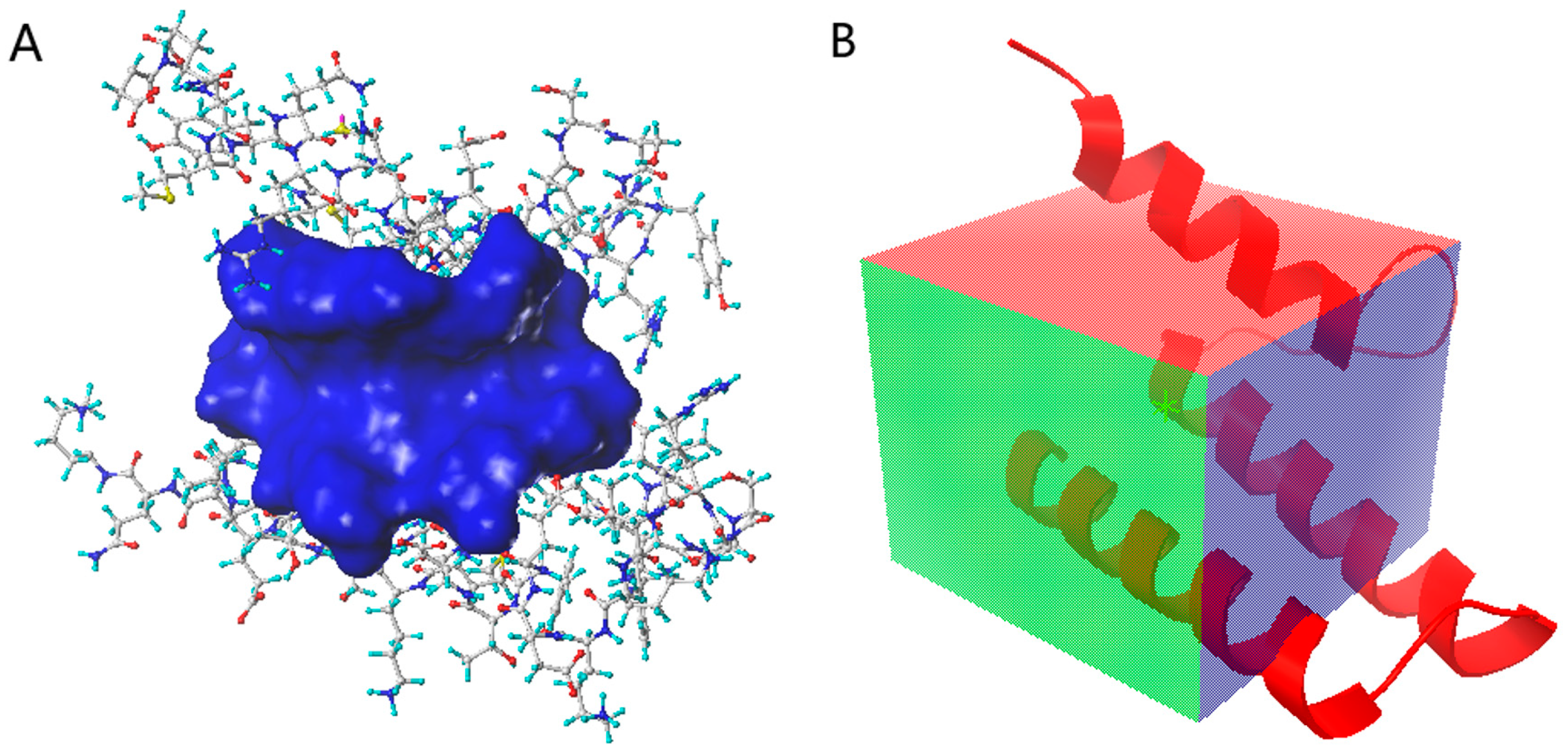
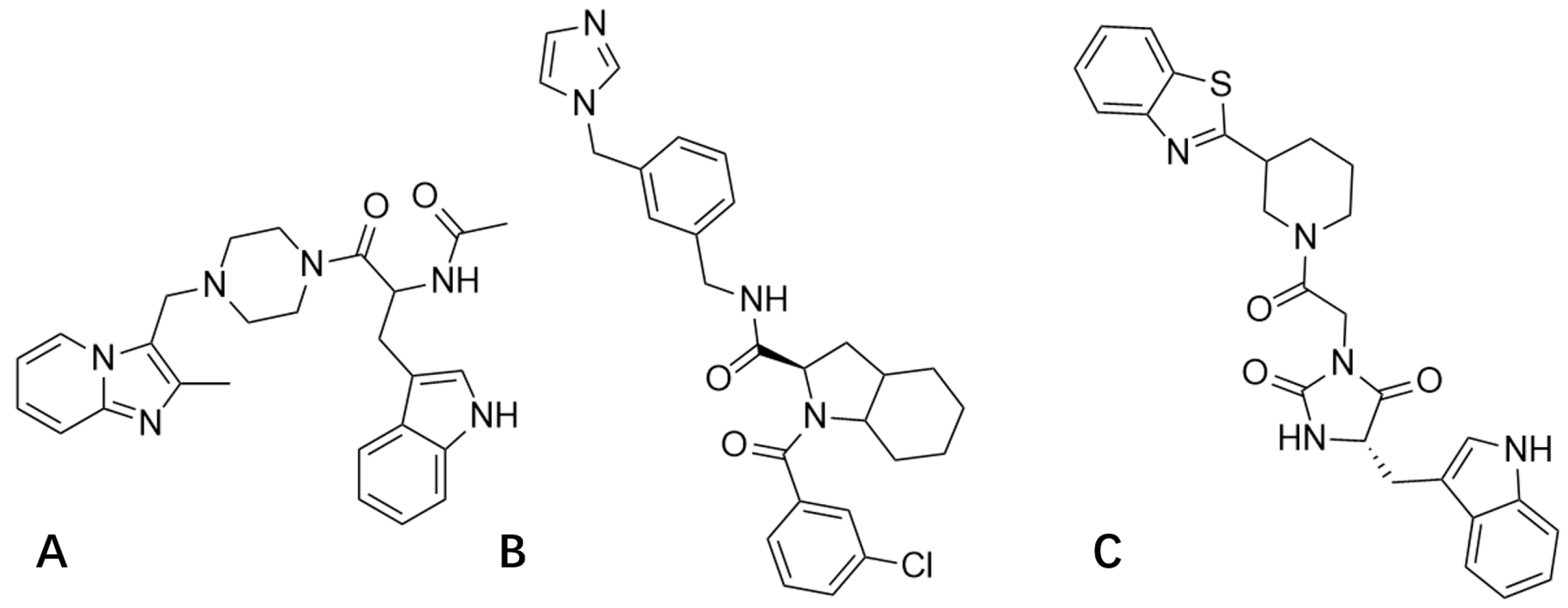
3.2. Molecular Docking
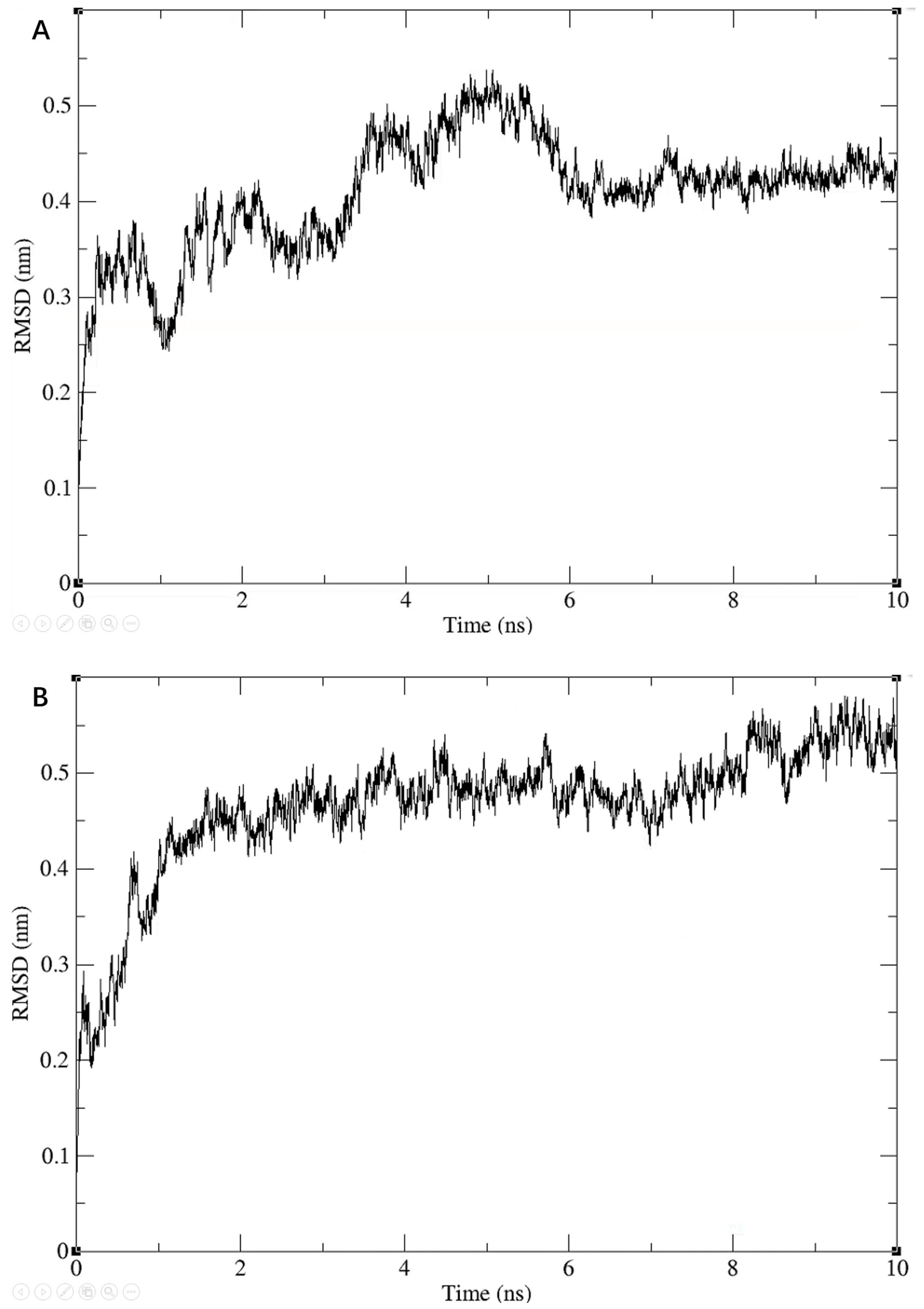
3.3. Molecular Dynamics
3.4. Calculation of the Binding Free Energy
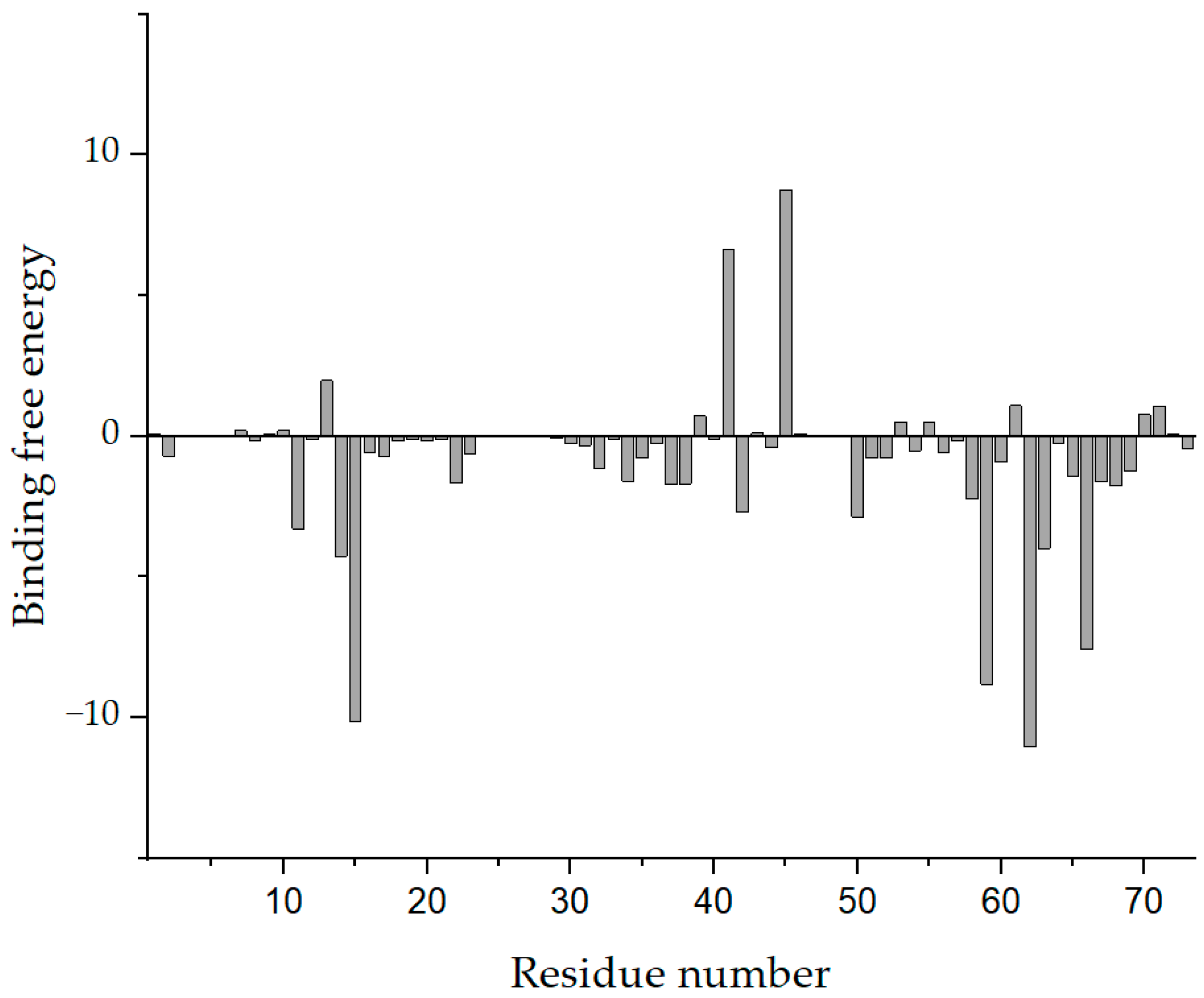
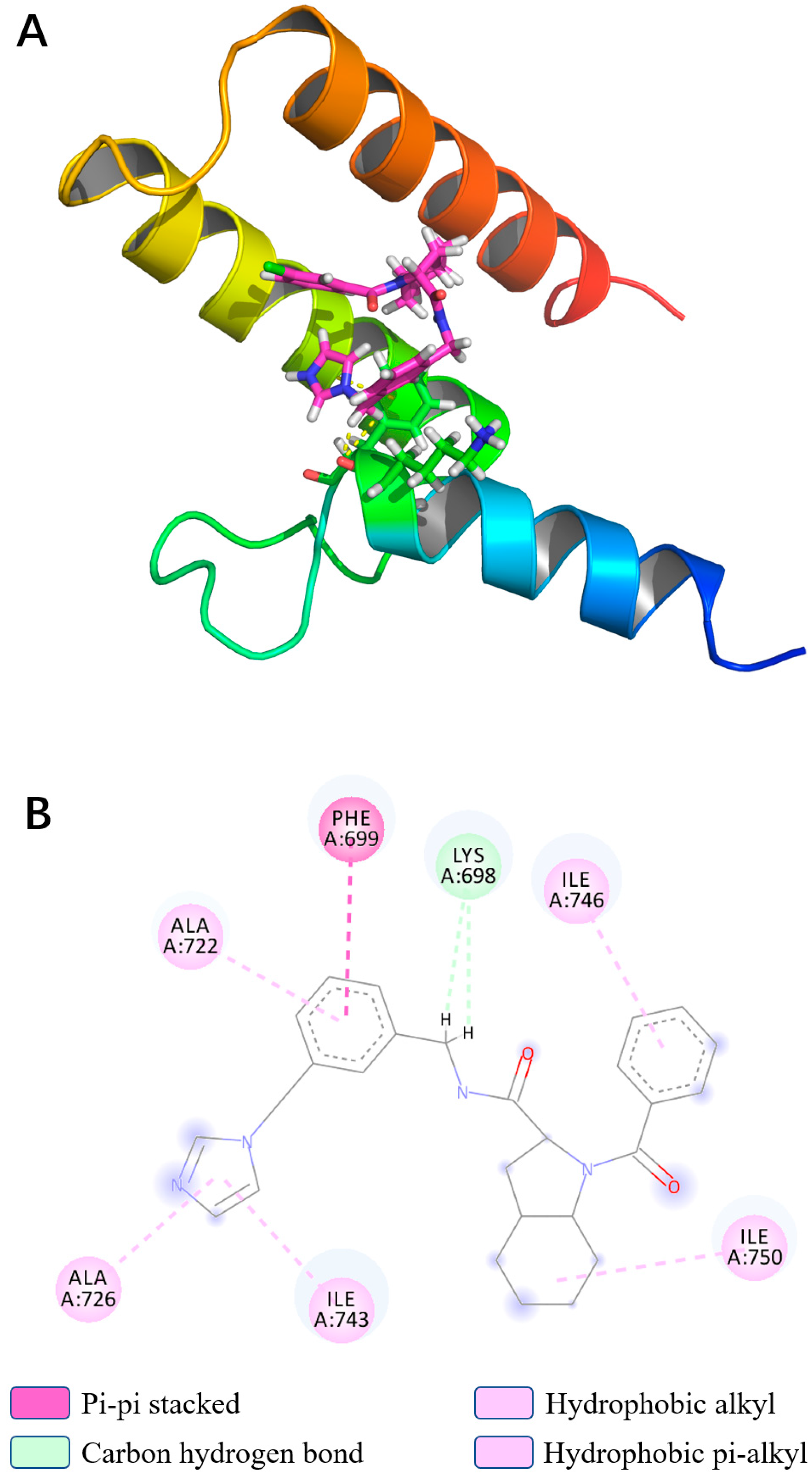
3.5. Binding Force Analysis
3.6. ADMET Predictions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Leung, Y.-H.; To, M.-K.; Lam, T.-S.; Yau, S.-W.; Leung, O.-S.; Chuang, S.-K. Epidemiology of human influenza A(H7N9) infection in Hong Kong. J. Microbiol. Immunol. Infect. 2017, 50, 183–188. [Google Scholar] [CrossRef]
- Dkhar, H.K.; Gopalsamy, A.; Loharch, S.; Kaur, A.; Bhutani, I.; Saminathan, K.; Bhagyaraj, E.; Chandra, V.; Swaminathan, K.; Agrawal, P.; et al. Discovery of Mycobacterium tuberculosis α-1,4-Glucan Branching Enzyme (GlgB) Inhibitors by Structure- and Ligand-based Virtual Screening. J. Biol. Chem. 2015, 290, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Forrest, H.L.; Webster, R.G. Perspectives on influenza evolution and the role of research. Anim. Health Res. Rev. 2010, 11, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Hay, A.J.; Gregory, V.; Douglas, A.R.; Lin, Y.P. The evolution of human influenza viruses. Philos. Trans. R. Soc. B Biol. Sci. 2001, 356, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Brooke Christopher, B.; Pierson Ted, C. Population Diversity and Collective Interactions during Influenza Virus Infection. J. Virol. 2017, 91, e01164-17. [Google Scholar]
- Matsuzaki, Y.; Katsushima, N.; Nagai, Y.; Shoji, M.; Itagaki, T.; Sakamoto, M.; Kitaoka, S.; Mizuta, K.; Nishimura, H. Clinical Features of Influenza C Virus Infection in Children. J. Infect. Dis. 2006, 193, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Asha, K.; Kumar, B. Emerging Influenza D Virus Threat: What We Know so Far! J. Clin. Med. 2019, 8, 192. [Google Scholar] [CrossRef]
- Uyeki, T.M.; Bernstein, H.H.; Bradley, J.S.; Englund, J.A.; File, T.M., Jr.; Fry, A.M.; Gravenstein, S.; Hayden, F.G.; Harper, S.A.; Hirshon, J.M.; et al. Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenza. Clin. Infect. Dis. 2019, 68, e1–e47. [Google Scholar] [CrossRef]
- Jin, Z.; Wang, Y.; Yu, X.-F.; Tan, Q.-Q.; Liang, S.-S.; Li, T.; Zhang, H.; Shaw, P.-C.; Wang, J.; Hu, C. Structure-based virtual screening of influenza virus RNA polymerase inhibitors from natural compounds: Molecular dynamics simulation and MM-GBSA calculation. Comput. Biol. Chem. 2020, 85, 107241. [Google Scholar] [CrossRef]
- Deyde, V.M.; Xu, X.; Bright, R.A.; Shaw, M.; Smith, C.B.; Zhang, Y.; Shu, Y.; Gubareva, L.V.; Cox, N.J.; Klimov, A.I. Surveillance of Resistance to Adamantanes among Influenza A(H3N2) and A(H1N1) Viruses Isolated Worldwide. J. Infect. Dis. 2007, 196, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Global Influenza Strategy 2019–2030. Geneva: World Health Organization; 2019. License: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) Data. Available online: http://apps.who.int/iris.
- Suzuki, Y.; Saito, R.; Zaraket, H.; Dapat, C.; Caperig-Dapat, I.; Suzuki, H. Rapid and Specific Detection of Amantadine-Resistant Influenza A Viruses with a Ser31Asn Mutation by the Cycling Probe Method. J. Clin. Microbiol. 2010, 48, 57–63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bright, R.A.; Shay, D.K.; Shu, B.; Cox, N.J.; Klimov, A.I. Adamantane Resistance Among Influenza A Viruses Isolated Early During the 2005–2006 Influenza Season in the United States. JAMA 2006, 295, 891–894. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.H.; Song, M.-S.; Lee, E.-Y.; Kim, Y.-I.; Kim, E.-H.; Park, S.-J.; Park, K.J.; Kwon, H.-I.; Pascua, P.N.Q.; Lim, G.-J.; et al. Profiling and Characterization of Influenza Virus N1 Strains Potentially Resistant to Multiple Neuraminidase Inhibitors. J. Virol. 2014, 89, 287–299. [Google Scholar] [CrossRef]
- Hussain, M.; Galvin, H.D.; Haw, T.Y.; Nutsford, A.N.; Husain, M. Drug resistance in influenza A virus: The epidemiology and management. Infect. Drug Resist. 2017, 10, 121–134. [Google Scholar] [CrossRef]
- Herz, C.; Stavnezer, E.; Krug, R.M.; Gurney, T. Influenza virus, an RNA virus, synthesizes its messenger RNA in the nucleus of infected cells. Cell 1981, 26, 391–400. [Google Scholar] [CrossRef]
- York, A.; Fodor, E. Biogenesis, assembly, and export of viral messenger ribonucleoproteins in the influenza A virus infected cell. RNA Biol. 2013, 10, 1274–1282. [Google Scholar] [CrossRef]
- Massari, S.; Goracci, L.; Desantis, J.; Tabarrini, O. Polymerase Acidic Protein–Basic Protein 1 (PA–PB1) Protein–Protein Interaction as a Target for Next-Generation Anti-influenza Therapeutics. J. Med. Chem. 2016, 59, 7699–7718. [Google Scholar] [CrossRef]
- Noble, E.; Cox, A.; Deval, J.; Kim, B. Endonuclease substrate selectivity characterized with full-length PA of influenza A virus polymerase. Virology 2012, 433, 27–34. [Google Scholar] [CrossRef]
- Trist, I.M.L.; Nannetti, G.; Tintori, C.; Fallacara, A.L.; Deodato, D.; Mercorelli, B.; Palù, G.; Wijtmans, M.; Gospodova, T.; Edink, E.; et al. 4,6-Diphenylpyridines as Promising Novel Anti-Influenza Agents Targeting the PA–PB1 Protein–Protein Interaction: Structure–Activity Relationships Exploration with the Aid of Molecular Modeling. J. Med. Chem. 2016, 59, 2688–2703. [Google Scholar] [CrossRef]
- Massari, S.; Nannetti, G.; Desantis, J.; Muratore, G.; Sabatini, S.; Manfroni, G.; Mercorelli, B.; Cecchetti, V.; Palù, G.; Cruciani, G.; et al. A Broad Anti-influenza Hybrid Small Molecule That Potently Disrupts the Interaction of Polymerase Acidic Protein–Basic Protein 1 (PA-PB1) Subunits. J. Med. Chem. 2015, 58, 3830–3842. [Google Scholar] [CrossRef]
- Guu, T.S.; Dong, L.; Wittung-Stafshede, P.; Tao, Y.J. Mapping the domain structure of the influenza A virus polymerase acidic protein (PA) and its interaction with the basic protein 1 (PB1) subunit. Virology 2008, 379, 135–142. [Google Scholar] [CrossRef]
- Sable, R.; Jois, S.D.S. Surfing the Protein-Protein Interaction Surface Using Docking Methods: Application to the Design of PPI Inhibitors. Molecules 2015, 20, 11569–11603. [Google Scholar] [CrossRef]
- Chase, G.; Wunderlich, K.; Reuther, P.; Schwemmle, M. Identification of influenza virus inhibitors which disrupt of viral polymerase protein–protein interactions. Methods 2011, 55, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.L.; Medcalf, L.; Elton, D.; Digard, P. Evidence that the C-terminal PB2-binding region of the influenza A virus PB1 protein is a discrete α-helical domain. FEBS Lett. 2007, 581, 5300–5306. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, T.; Adyshev, D.M.; Kobayashi, M.; Iwata, A.; Ishihama, A. Molecular Assembly of the Influenza Virus RNA Polymerase: Determination of the Subunit-Subunit Contact Sites. J. Gen. Virol. 1996, 77, 2149–2157. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.; Zurcher, T.; Ortin, J. Identification of Two Separate Domains in the Influenza Virus PB1 Protein Involved in the Interaction with the PB2 and PA Subunits: A Model for the Viral RNA Polymerase Structure. Nucleic Acids Res. 1996, 24, 4456–4463. [Google Scholar] [CrossRef]
- Perales, B.; de la Luna, S.; Palacios, I.; Ortin, J. Mutational analysis identifies functional domains in the influenza A virus PB2 polymerase subunit. J. Virol. 1996, 70, 1678–1686. [Google Scholar] [CrossRef] [PubMed]
- Reuther, P.; Mänz, B.; Brunotte, L.; Schwemmle, M.; Wunderlich, K. Targeting of the Influenza A Virus Polymerase PB1-PB2 Interface Indicates Strain-Specific Assembly Differences. J. Virol. 2011, 85, 13298–13309. [Google Scholar] [CrossRef]
- Bárcena, J.; Ochoa, M.; de la Luna, S.; Melero, J.A.; Nieto, A.; Ortín, J.; Portela, A. Monoclonal antibodies against influenza virus PB2 and NP polypeptides interfere with the initiation step of viral mRNA synthesis in vitro. J. Virol. 1994, 68, 6900–6909. [Google Scholar] [CrossRef]
- Ochoa, M.; Bárcena, J.; de la Luna, S.; Melero, J.; Douglas, A.; Nieto, A.; Ortín, J.; Skehel, J.; Portela, A. Epitope mapping of cross-reactive monoclonal antibodies specific for the influenza A virus PA and PB2 polypeptides. Virus Res. 1995, 37, 305–315. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015.
- Sugiyama, K.; Obayashi, E.; Kawaguchi, A.; Suzuki, Y.; Tame, J.R.H.; Nagata, K.; Park, S.-Y. Structural insight into the essential PB1–PB2 subunit contact of the influenza virus RNA polymerase. EMBO J. 2009, 28, 1803–1811. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G. Open Babel: An open chemical toolbox. J. Chemininform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15–Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Berendsen, H.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Qiu, Y.; Baron, R.; Molinero, V. Coarse-Graining of TIP4P/2005, TIP4P-Ew, SPC/E, and TIP3P to Monatomic Anisotropic Water Models Using Relative Entropy Minimization. J. Chem. Theory Comput. 2014, 10, 4104–4120. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE-AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Grubmüller, H.; Heller, H.; Windemuth, A.; Schulten, K. Generalized Verlet Algorithm for Efficient Molecular Dynamics Simulations with Long-range Interactions. Mol. Simul. 1991, 6, 121–142. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2017, 27, 112–128. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB | Favored | Allowed | Outliers |
|---|---|---|---|
| 2ZTT | 201 (96%) | 9 (4%) | 0 (0%) |
| 3A1G | 209 (98%) | 4 (2%) | 1 (0%) |
| Compound | Score (Sybyl) | Binding Energies (Vina) | Score (Discovery Studio) |
|---|---|---|---|
| Compound 6320 | 5.4247 | −8.5 | 100.215 |
| Compound 13815 | 6.4074 | −8.1 | 100.972 |
| Compound 8770 | 6.6121 | −8.1 | 91.1954 |
| Compound 979 | 6.2028 | −7.9 | 103.837 |
| Compound 15672 | 6.2651 | −7.9 | 101.386 |
| Compound 11902 | 7.0193 | −7.8 | 117.427 |
| Compound 7679 | 6.3579 | −7.8 | 111.038 |
| Compound 4804 | 6.4706 | −7.8 | 110.254 |
| Compound 3206 | 7.3773 | −7.8 | 109.4 |
| Compound 637 | 6.0599 | −7.8 | 104.964 |
| Compound 5068 | 6.1561 | −7.8 | 104.127 |
| Compound 17948 | 6.3089 | −7.8 | 104.063 |
| Compound 13816 | 5.2263 | −7.8 | 100.972 |
| Compound 3165 | 7.8442 | −7.8 | 100.56 |
| Compound 6322 | 5.6455 | −7.8 | 99.0563 |
| Compound 12538 | 6.5092 | −7.7 | 120.272 |
| Compound 4808 | 7.5041 | −7.7 | 110.254 |
| Compound 15144 | 7.8586 | −7.7 | 109.751 |
| Compound 8836 | 6.4969 | −7.7 | 105.816 |
| Compound 15026 | 6.6115 | −7.7 | 105.675 |
| Compound 11365 | 7.7766 | −7.7 | 105.223 |
| Compound 4560 | 6.5334 | −7.7 | 103.375 |
| Compound 11915 | 6.8264 | −7.7 | 103.172 |
| Compound 8721 | 7.5264 | −7.7 | 101.89 |
| Compound 6158 | 6.5414 | −7.7 | 100.276 |
| Compound 13727 | 6.8292 | −7.7 | 97.5998 |
| Compound 5201 | 6.4911 | −7.7 | 97.0757 |
| Compound 12269 | 6.0082 | −7.7 | 96.9189 |
| Compound 1451 | 5.4295 | −7.7 | 96.7586 |
| Compound 118 | 5.4985 | −7.7 | 92.4782 |
| Compound 11904 | 7.6596 | −7.6 | 117.427 |
| Compound 8631 | 5.9089 | −7.6 | 111.328 |
| Compound 943 | 6.0759 | −7.6 | 110.615 |
| Compound 7267 | 6.469 | −7.6 | 108.918 |
| Compound 13884 | 6.6642 | −7.6 | 108.747 |
| Compound 9555 | 7.4359 | −7.6 | 106.817 |
| Compound 9561 | 7.1219 | −7.6 | 106.817 |
| Compound 19179 | 5.9982 | −7.6 | 105.181 |
| Compound 13047 | 6.6693 | −7.6 | 104.76 |
| Compound 9599 | 6.2425 | −7.6 | 103.66 |
| Compound 4108 | 5.6692 | −7.6 | 101.636 |
| Compound 6159 | 6.5045 | −7.6 | 100.276 |
| Compound 10747 | 6.4367 | −7.6 | 99.8972 |
| Compound 10746 | 6.3898 | −7.6 | 99.8972 |
| Compound 1481 | 7.3196 | −7.6 | 99.869 |
| Compound 13726 | 5.6056 | −7.6 | 99.346 |
| Compound 9150 | 5.5108 | −7.6 | 98.0525 |
| Compound 5609 | 6.8803 | −7.6 | 97.5804 |
| Compound 12672 | 6.9693 | −7.6 | 97.5469 |
| Compound 8636 | 7.6168 | −7.6 | 97.3231 |
| Compound 8632 | 6.4931 | −7.5 | 111.328 |
| Compound 10818 | 6.6349 | −7.5 | 110.749 |
| Compound 2100 | 7.1846 | −7.5 | 110.593 |
| Compound 5242 | 6.8628 | −7.5 | 110.566 |
| Compound 6845 | 5.7634 | −7.5 | 109.107 |
| Compound 7266 | 5.8177 | −7.5 | 108.918 |
| Compound 13888 | 6.9957 | −7.5 | 108.747 |
| Compound 9012 | 7.4817 | −7.5 | 108.074 |
| Compound 1095 | 6.5534 | −7.5 | 105.985 |
| Compound 1832 | 7.5959 | −7.5 | 103.958 |
| Compound 2510 | 8.1886 | −7.5 | 103.477 |
| Compound 1947 | 6.3243 | −7.5 | 101.659 |
| Compound 8626 | 5.7178 | −7.5 | 100.117 |
| Compound 5611 | 6.3159 | −7.5 | 98.6665 |
| Compound 104 | 6.3199 | −7.5 | 98.2362 |
| Compound 105 | 6.2256 | −7.5 | 98.2362 |
| Compound 9149 | 5.8438 | −7.5 | 98.0525 |
| Compound 10315 | 6.0586 | −7.5 | 97.5558 |
| Compound 10316 | 5.8198 | −7.5 | 97.5558 |
| Compound 9392 | 5.9734 | −7.5 | 97.3548 |
| Compound 7142 | 5.2493 | −7.5 | 97.0345 |
| Compound 15055 | 7.1738 | −7.5 | 95.3654 |
| Compound 6657 | 5.2667 | −7.5 | 93.2045 |
| Compound 8194 | 5.5902 | −7.5 | 91.8273 |
| Compound 15053 | 6.5572 | −7.5 | 90.1132 |
| Compound 2289 | 6.4862 | −7.4 | 113.683 |
| Compound 7678 | 5.6392 | −7.4 | 111.038 |
| Compound 2170 | 8.3252 | −7.40 | 110.926 |
| Compound 9481 | 7.9091 | −7.4 | 108.32 |
| Compound 15029 | 6.2396 | −7.4 | 105.675 |
| Compound 11630 | 5.4344 | −7.4 | 104.942 |
| Compound 13048 | 7.7122 | −7.4 | 104.76 |
| Compound 13049 | 7.67 | −7.4 | 104.76 |
| Compound 8224 | 5.02 | −7.4 | 104.605 |
| Compound 1831 | 7.7449 | −7.4 | 103.958 |
| Compound 1058 | 6.2938 | −7.4 | 103.642 |
| Compound 2638 | 6.1372 | −7.4 | 102.785 |
| Compound 3836 | 5.926 | −7.4 | 102.65 |
| Compound 2950 | 5.833 | −7.4 | 99.3493 |
| Compound 7656 | 6.9155 | −7.4 | 98.4305 |
| Compound 120 | 5.7108 | −7.4 | 97.1121 |
| Compound 10134 | 6.1476 | −7.4 | 96.6006 |
| Compound 1242 | 5.4844 | −7.3 | 109.72 |
| Compound 3813 | 7.5651 | −7.3 | 107.587 |
| Compound 12305 | 5.3583 | −7.3 | 98.7621 |
| Compound 2046 | 6.6877 | −7.3 | 98.6504 |
| Compound 5098 | 7.0632 | −7.3 | 98.1682 |
| Compound 11824 | 5.3915 | −7.3 | 98.022 |
| Compound 11612 | 7.2281 | −7.3 | 94.3189 |
| Compound 2158 | 7.3805 | −7.3 | 91.2644 |
| Compound No. | Binding Free Energy | PB Energy | SA Energy | Electrostatic Attraction | Van der Waals Force |
|---|---|---|---|---|---|
| 3206 | −128.049 | 89.613 | −21.492 | −39.663 | −156.507 |
| 4808 | −122.761 | 62.573 | −20.492 | 1.721 | −166.564 |
| 15144 | −106.93 | 30.743 | −16.627 | −9.602 | −111.444 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, Y.; Long, S.; Cao, S. Molecular Docking and Virtual Screening of an Influenza Virus Inhibitor That Disrupts Protein–Protein Interactions. Viruses 2021, 13, 2229. https://doi.org/10.3390/v13112229
Ren Y, Long S, Cao S. Molecular Docking and Virtual Screening of an Influenza Virus Inhibitor That Disrupts Protein–Protein Interactions. Viruses. 2021; 13(11):2229. https://doi.org/10.3390/v13112229
Chicago/Turabian StyleRen, Yixin, Sihui Long, and Shuang Cao. 2021. "Molecular Docking and Virtual Screening of an Influenza Virus Inhibitor That Disrupts Protein–Protein Interactions" Viruses 13, no. 11: 2229. https://doi.org/10.3390/v13112229
APA StyleRen, Y., Long, S., & Cao, S. (2021). Molecular Docking and Virtual Screening of an Influenza Virus Inhibitor That Disrupts Protein–Protein Interactions. Viruses, 13(11), 2229. https://doi.org/10.3390/v13112229