
Spike Proteins of SARS-CoV-2 Induce Pathological Changes in Molecular Delivery and Metabolic Function in the Brain Endothelial Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Immunofluorescence Assay (IFA)
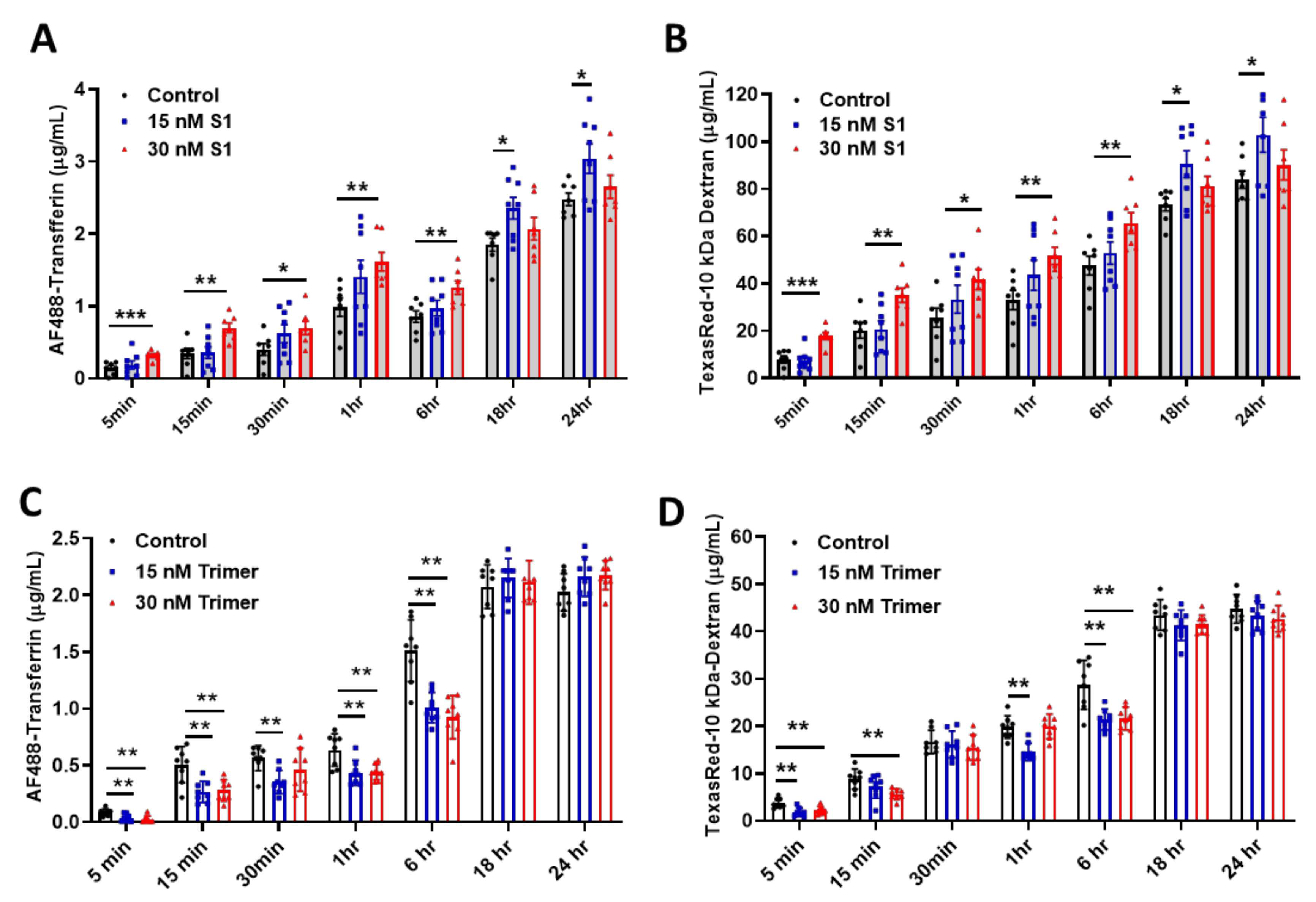
2.4. In Vitro BBB Permeability Assay
2.5. Live-Cell Metabolic Assay
2.6. TUNEL Apotosis Assay
2.7. Statistical Analysis
3. Results
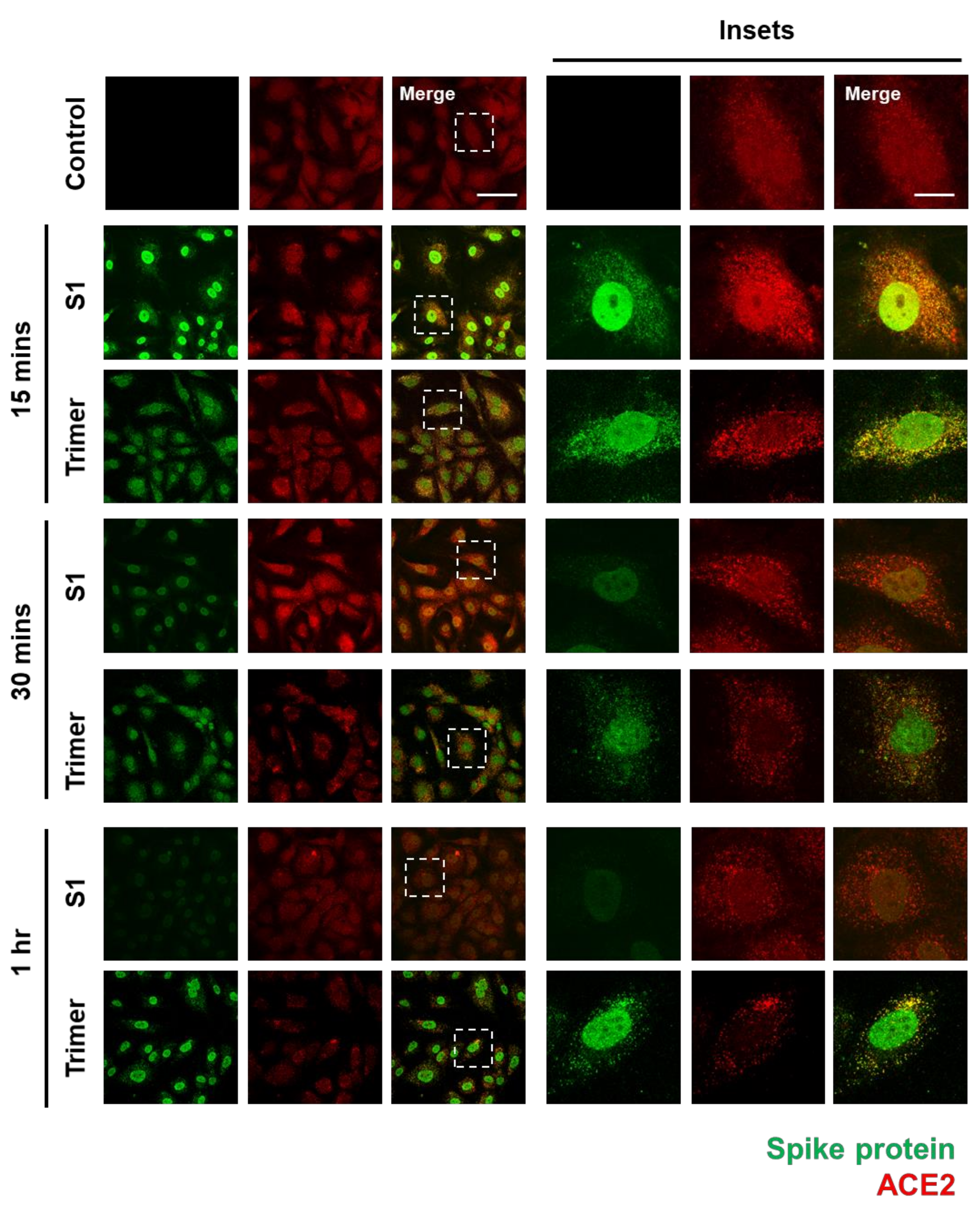
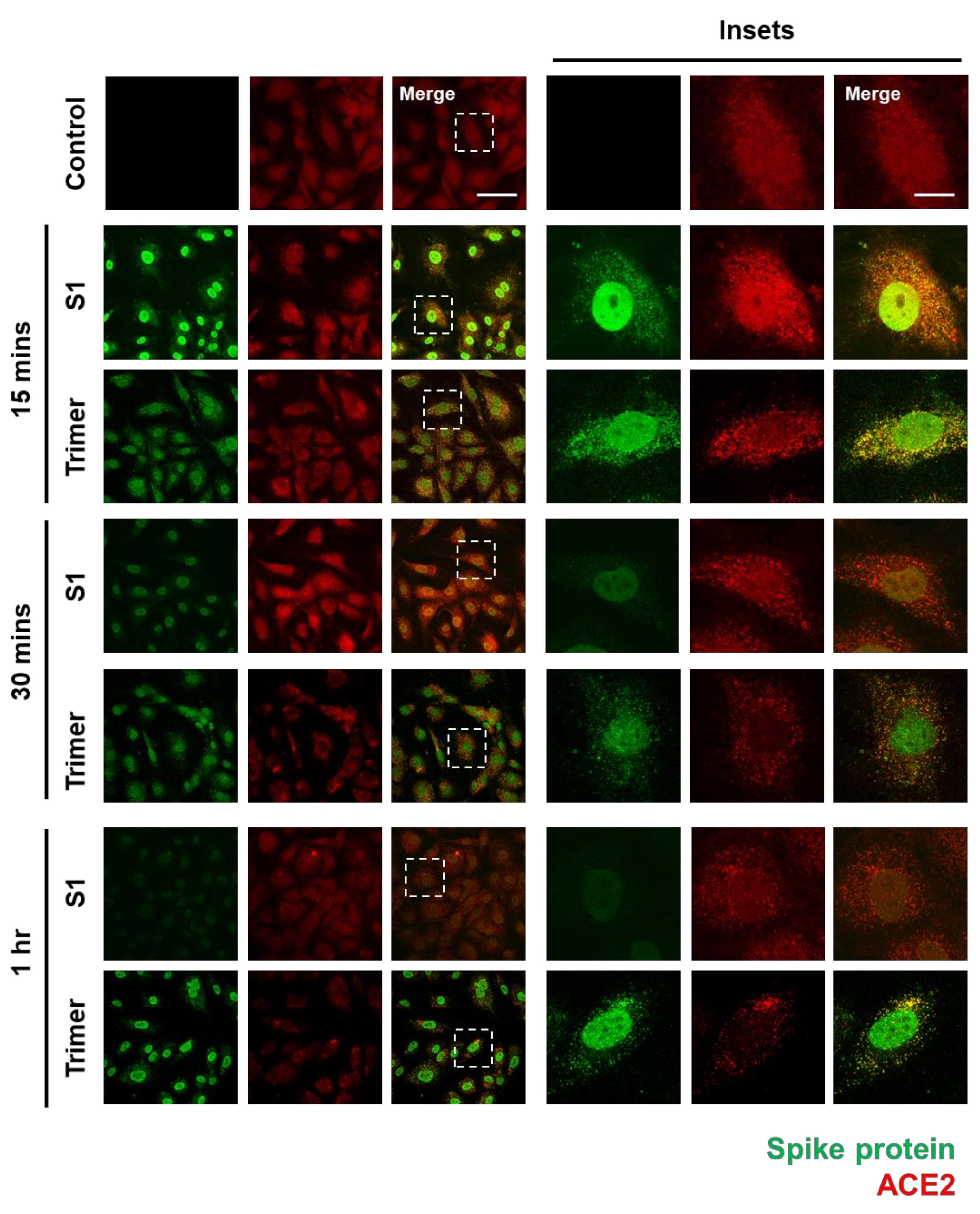
3.1. Spike Protein of SARS-CoV-2 Enters the Brain ECs and Shows Strong Nuclear Localization
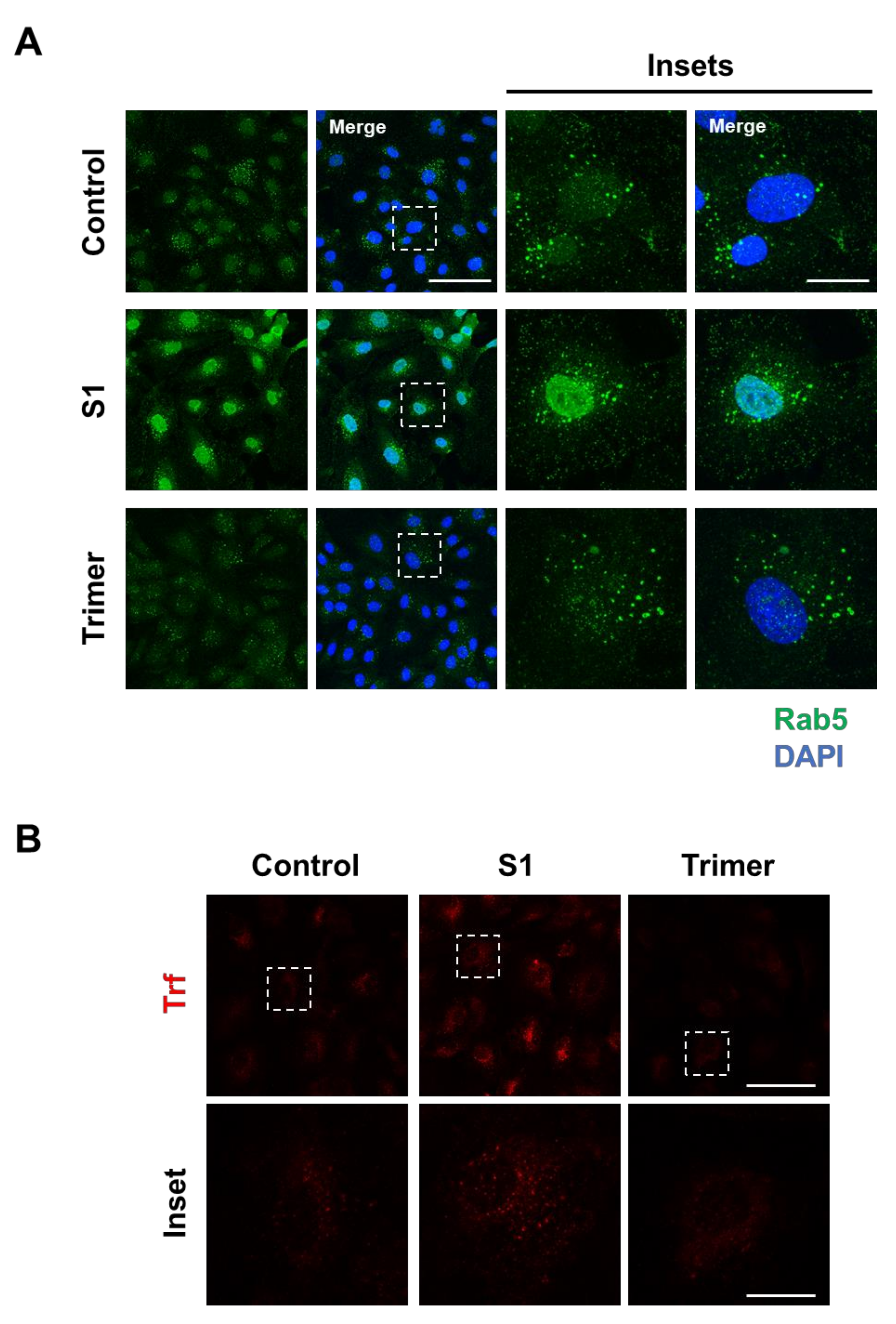
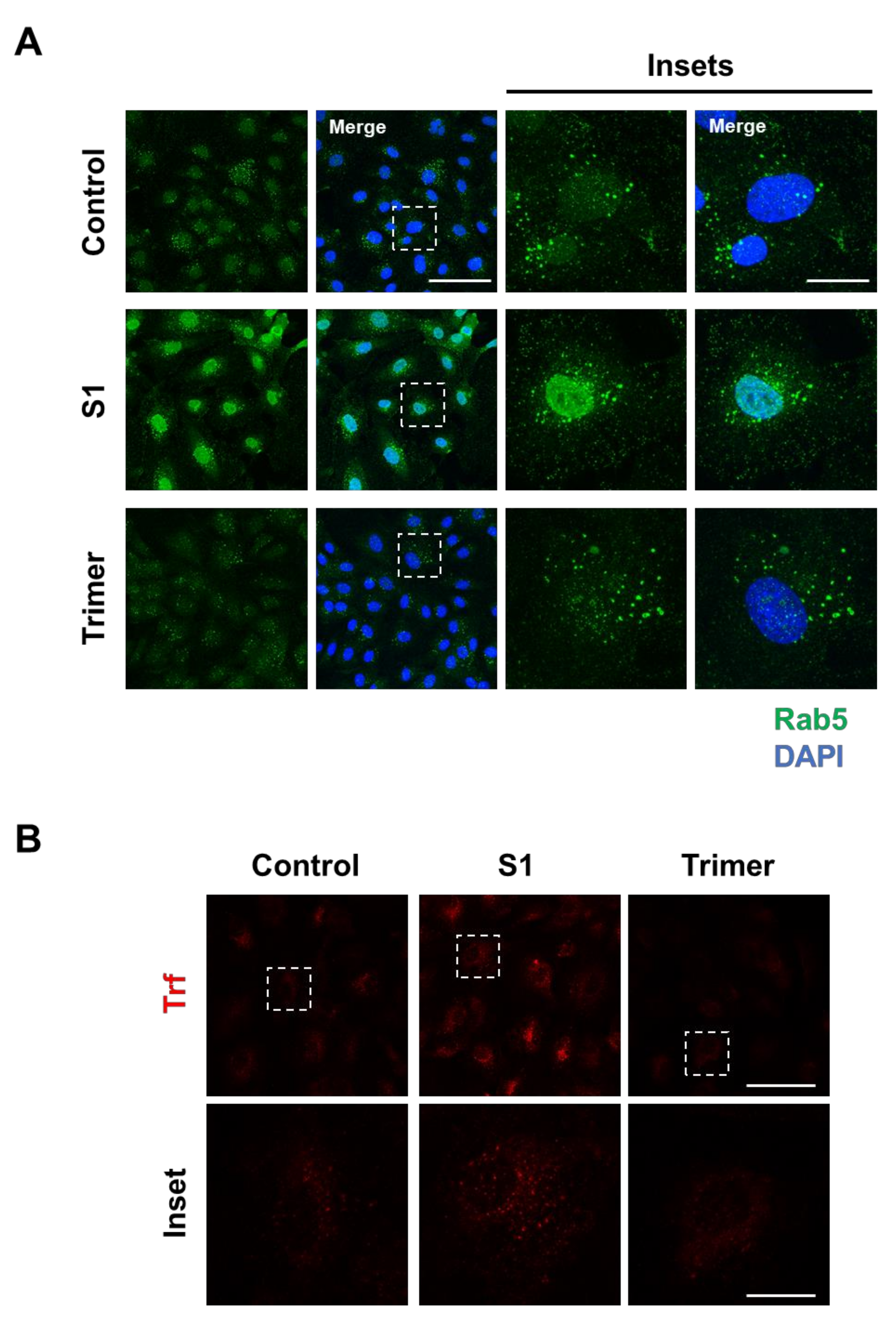
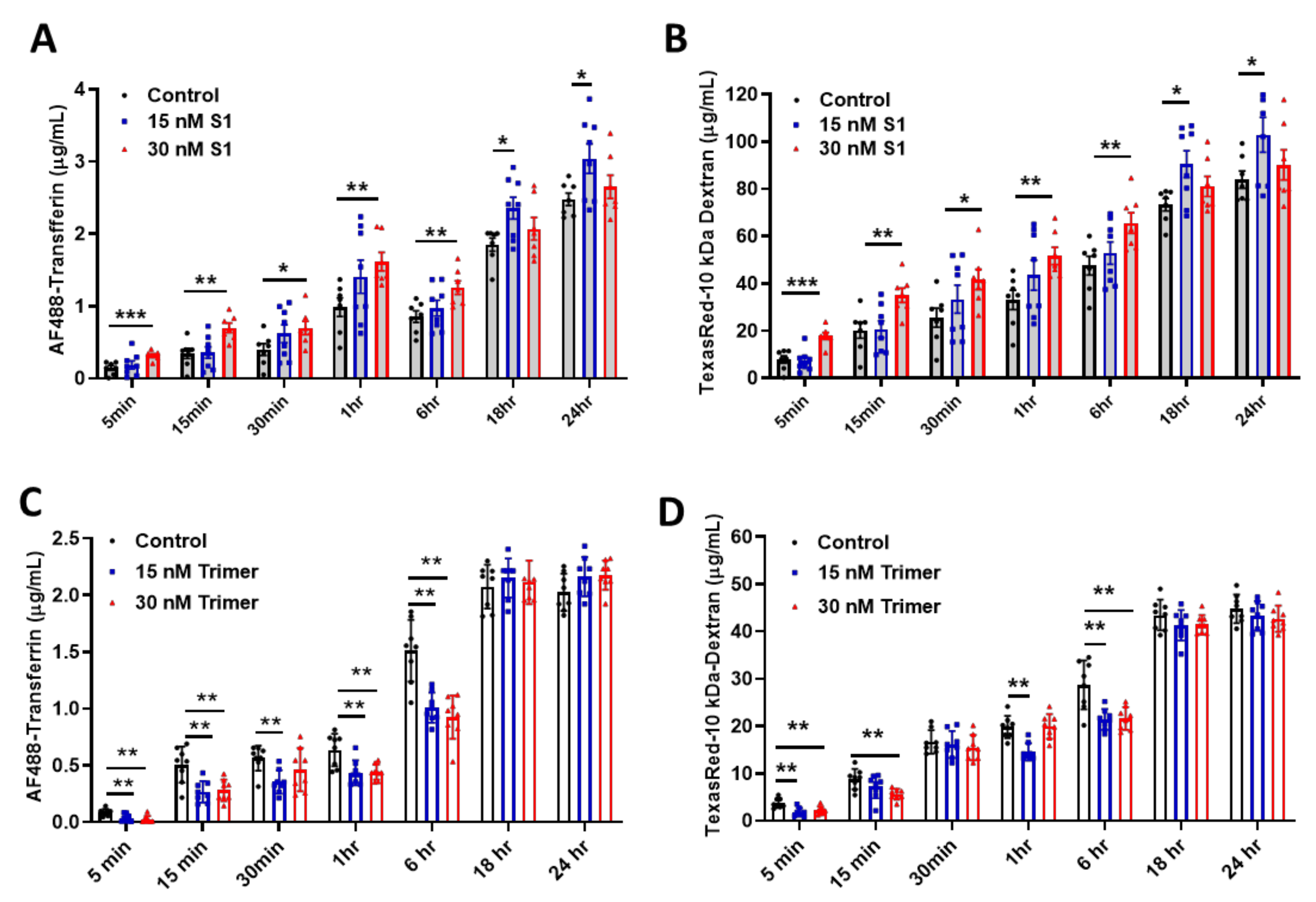
3.2. Spike Protein of SARS-CoV-2 Activates Endosomal Trafficking Pathway in the Brain ECs
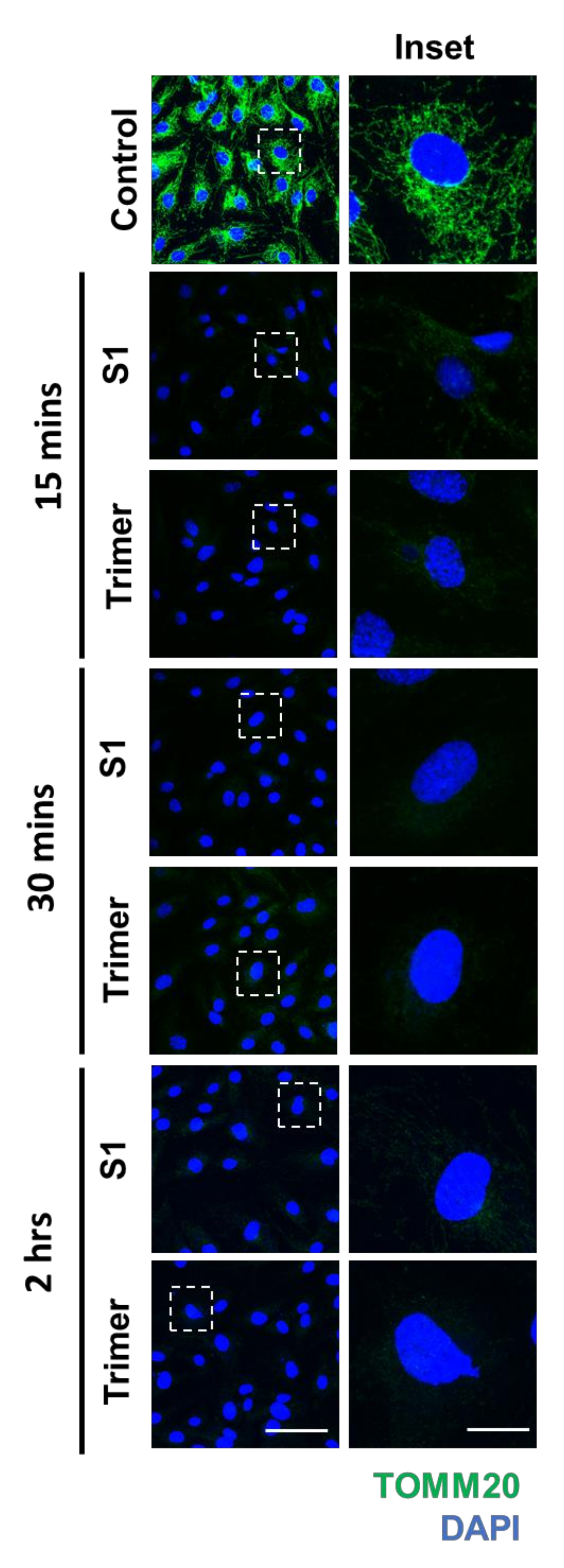
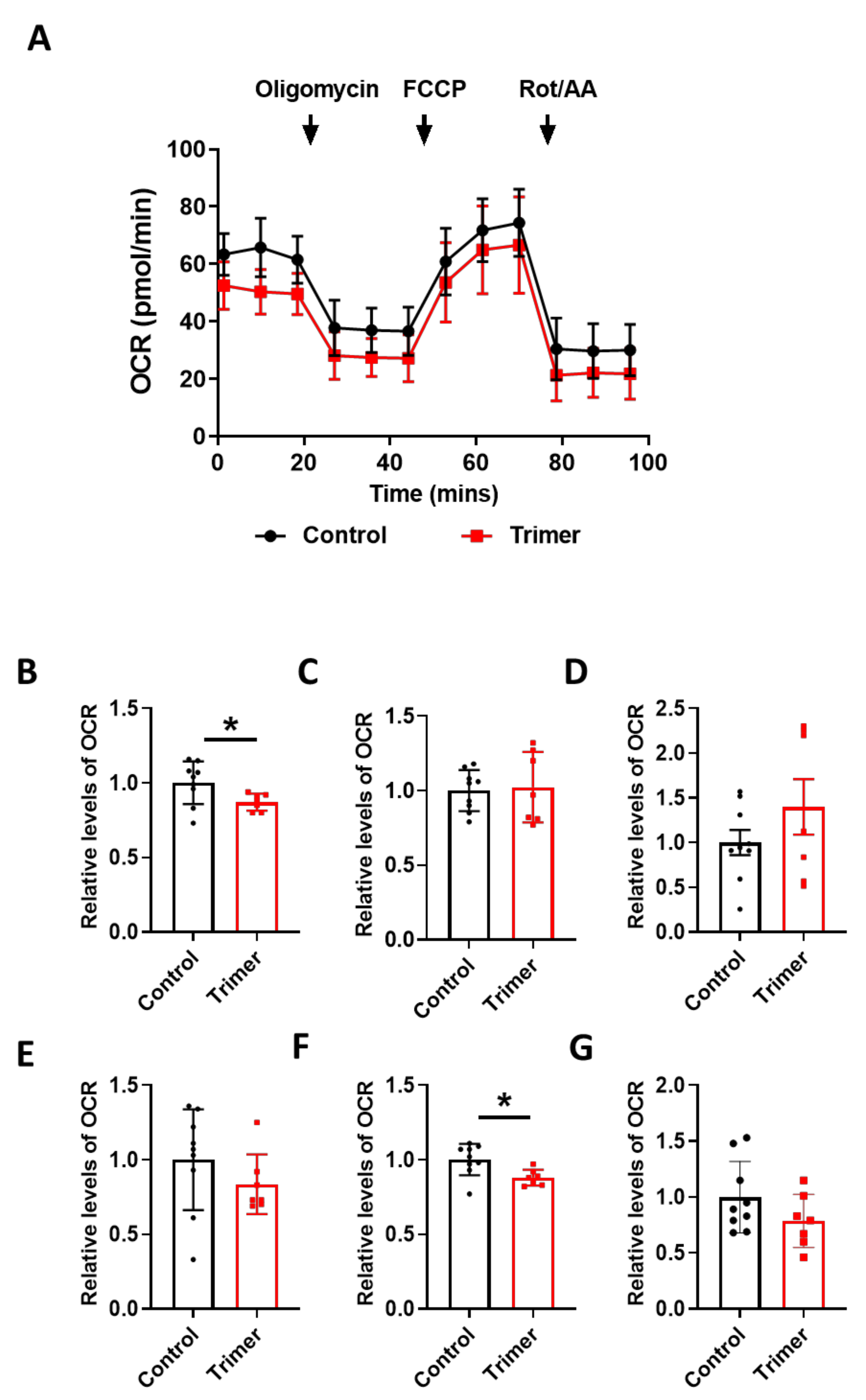
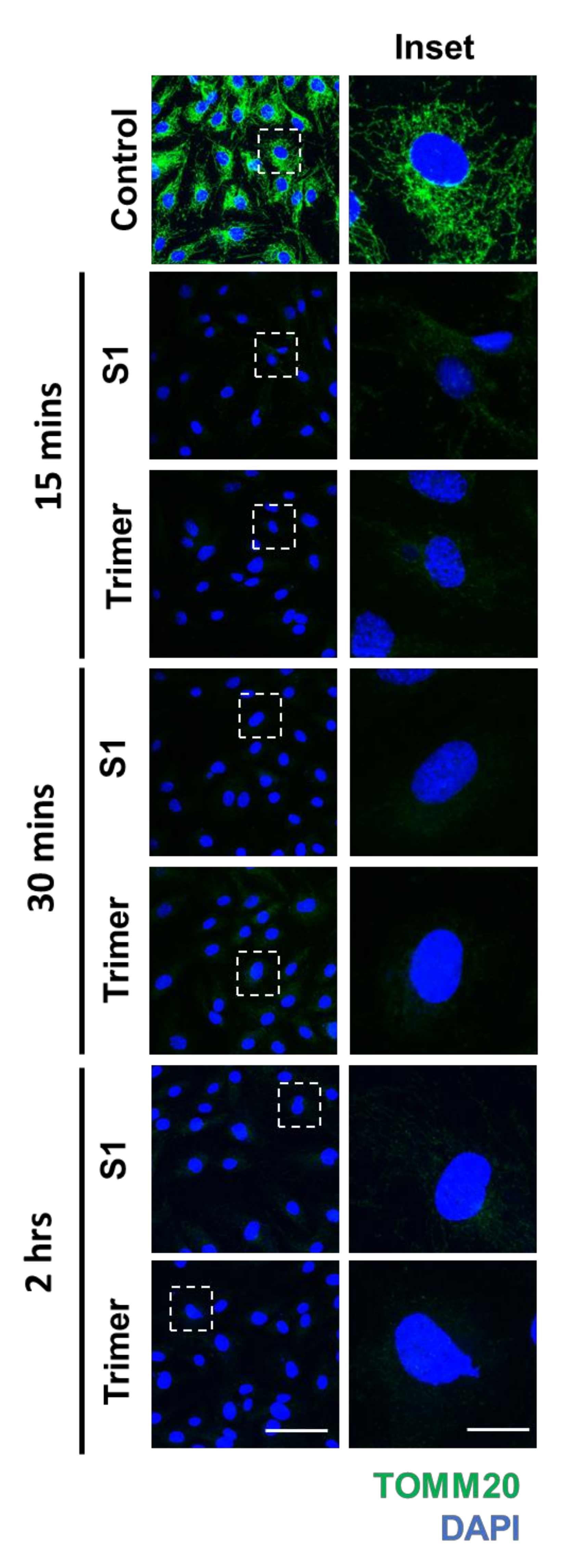
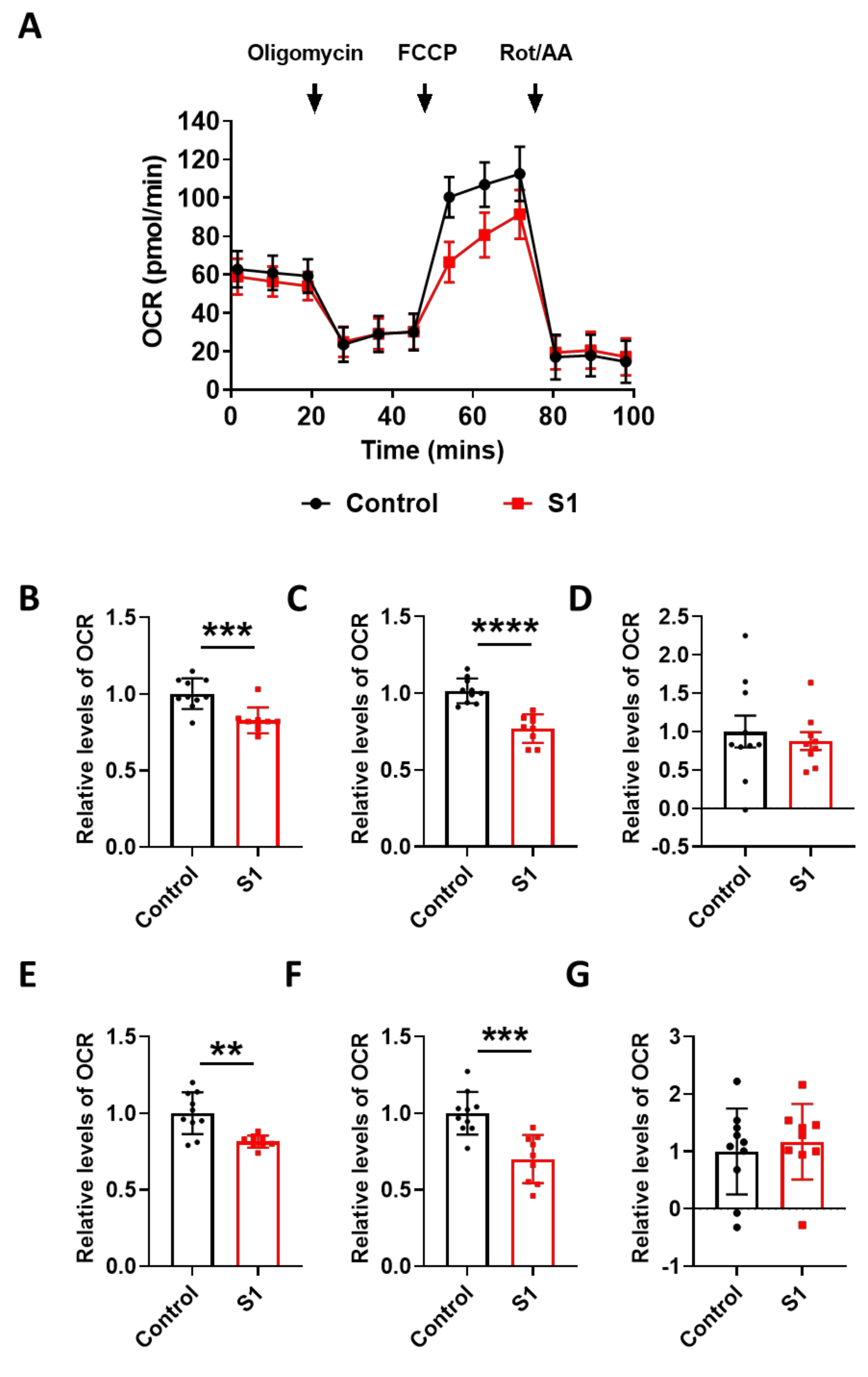
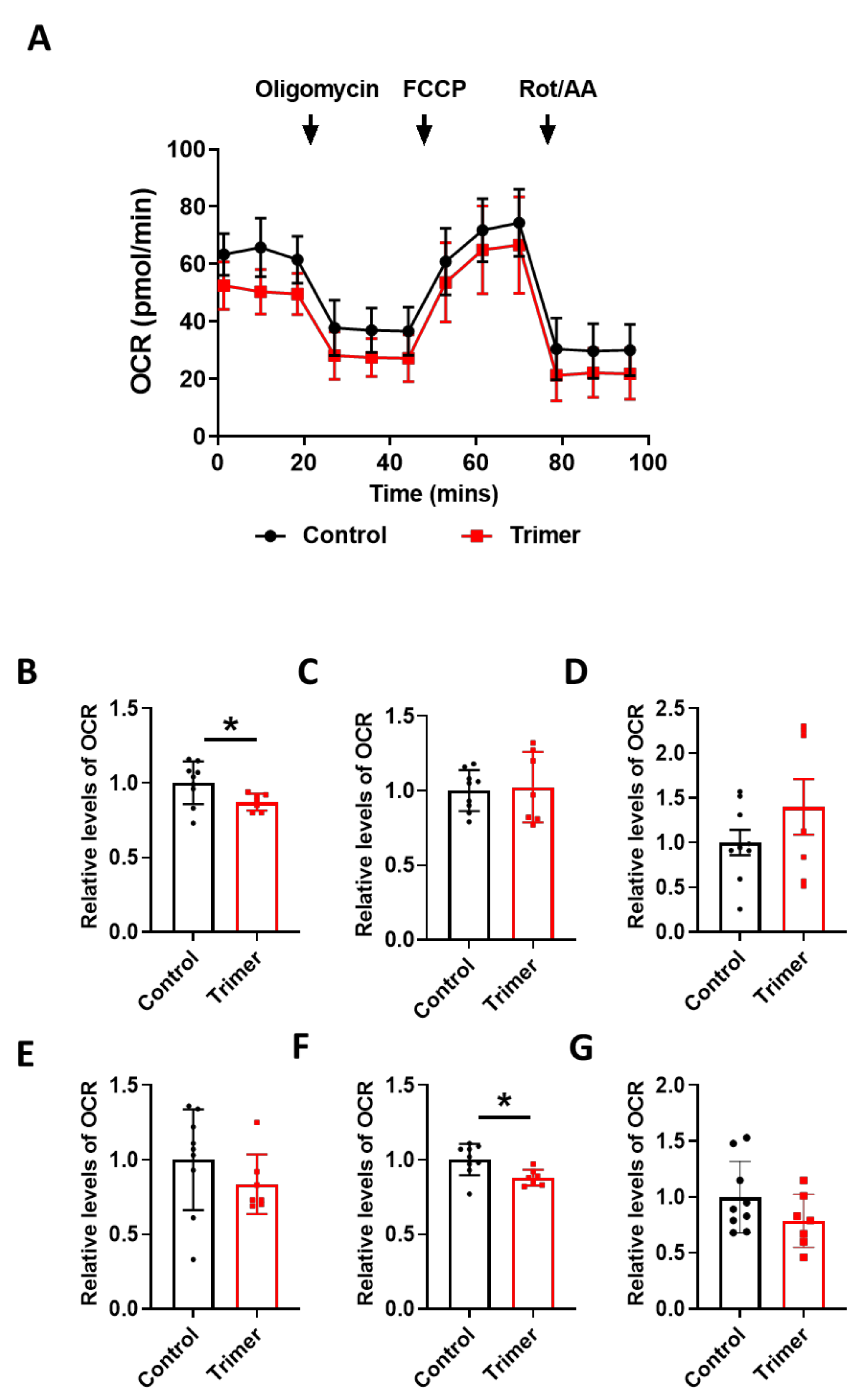
3.3. Spike Protein of SARS-CoV-2 Induces Damage to the Mitochondria
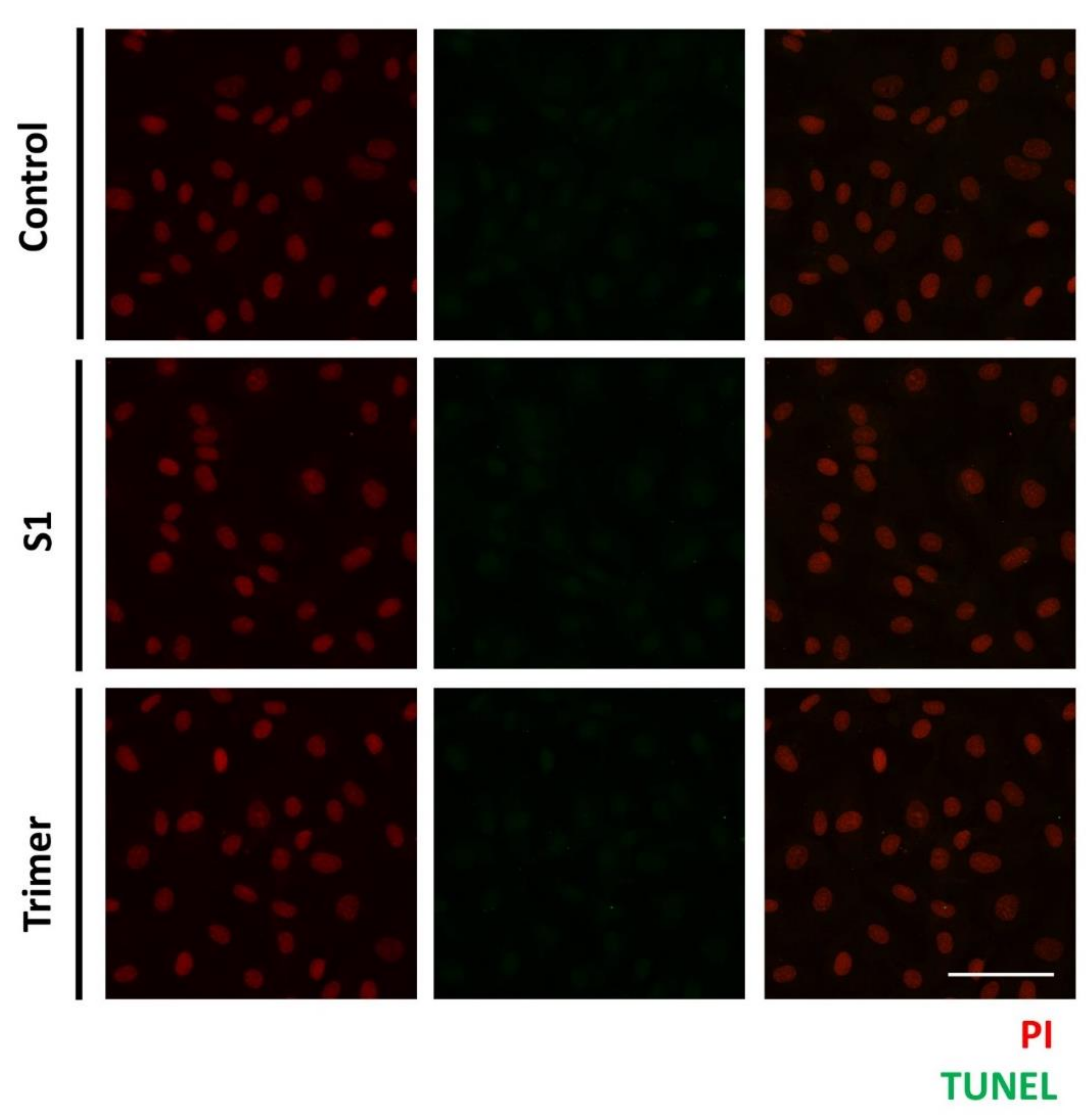
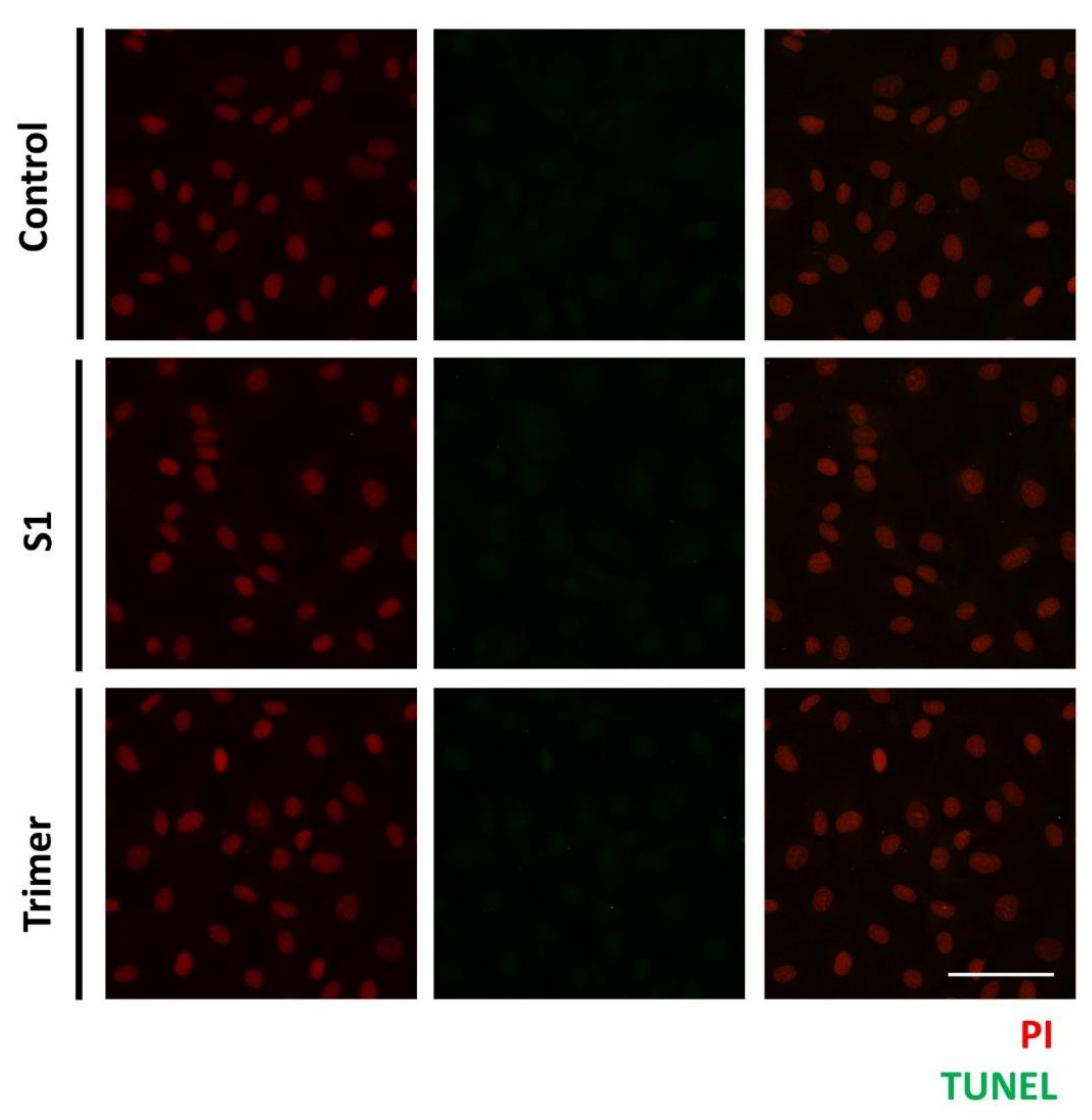
3.4. Spike Proteins of SARS-CoV-2 Do Not Induce Apoptotic Changes in the Primary Human Brain Endothelial Cell
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Xiong, J.; Lipsitz, O.; Nasri, F.; Lui, L.M.W.; Gill, H.; Phan, L.; Chen-Li, D.; Iacobucci, M.; Ho, R.; Majeed, A.; et al. Impact of COVID-19 pandemic on mental health in the general population: A systematic review. J. Affect. Disord. 2020, 277, 55–64. [Google Scholar] [CrossRef]
- WHO. Coronavirus Disease (COVID-19) Dashboard: World Health Organization. 2021. Available online: https://covid19.who.int/ (accessed on 5 October 2021).
- Mo, P.; Xing, Y.; Xiao, Y.; Deng, L.; Zhao, Q.; Wang, H.; Xiong, Y.; Cheng, Z.; Gao, S.; Liang, K.; et al. Clinical characteristics of refractory COVID-19 pneumonia in Wuhan, China. Clin. Infect. Dis. 2020, ciaa270. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Tang, J.; Wei, F. Updated understanding of the outbreak of 2019 novel coronavirus (2019-nCoV) in Wuhan, China. J. Med. Virol. 2020, 92, 441–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shereen, M.A.; Khan, S.; Kazmi, A.; Bashir, N.; Siddique, R. COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses. J. Adv. Res. 2020, 24, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Redondo, N.; Zaldívar-López, S.; Garrido, J.J.; Montoya, M. SARS-CoV-2 Accessory Proteins in Viral Pathogenesis: Knowns and Unknowns. Front. Immunol. 2021, 12, 708264. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Bhalla, V.; Blish, C.A.; South, A.M. A historical perspective on ACE2 in the COVID-19 era. J. Hum. Hypertens. 2020, 35, 935–939. [Google Scholar]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef]
- Keaney, J.; Campbell, M. The dynamic blood–brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef]
- Wolburg, H.; Noell, S.; Mack, A.; Wolburg-Buchholz, K.; Fallier-Becker, P. Brain endothelial cells and the glio-vascular complex. Cell Tissue Res. 2009, 335, 75–96. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.; Liddelow, S.; Dziegielewska, K. Barrier Mechanisms in the Developing Brain. Front. Pharmacol. 2012, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Feustel, S.M.; Meissner, M.; Liesenfeld, O. Toxoplasma gondii and the blood-brain barrier. Virulence 2012, 3, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Ivey, N.S.; MacLean, A.G.; Lackner, A.A. Acquired immunodeficiency syndrome and the blood-brain barrier. J. Neurovirol. 2009, 15, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzini, A.; Padovani, A. Lifting the mask on neurological manifestations of COVID-19. Nat. Rev. Neurol. 2020, 16, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Varatharaj, A.; Thomas, N.; Ellul, M.A.; Davies, N.W.S.; Pollak, T.A.; Tenorio, E.L.; Sultan, M.; Easton, A.; Breen, G.; Zandi, M.; et al. Neurological and neuropsychiatric complications of COVID-19 in 153 patients: A UK-wide surveillance study. Lancet Psychiatry 2020, 7, 875–882. [Google Scholar] [CrossRef]
- Abdel-Mannan, O.; Eyre, M.; Löbel, U.; Bamford, A.; Eltze, C.; Hameed, B.; Hemingway, C.; Hacohen, Y. Neurologic and Radiographic Findings Associated With COVID-19 Infection in Children. JAMA Neurol. 2020, 77, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, P.G.; Wong, S.; Al Mulhim, N.; Palmer, L.E.; Miners, J.S. Angiotensin-converting enzyme 2 is reduced in Alzheimer’s disease in association with increasing amyloid-β and tau pathology. Alzheimer’s Res. Ther. 2016, 8, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamming, I.; Timens, W.; Bulthuis, M.; Lely, A.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.; Schuepbach, R.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Zhang, H.; Penninger, J.M.; Li, Y.; Zhong, N.; Slutsky, A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: Molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020, 46, 586–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainsworth, A.H.; Oommen, A.T.; Bridges, L.R. Endothelial cells and human cerebral small vessel disease. Brain Pathol. 2015, 25, 44–50. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.K.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood-brain transport is impaired with age by a shift in transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial Crisis in Cerebrovascular Endothelial Cells Opens the Blood-Brain Barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, C.Z.; Swaroop, M.; Xu, M.; Wang, L.; Lee, J.; Wang, A.Q.; Pradhan, M.; Hagen, N.; Chen, L.; et al. Heparan sulfate assists SARS-CoV-2 in cell entry and can be targeted by approved drugs in vitro. Cell Discov. 2020, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Villaseñor, R.; Lampe, J.; Schwaninger, M.; Collin, L. Intracellular transport and regulation of transcytosis across the blood–brain barrier. Cell. Mol. Life Sci. 2019, 76, 1081–1092. [Google Scholar] [CrossRef] [Green Version]
- Preston, J.; Abbott, N.; Begley, D. Transcytosis of Macromolecules at the Blood–Brain Barrier. Adv. Pharmacol. 2014, 71, 147–163. [Google Scholar]
- Moreno-Altamirano, M.M.B.; Kolstoe, S.E.; Sánchez-García, F.J. Virus Control of Cell Metabolism for Replication and Evasion of Host Immune Responses. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Everett, H.; McFadden, G. Viruses and Apoptosis: Meddling with Mitochondria. Virology 2001, 288, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Syed, G.H.; Kim, S.-J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [Green Version]
- Scozzi, D.; Cano, M.; Ma, L.; Zhou, D.; Zhu, J.H.; O’Halloran, J.A.; Goss, C.; Rauseo, A.M.; Liu, Z.; Peritore, V.; et al. Circulating mitochondrial DNA is an early indicator of severe illness and mortality from COVID-19. JCI Insight 2021, 6, e143299. [Google Scholar]
- Tang, X.; Luo, Y.-X.; Chen, H.-Z.; Liu, D.-P. Mitochondria, endothelial cell function, and vascular diseases. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef]
- Caja, S.; Enríquez, J.A. Mitochondria in endothelial cells: Sensors and integrators of environmental cues. Redox Biol. 2017, 12, 821–827. [Google Scholar] [CrossRef]
- Kyrylkova, K.; Kyryachenko, S.; Leid, M.; Kioussi, C. Detection of apoptosis by TUNEL assay. Methods Mol. Biol. 2012, 887, 41–47. [Google Scholar]
- Pilotto, A.; Cristillo, V.; Piccinelli, S.C.; Zoppi, N.; Bonzi, G.; Sattin, D. COVID-19 severity impacts on long-term neurological manifestation after hospitalisation. medRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Berlit, P.; Bösel, J.; Gahn, G.; Isenmann, S.; Meuth, S.G.; Nolte, C.H.; Pawlitzki, M.; Rosenow, F.; Schoser, B.; Thomalla, G.; et al. “Neurological manifestations of COVID-19”—Guideline of the German society of neurology. Neurol. Res. Pract. 2020, 2, 51. [Google Scholar] [CrossRef]
- Kaul, M. HIV-1 associated dementia: Update on pathological mechanisms and therapeutic approaches. Curr. Opin. Neurol. 2009, 22, 315–320. [Google Scholar] [CrossRef] [Green Version]
- Al-Obaidi, M.M.J.; Desa, M.N.M. Mechanisms of Blood Brain Barrier Disruption by Different Types of Bacteria, and Bacterial–Host Interactions Facilitate the Bacterial Pathogen Invading the Brain. Cell. Mol. Neurobiol. 2018, 38, 1349–1368. [Google Scholar] [CrossRef] [PubMed]
- Forrester, J.V.; McMenamin, P.G.; Dando, S.J. CNS infection and immune privilege. Nat. Rev. Neurosci. 2018, 19, 655–671. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Gao, G.F. Viral targets for vaccines against COVID-19. Nat. Rev. Immunol. 2021, 21, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Oldendorf, W.H.; Brown, W.J. Greater Number of Capillary Endothelial Cell Mitochondria in Brain Than in Muscle. Proc. Soc. Exp. Biol. Med. 1975, 149, 736–738. [Google Scholar] [CrossRef]
- Rogers, J.P.; Watson, C.J.; Badenoch, J.; Cross, B.; Butler, M.; Song, J.; Hafeez, D.; Morrin, H.; Rengasamy, E.R.; Thomas, L.; et al. Neurology and neuropsychiatry of COVID-19: A systematic review and meta-analysis of the early literature reveals frequent CNS manifestations and key emerging narratives. J. Neurol. Neurosurg. Psychiatry 2021, 92, 932–941. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Sano, K.; Tan, T.S.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; et al. SARS-CoV-2 D614G spike mutation increases entry efficiency with enhanced ACE2-binding affinity. Nat. Commun. 2021, 12, 848. [Google Scholar] [CrossRef]
- Cohen, S.R.; Prussick, L.; Kahn, J.S.; Gao, D.X.; Radfar, A.; Rosmarin, D. Leukocytoclastic vasculitis flare following the COVID-19 vaccine. Int. J. Dermatol. 2021, 60, 1032–1033. [Google Scholar] [CrossRef]
- Gillion, V.; Jadoul, M.; Demoulin, N.; Aydin, S.; Devresse, A. Granulomatous vasculitis after the AstraZeneca anti-SARS-CoV-2 vaccine. Kidney Int. 2021, 100, 706–707. [Google Scholar] [CrossRef]
- Maramattom, B.V.; Krishnan, P.; Paul, R.; Padmanabhan, S.; Cherukudal Vishnu Nampoothiri, S.; Syed, A.A.; Mangat, H.S. Guillain-Barré Syndrome following ChAdOx1-S/nCoV-19 Vaccine. Ann. Neurol. 2021, 90, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Zhang, J.; Schiavon, C.R.; He, M.; Chen, L.; Shen, H. SARS-CoV-2 Spike Protein Impairs Endothelial Function via Downregulation of ACE 2. Circ. Res. 2021, 128, 1323–1326. [Google Scholar] [CrossRef] [PubMed]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A.; Bullock, T.A.; McGary, H.M.; Khan, J.A.; Razmpour, R.; Hale, J.F.; Galie, P.A.; Potula, R.; et al. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- García-Pérez, B.E.; González-Rojas, J.A.; Salazar, M.I.; Torres-Torres, C.; Castrejón-Jiménez, N.S. Taming the Autophagy as a Strategy for Treating COVID-19. Cells 2020, 9, 2679. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.S.; Jeon, M.-T.; Kim, K.-S.; Lee, S.; Kim, S.; Kim, D.-G. Spike Proteins of SARS-CoV-2 Induce Pathological Changes in Molecular Delivery and Metabolic Function in the Brain Endothelial Cells. Viruses 2021, 13, 2021. https://doi.org/10.3390/v13102021
Kim ES, Jeon M-T, Kim K-S, Lee S, Kim S, Kim D-G. Spike Proteins of SARS-CoV-2 Induce Pathological Changes in Molecular Delivery and Metabolic Function in the Brain Endothelial Cells. Viruses. 2021; 13(10):2021. https://doi.org/10.3390/v13102021
Chicago/Turabian StyleKim, Eun Seon, Min-Tae Jeon, Kyu-Sung Kim, Suji Lee, Suji Kim, and Do-Geun Kim. 2021. "Spike Proteins of SARS-CoV-2 Induce Pathological Changes in Molecular Delivery and Metabolic Function in the Brain Endothelial Cells" Viruses 13, no. 10: 2021. https://doi.org/10.3390/v13102021
APA StyleKim, E. S., Jeon, M.-T., Kim, K.-S., Lee, S., Kim, S., & Kim, D.-G. (2021). Spike Proteins of SARS-CoV-2 Induce Pathological Changes in Molecular Delivery and Metabolic Function in the Brain Endothelial Cells. Viruses, 13(10), 2021. https://doi.org/10.3390/v13102021