
An Opportunistic Survey Reveals an Unexpected Coronavirus Diversity Hotspot in North America

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. SARS-CoV-2 Detection
2.2. Pan-Coronavirus Testing
2.3. Phylogenetic Analysis
3. Results
3.1. Overall Findings
3.2. Tissue Distribution of Coronaviruses
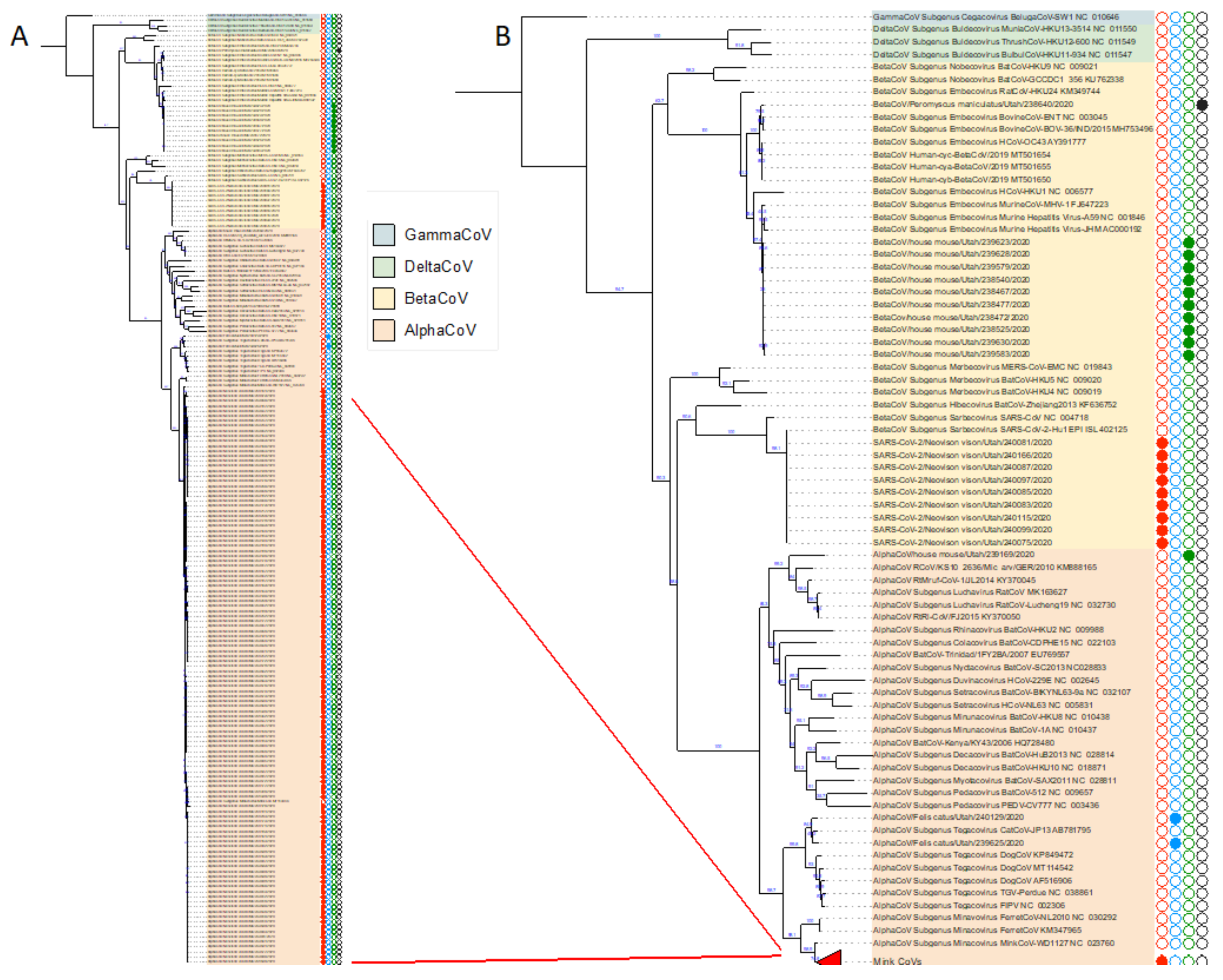
3.3. Phylogenetic Analysis
3.4. Coinfections
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimers
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO-convened global study of origins of SARS-CoV-2: China Part. 120 pp.. Available online: https://www.who.int/publications/i/item/who-convened-global-study-of-origins-of-sars-cov-2-china-part (accessed on 16 August 2021).
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. SARS Working Group. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Coleman, C.M.; Frieman, M.B. Emergence of the Middle East respiratory syndrome coronavirus. PLoS Pathog. 2013, 9, e1003595. [Google Scholar] [CrossRef]
- Ghai, R.R.; Carpenter, A.; Liew, A.Y.; Martin, K.B.; Herring, M.K.; Gerber, S.I.; Hall, A.J.; Sleeman, J.M.; VonDobschuetz, S.; Barton Behravesh, C. Animal reservoirs and hosts for emerging alphacoronaviruses and betacoronaviruses. Emerg Infect. Dis 2021, 27, 1015–1022. [Google Scholar] [CrossRef]
- Turlewicz-Podbielska, H.; Pomorska-Mól, M. Porcine coronaviruses: Overview of the state of the art. Virol Sinica 2021, 15, 1–19. [Google Scholar] [CrossRef]
- Parrlberg, P.L. Updated estimated economic welfare impacts of Procine Epidemic Diarrhea Virus (PEDV); Working Paper 14-4; Perdue University: West Lafayette, IN, USA, 2014; pp. 1–38. [Google Scholar]
- Stout, A.E.; Guo, Q.; Millet, J.K.; de Matos, R.; Whittaker, G.R. Coronaviruses associated with the superfamily Musteloidea. mBio 2021, 12, e02873-20. [Google Scholar] [CrossRef]
- World Organization for Animal Health. Disease Data Collection. Available online: https://www.oie.int/en/alerts-disease-information/ (accessed on 16 August 2021).
- Shriner, S.A.; Ellis, J.W.; Root, J.J.; Roug, A.; Stopak, S.R.; Wiscomb, G.W.; Zierenberg, J.R.; Ip, H.S.; Torchetti, M.K.; DeLiberto, T.J. SARS-CoV-2 exposure in escaped mink, Utah, USA. Emerg Infect. Dis. 2021, 27, 988–990. [Google Scholar] [CrossRef] [PubMed]
- Docherty, D.E.; Slota, P.G. Use of Muscovy duck embryo fibroblasts for the isolation of viruses from wild birds. J. Tissue Cult. Meth. 1988, 11, 165–170. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Sakthivel, S.K.; Whitaker, B.; Murray, J.; Kamili, S.; Lynch, B.; Malapati, L.; Burke, S.A.; Harcourt, J.; et al. US CDC real-time reverse transcription PCR panel for detection of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg Infect. Dis 2020, 26, 1654–1665. [Google Scholar] [CrossRef]
- Hu, H.; Jung, K.; Wang, Q.; Saif, L.J.; Vlasova, A.N. Development of a one-step RT-PCR assay for detection of pancoronaviruses (α-, β-, γ-, and δ-coronaviruses) using newly designed degenerate primers for porcine and avian fecal samples. J. Virol. Methods 2018, 256, 116–122. [Google Scholar] [CrossRef]
- Falcón, A.; Vázquez-Morón, S.; Casas, I.; Aznar, C.; Ruiz, G.; Pozo, F.; Perez-Breña, P.; Juste, J.; Ibáñez, C.; Garin, I.; et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol 2011, 156, 1883–1890. [Google Scholar] [CrossRef]
- Oreshkova, N.; Molenaar, R.J.; Vreman, S.; Harders, F.; Oude Munnink, B.B.; Hakze-van der Honing, R.W.; Gerhards, N.; Tolsma, P.; Bouwstra, R.; Sikkema, R.S.; et al. SARS-CoV-2 infection in farmed minks, the Netherlands, April and May 2020. Euro Surveill. 2020, 25, 2001005. [Google Scholar] [CrossRef]
- Larsen, H.D.; Fonager, J.; Lomholt, F.K.; Dalby, T.; Benedetti, G.; Kristensen, B.; Urth, T.R.; Rasmussen, M.; Lassaunière, R.; Rasmussen, T.B.; et al. Preliminary report of an outbreak of SARS-CoV-2 in mink and mink farmers associated with community spread, Denmark, June to November 2020. Euro Surveil. 2021, 26, 2100009. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2021, 371, 172–177. [Google Scholar] [CrossRef]
- Vlasova, A.N.; Halpin, R.; Wang, S.; Ghedin, E.; Spiro, D.J.; Saif, L.J. Molecular characterization of a new species in the genus Alphacoronavirus associated with mink epizootic catarrhal gastroenteritis. J. Gen. Virol 2011, 6, 1369–1379. [Google Scholar] [CrossRef]
- Wang, W.; Lin, X.D.; Guo, W.P.; Zhou, R.H.; Wang, M.R.; Wang, C.Q.; Ge, S.; Mei, S.H.; Li, M.H.; Shi, M.; et al. Discovery, diversity and evolution of novel coronaviruses sampled from rodents in China. Virology 2015, 474, 19–27. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- Ge, X.Y.; Yang, W.H.; Zhou, J.H.; Li, B.; Zhang, W.; Shi, Z.L.; Zhang, Y.Z. Detection of alpha- and betacoronaviruses in rodents from Yunnan, China. Virol J. 2017, 14, 98. [Google Scholar] [CrossRef]
- Tsoleridis, T.; Onianwa, O.; Horncastle, E.; Dayman, E.; Zhu, M.; Danjittrong, T.; Wachtl, M.; Behnke, J.M.; Chapman, S.; Strong, V.; et al. Discovery of novel alphacoronaviruses in European rodents and shrews. Viruses 2016, 8, 84. [Google Scholar] [CrossRef]
- Martin, H.D.; Zeidner, N.S. Concomitant cryptosporidia, coronavirus and parvovirus infection in a raccoon (Procyon lotor). J. Wildl. Dis. 1992, 28, 113–115. [Google Scholar] [CrossRef]
- Aoki, E.; Soma, T.; Yokoyama, M.; Matsubayashi, M.; Sasai, K. Surveillance for antibodies against six canine viruses in wild raccoons (Procyon lotor) in Japan. J. Wildl. Dis. 2017, 53, 761–768. [Google Scholar] [CrossRef]
- Bosco-Lauth, A.M.; Root, J.J.; Porter, S.M.; Walker, A.E.; Guilbert, L.; Hawvermale, D.; Pepper, A.; Maison, R.M.; Hartwig, A.E.; Gordy, P.; et al. Peridomestic mammal susceptibility to Severe Acute Respiratory Syndrome Coronavirus 2 infection. Emerg. Infect. Dis 2021, 27, 2073–2080. [Google Scholar] [CrossRef]
- Halfmann, P.J.; Hatta, M.; Chiba, S.; Maemura, T.; Fan, S.; Takeda, M.; Kinoshita, N.; Hattori, S.I.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; et al. Transmission of SARS-CoV-2 in Domestic Cats. N. Engl. J. Med. 2020, 383, 592–594. [Google Scholar] [CrossRef]
- Newman, A.; Smith, D.; Ghai, R.R.; Wallace, R.M.; Torchetti, M.K.; Loiacono, C.; Murrell, L.S.; Carpenter, A.; Moroff, S.; Rooney, J.A.; et al. First Reported Cases of SARS-CoV-2 Infection in Companion Animals - New York, March-April 2020. Morb. Mortal. Wkly Rep. 2020, 69, 710–713. [Google Scholar] [CrossRef]
- McAloose, D.; Laverack, M.; Wang, L.; Killian, M.L.; Caserta, L.C.; Yuan, F.; Mitchell, P.K.; Queen, K.; Mauldin, M.R.; Cronk, B.D.; et al. From People to Panthera: Natural SARS-CoV-2 Infection in Tigers and Lions at the Bronx Zoo. mBio 2020, 11, e02220-02220. [Google Scholar] [CrossRef]
- Körner, R.W.; Majjouti, M.; Alcazar, M.A.A.; Mahabir, E. Of mice and men: The coronavirus MHV and mouse models as a translational approach to understand SARS-CoV-2. Viruses 2020, 12, 880. [Google Scholar] [CrossRef]
- De Albuquerque, N.; Baig, E.; Ma, X.; Zhang, J.; He, W.; Rowe, A.; Habal, M.; Liu, M.; Shalev, I.; Downey, G.P.; et al. Murine hepatitis virus strain 1 produces a clinically relevant model of severe acute respiratory syndrome in A/J mice. J. Virol. 2006, 80, 10382–10394. [Google Scholar] [CrossRef]
- Becker, S.D.; Bennett, M.; Stewart, J.P.; Hurst, J.L. Serological survey of virus infection among wild house mice (Mus domesticus) in the UK. Lab. Anim. 2007, 41, 229–238. [Google Scholar] [CrossRef]
- Parker, S.E.; Malone, S.; Bunte, R.M.; Smith, A.L. Infectious diseases in wild mice (Mus musculus) collected on and around the University of Pennsylvania (Philadelphia) campus. Comp. Med. 2009, 59, 424–430. [Google Scholar] [PubMed]
- Drexler, J.F.; Corman, V.M.; Lukashev, A.N.; van den Brand, J.M.; Gmyl, A.P.; Brünink, S.; Rasche, A.; Seggewiβ, N.; Feng, H.; et al.; Hepatovirus Ecology Consortium Evolutionary origins of hepatitis A virus in small mammals. Proc. Natl. Acad. Sci. USA 2015, 112, 15190–15195. [Google Scholar] [CrossRef]
- McIver, D.J.; Silithammavong, S.; Theppangna, W.; Gillis, A.; Douangngeun, B.; Khammavong, K.; Singhalath, S.; Duong, V.; Buchy, P.; Olson, S.H.; et al. Coronavirus surveillance of wildlife in the Lao People’s Democratic Republic detects viral RNA in rodents. Arch. Virol. 2020, 165, 1869–1875. [Google Scholar] [CrossRef]
- Monchatre-Leroy, E.; Boué, F.; Boucher, J.M.; Renault, C.; Moutou, F.; Ar Gouilh, M.; Umhang, G. Identification of alpha and beta coronavirus in wildlife species in France: Bats, rodents, rabbits, and hedgehogs. Viruses 2017, 9, 364. [Google Scholar] [CrossRef]
- Silverman, J.; Paturzo, F.; Smith, A.L. Effects of experimental infection of the deer mouse (Peromyscus maniculatus) with mouse hepatitis virus. Lab. Anim. Sci. 1982, 32, 273–274. [Google Scholar] [PubMed]
- Gruetzmacher, K.; Karesh, W.B.; Amuasi, J.H.; Arshad, A.; Farlow, A.; Gabrysch, S.; Jetzkowitz, J.; Lieberman, S.; Palmer, C.; Winkler, A.S.; et al. The Berlin principles on one health - Bridging global health and conservation. Sci. Total Environ. 2021, 764, 142919. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Species | Total | Negative | Positive | % Pos |
|---|---|---|---|---|
| Cat | 2 | 0 | 2 | 100% |
| Mink | 252 | 23 | 229 | 91% |
| Mouse, Deer | 47 | 43 | 4 | 9% |
| Mouse, House | 51 | 30 | 21 | 41% |
| Raccoon | 6 | 4 | 2 | 33% |
| Skunk, Striped | 7 | 4 | 3 | 43% |
| Total | 365 | 104 | 261 | 72% |
| Tissue | Total | Negative | Positive | % Pos |
|---|---|---|---|---|
| Colon/Rectum | 89 | 68 | 21 | 24% |
| Heart/Kidney | 90 | 90 | 0 | - |
| Liver | 48 | 48 | 0 | - |
| Liver/Spleen | 42 | 42 | 0 | - |
| Lung | 96 | 85 | 11 | 11% |
| Small Intestine | 89 | 89 | 0 | - |
| Trachea | 5 | 5 | 0 | - |
| Total | 459 | 427 | 32 | 7% |
| Species | AlphaCoV | BetaCoV | Sequenced | Unk |
|---|---|---|---|---|
| Cat | 2 | 0 | 2 | 0 |
| Mink | 123 | 74 | 131 | 60 |
| Mouse, Deer | 0 | 1 | 1 | 3 |
| Mouse, House | 2 | 13 | 15 | 6 |
| Raccoon | 0 | 0 | 0 | 2 |
| Skunk, Striped | 0 | 0 | 0 | 3 |
| Total | 127 | 88 | 149 | 74 |
| Animal ID | Common Name | Scientific Name | Sex | Age | SARS-CoV-2 | Second Strain |
|---|---|---|---|---|---|---|
| 46844-098 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-100 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-105 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-118 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-124 | Mink | Neovison vison | M | UNK | Pos | AlphaCoV (MinkCoV) |
| 46844-125 | Mink | Neovison vison | F | J | Pos | AlphaCoV (MinkCoV) |
| 46844-151 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-154 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-155 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-164 | Mink | Neovison vison | F | J | Pos | AlphaCoV (MinkCoV) |
| 46844-165 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-168 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-169 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-177 | Mink | Neovison vison | F | J | Pos | AlphaCoV (MinkCoV) |
| 46844-202 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-208 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-212 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-214 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-217 | Mink | Neovison vison | F | A | Pos | AlphaCoV (MinkCoV) |
| 46844-220 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-221 | Mink | Neovison vison | F | UNK | Pos | AlphaCoV (MinkCoV) |
| 46844-236 | Mink | Neovison vison | UNK | UNK | Pos | AlphaCoV (MinkCoV) |
| 46844-243 | Mink | Neovison vison | M | J | Pos | AlphaCoV (MinkCoV) |
| 46844-248 | Mink | Neovison vison | M | A | Pos | AlphaCoV (MinkCoV) |
| 46844-257 | Mink | Neovison vison | UNK | UNK | Pos | AlphaCoV (MinkCoV) |
| 46844-291 | Mink | Neovison vison | F | A | Pos | AlphaCoV (MinkCoV) |
| 46844-297 | Mink | Neovison vison | F | A | Pos | AlphaCoV (MinkCoV) |
| 46844-103 | Mink | Neovison vison | F | J | Pos | Pan-CoV Equivocal |
| 46844-111 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-115 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-188 | Mink | Neovison vison | F | J | Pos | Pan-CoV Equivocal |
| 46844-203 | Mink | Neovison vison | M | A | Pos | Pan-CoV Equivocal |
| 46844-210 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-226 | Mink | Neovison vison | F | UNK | Pos | Pan-CoV Equivocal |
| 46844-249 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-259 | Mink | Neovison vison | UNK | UNK | Pos | Pan-CoV Equivocal |
| 46844-260 | Mink | Neovison vison | UNK | UNK | Pos | Pan-CoV Equivocal |
| 46844-261 | Mink | Neovison vison | UNK | UNK | Pos | Pan-CoV Equivocal |
| 46844-262 | Mink | Neovison vison | F | UNK | Pos | Pan-CoV Equivocal |
| 46844-267 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-281 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-282 | Mink | Neovison vison | F | A | Pos | Pan-CoV Equivocal |
| 46844-283 | Mink | Neovison vison | M | A | Pos | Pan-CoV Equivocal |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ip, H.S.; Griffin, K.M.; Messer, J.D.; Winzeler, M.E.; Shriner, S.A.; Killian, M.L.; K. Torchetti, M.; DeLiberto, T.J.; Amman, B.R.; Cossaboom, C.M.; et al. An Opportunistic Survey Reveals an Unexpected Coronavirus Diversity Hotspot in North America. Viruses 2021, 13, 2016. https://doi.org/10.3390/v13102016
Ip HS, Griffin KM, Messer JD, Winzeler ME, Shriner SA, Killian ML, K. Torchetti M, DeLiberto TJ, Amman BR, Cossaboom CM, et al. An Opportunistic Survey Reveals an Unexpected Coronavirus Diversity Hotspot in North America. Viruses. 2021; 13(10):2016. https://doi.org/10.3390/v13102016
Chicago/Turabian StyleIp, Hon S., Kathryn M. Griffin, Jeffrey D. Messer, Megan E. Winzeler, Susan A. Shriner, Mary Lea Killian, Mia K. Torchetti, Thomas J. DeLiberto, Brian R. Amman, Caitlin M. Cossaboom, and et al. 2021. "An Opportunistic Survey Reveals an Unexpected Coronavirus Diversity Hotspot in North America" Viruses 13, no. 10: 2016. https://doi.org/10.3390/v13102016
APA StyleIp, H. S., Griffin, K. M., Messer, J. D., Winzeler, M. E., Shriner, S. A., Killian, M. L., K. Torchetti, M., DeLiberto, T. J., Amman, B. R., Cossaboom, C. M., Harvey, R. R., Wendling, N. M., Rettler, H., Taylor, D., Towner, J. S., Barton Behravesh, C., & Blehert, D. S. (2021). An Opportunistic Survey Reveals an Unexpected Coronavirus Diversity Hotspot in North America. Viruses, 13(10), 2016. https://doi.org/10.3390/v13102016