
“Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review


Abstract
1. Introduction
2. Repressors of Gene Silencing, Viral Transactivators, and Host Evasion Factors
2.1. RL2 or α0 (ICP0)
2.2. UL46 and UL47 (VP11/12 and VP13/14)
2.3. UL49 (VP22)
2.4. US1 (ICP22)
3. Host Evasion Factors
3.1. RL1 or γ134.5 (ICP34.5)
3.2. US12 (ICP47)
4. Nucleic Acid Metabolism and Endonucleases
4.1. UL2, UL12, UL12.5, UL50
4.2. UL39 and UL40 (RR1 and RR2)
4.3. UL41 (vhs)
5. Viral Kinases
5.1. UL23 (TK)
5.2. US3 and US3.5
5.3. UL13
6. Virion Morphogenesis, Egress, Cell-to-Cell Spread, and Host Evasion
6.1. UL3 and UL4
6.2. UL7 and UL51
6.3. UL10 and UL49.5 (gM and gN)
6.4. UL11
6.5. UL16
6.6. UL53 (gK) and UL20
6.7. UL21
6.8. UL24
6.9. UL31 and UL34
6.10. UL35 (VP26)
6.11. UL43
6.12. UL44 (gC)
6.13. UL45
6.14. UL55 and UL56
6.15. US2
6.16. US4 (gG)
6.17. US5 (gJ)
6.18. gE/gI (US8/US7), US9
6.19. US8.5
6.20. US10
6.21. US11
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- McGeoch, D.J.; Dolan, A.; Ralph, A.C. Toward a comprehensive phylogeny for mammalian and avian herpesviruses. J. Virol. 2000, 74, 10401–10406. [Google Scholar] [CrossRef] [PubMed]
- Underdown, S.J.; Kumar, K.; Houldcroft, C. Network analysis of the hominin origin of Herpes Simplex virus 2 from fossil data. Virus Evol. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley, P. Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 1. [Google Scholar]
- Mador, N.; Goldenberg, D.; Cohen, O.; Panet, A.; Steiner, I. Herpes simplex virus type 1 latency-associated transcripts suppress viral replication and reduce immediate-early gene mRNA levels in a neuronal cell line. J. Virol. 1998, 72, 5067–5075. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Sa e Silva, M.; Jaber, T.; Vitvitskaia, O.; Li, S.; Henderson, G.; Jones, C. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J. Virol. 2009, 83, 9131–9139. [Google Scholar] [CrossRef] [PubMed]
- Croen, K.D.; Ostrove, J.M.; Dragovic, L.J.; Smialek, J.E.; Straus, S.E. Latent herpes simplex virus in human trigeminal ganglia. Detection of an immediate early gene “anti-sense” transcript by in situ hybridization. N. Engl. J. Med. 1987, 317, 1427–1432. [Google Scholar] [CrossRef]
- Mador, N.; Panet, A.; Latchman, D.; Steiner, I. Expression and splicing of the latency-associated transcripts of herpes simplex virus type 1 in neuronal and non-neuronal cell lines. J. Biochem. 1995, 117, 1288–1297. [Google Scholar] [CrossRef]
- Garber, D.A.; Schaffer, P.A.; Knipe, D.M. A LAT-associated function reduces productive-cycle gene expression during acute infection of murine sensory neurons with herpes simplex virus type 1. J. Virol. 1997, 71, 5885–5893. [Google Scholar] [CrossRef]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [CrossRef]
- Wagner, E.K.; Bloom, D.C. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 1997, 10, 419–443. [Google Scholar] [CrossRef]
- Ahmed, M.; Lock, M.; Miller, C.G.; Fraser, N.W. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J. Virol. 2002, 76, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Henderson, G.; Peng, W.; Jin, L.; Perng, G.-C.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J. Neurovirol. 2002, 8, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Inman, M.; Perng, G.-C.; Henderson, G.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J. Virol. 2001, 75, 3636–3646. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Peng, W.; Perng, G.-C.; Brick, D.J.; Nesburn, A.B.; Jones, C.; Wechsler, S.L. Identification of herpes simplex virus type 1 latency-associated transcript sequences that both inhibit apoptosis and enhance the spontaneous reactivation phenotype. J. Virol. 2003, 77, 6556–6561. [Google Scholar] [CrossRef] [PubMed]
- Perng, G.C.; Ghiasi, H.; Slanina, S.M.; Nesburn, A.B.; Wechsler, S.L. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. J. Virol. 1996, 70, 976–984. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Henderson, G.; Perng, G.-C.; Nesburn, A.B.; Wechsler, S.L.; Jones, C. The gene that encodes the herpes simplex virus type 1 latency-associated transcript influences the accumulation of transcripts (Bcl-xL and Bcl-xS) that encode apoptotic regulatory proteins. J. Virol. 2003, 77, 10714–10718. [Google Scholar] [CrossRef]
- Perng, G.-C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [CrossRef]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis: Sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA 1975, 72, 1276–1280. [Google Scholar] [CrossRef]
- Longnecker, R.; Roizman, B. Clustering of genes dispensable for growth in culture in the S component of the HSV-1 genome. Science 1987, 236, 573–576. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Dolan, A.; Frame, M.C. DNA sequence of the region in the genome of herpes simplex virus type 1 containing the exonuclease gene and neighbouring genes. Nucleic Acids Res. 1986, 14, 3435–3448. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Toh, Y.; Tanaka, S.; Liu, Y.; Hidaka, Y.; Mori, R. Molecular characterization of naturally occuring glycoprotein C-negative herpes simplex virus type 1. Arch. Virol. 1993, 129, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, Y.; Sakuma, S.; Kumano, Y.; Minagawa, H.; Mori, R. Characterization of glycoprotein C-negative mutants of herpes simplex virus type 1 isolated from a patient with keratitis. Arch. Virol. 1990, 113, 195–207. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc. Natl. Acad. Sci. USA 2014, 111, E611-7. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, T.; Kalamvoki, M. Impaired STING pathway in human osteosarcoma U2OS cells contributes to the growth of ICP0-null mutant herpes simplex virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Ackermann, M.; Braun, D.K.; Pereira, L.; Roizman, B. Characterization of herpes simplex virus 1 alpha proteins 0, 4, and 27 with monoclonal antibodies. J. Virol. 1984, 52, 108–118. [Google Scholar] [CrossRef]
- Stow, N.D.; Stow, E.C. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 1986, 67, 2571–2585. [Google Scholar] [CrossRef]
- Sacks, W.R.; Schaffer, P.A. Deletion mutants in the gene encoding the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol. 1987, 61, 829–839. [Google Scholar] [CrossRef]
- Yao, F.; Schaffer, P.A. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 1995, 69, 6249–6258. [Google Scholar] [CrossRef]
- Chen, J.; Silverstein, S. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 1992, 66, 2916–2927. [Google Scholar] [CrossRef]
- Everett, R.D.; Boutell, C.; Orr, A. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 2004, 78, 1763–1774. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. Role of herpes simplex virus ICP0 in the transactivation of genes introduced by infection or transfection: A reappraisal. J. Virol. 2010, 84, 4222–4228. [Google Scholar] [CrossRef] [PubMed]
- Alandijany, T.; Roberts, A.P.E.; Conn, K.L.; Loney, C.; McFarlane, S.; Orr, A.; Boutell, C. Distinct temporal roles for the promyelocytic leukaemia (PML) protein in the sequential regulation of intracellular host immunity to HSV-1 infection. PLoS Pathog. 2018, 14, e1006927. [Google Scholar] [CrossRef]
- Sandri-Goldin, R.M.; Sekulovich, R.E.; Leary, K. The alpha protein ICP0 does not appear to play a major role in the regulation of herpes simplex virus gene expression during infection in tissue culture. Nucleic Acids Res. 1987, 15, 905–919. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moriuchi, H.; Moriuchi, M.; Straus, S.E.; Cohen, J.I. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J. Virol. 1993, 67, 4290–4295. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.; Orr, A.; Elliott, M. The equine herpesvirus 1 gene 63 RING finger protein partially complements Vmw110, its herpes simplex virus type 1 counterpart. J. Gen. Virol. 1995, 76, 2369–2374. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Boutell, C.; McNair, C.; Grant, L.; Orr, A. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J. Virol. 2010, 84, 3476–3487. [Google Scholar] [CrossRef]
- Quinlan, M.P.; Knipe, D.M. Stimulation of expression of a herpes simplex virus DNA-binding protein by two viral functions. Mol. Cell. Biol. 1985, 5, 957–963. [Google Scholar] [CrossRef]
- Knipe, D.M.; Smith, J.L. A mutant herpesvirus protein leads to a block in nuclear localization of other viral proteins. Mol. Cell. Biol. 1986, 6, 2371–2381. [Google Scholar] [CrossRef]
- Gelman, I.H.; Silverstein, S. Identification of immediate early genes from herpes simplex virus that transactivate the virus thymidine kinase gene. Proc. Natl. Acad. Sci. USA 1985, 82, 5265–5269. [Google Scholar] [CrossRef]
- Mosca, J.D.; Bednarik, D.P.; Raj, N.B.; Rosen, C.A.; Sodroski, J.G.; Haseltine, W.A.; Hayward, G.S.; Pitha, P.M. Activation of human immunodeficiency virus by herpesvirus infection: Identification of a region within the long terminal repeat that responds to a trans-acting factor encoded by herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 1987, 84, 7408–7412. [Google Scholar] [CrossRef] [PubMed]
- Ostrove, J.M.; Leonard, J.; Weck, K.E.; Rabson, A.B.; Gendelman, H.E. Activation of the human immunodeficiency virus by herpes simplex virus type 1. J. Virol. 1987, 61, 3726–3732. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, D.J.; Weller, S.K. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: Isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 1988, 62, 196–205. [Google Scholar] [CrossRef] [PubMed]
- McCusker, C.T.; Bacchetti, S. The responsiveness of human papillomavirus upstream regulatory regions to herpes simplex virus immediate early proteins. Virus Res. 1988, 11, 199–207. [Google Scholar] [CrossRef]
- Gius, D.; Laimins, L.A. Activation of human papillomavirus type 18 gene expression by herpes simplex virus type 1 viral transactivators and a phorbol ester. J. Virol. 1989, 63, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.M.; Rabson, A.B.; Straus, S.E.; Ostrove, J.M. Transactivation of the HIV-1 LTR by HSV-1 immediate-early genes. Virology 1992, 186, 788–791. [Google Scholar] [CrossRef]
- Nabel, G.J.; Rice, S.A.; Knipe, D.M.; Baltimore, D. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science 1988, 239, 1299–1302. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castaño-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef]
- O’Hare, P.; Hayward, G.S. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J. Virol. 1985, 56, 723–733. [Google Scholar] [CrossRef]
- Cai, W.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 1992, 66, 2904–2915. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Barlow, P.; Milner, A.; Luisi, B.; Orr, A.; Hope, G.; Lyon, D. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif of an alpha herpes virus protein family. J. Mol. Biol. 1993, 234, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- Grant, K.; Grant, L.; Tong, L.; Boutell, C. Depletion of intracellular zinc inhibits the ubiquitin ligase activity of viral regulatory protein ICP0 and restricts herpes simplex virus 1 replication in cell culture. J. Virol. 2012, 86, 4029–4033. [Google Scholar] [CrossRef] [PubMed]
- Lium, E.K.; Silverstein, S. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J. Virol. 1997, 71, 8602–8614. [Google Scholar] [CrossRef]
- Boutell, C.; Orr, A.; Everett, R.D. PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 2003, 77, 8686–8694. [Google Scholar] [CrossRef]
- Boutell, C.; Everett, R.D. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. J. Biol. Chem. 2003, 278, 36596–36602. [Google Scholar] [CrossRef]
- Boutell, C.; Canning, M.; Orr, A.; Everett, R.D. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 2005, 79, 12342–12354. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 2003, 100, 8963–8968. [Google Scholar] [CrossRef]
- Vanni, E.; Gatherer, D.; Tong, L.; Everett, R.D.; Boutell, C. Functional characterization of residues required for the herpes simplex virus 1 E3 ubiquitin ligase ICP0 to interact with the cellular E2 ubiquitin-conjugating enzyme UBE2D1 (UbcH5a). J. Virol. 2012, 86, 6323–6333. [Google Scholar] [CrossRef]
- Maul, G.G.; Guldner, H.H.; Spivack, J.G. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 1993, 74, 2679–2690. [Google Scholar] [CrossRef]
- Mullen, M.A.; Ciufo, D.M.; Hayward, G.S. Mapping of intracellular localization domains and evidence for colocalization interactions between the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J. Virol. 1994, 68, 3250–3266. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Maul, G.G. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 1994, 13, 5062–5069. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J.; Orr, A.; Preston, C.M. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 2007, 81, 10991–11004. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.; Corpet, A.; Roubille, S.; Maroui, M.A.; Poccardi, N.; Rousseau, A.; Kleijwegt, C.; Binda, O.; Texier, P.; Sawtell, N.; et al. Promyelocytic leukemia (PML) nuclear bodies (NBs) induce latent/quiescent HSV-1 genomes chromatinization through a PML NB/Histone H3.3/H3.3 Chaperone Axis. PLoS Pathog. 2018, 14, e1007313. [Google Scholar] [CrossRef]
- Maroui, M.A.; Callé, A.; Cohen, C.; Streichenberger, N.; Texier, P.; Takissian, J.; Rousseau, A.; Poccardi, N.; Welsch, J.; Corpet, A.; et al. Latency entry of herpes simplex Virus 1 is determined by the interaction of its genome with the nuclear environment. PLoS Pathog. 2016, 12, e1005834. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sawtell, N.M. Evidence that the herpes simplex virus type 1 ICP0 protein does not initiate reactivation from latency in vivo. J. Virol. 2006, 80, 10919–10930. [Google Scholar] [CrossRef]
- Cai, W.; Astor, T.L.; Liptak, L.M.; Cho, C.; Coen, D.M.; Schaffer, P.A. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 1993, 67, 7501–7512. [Google Scholar] [CrossRef]
- Roizman, B. The checkpoints of viral gene expression in productive and latent infection: The role of the HDAC/CoREST/LSD1/REST repressor complex. J. Virol. 2011, 85, 7474–7482. [Google Scholar] [CrossRef]
- Härle, P.; Sainz, B.J.; Carr, D.J.J.; Halford, W.P. The immediate-early protein, ICP0, is essential for the resistance of herpes simplex virus to interferon-alpha/beta. Virology 2002, 293, 295–304. [Google Scholar] [CrossRef]
- Halford, W.P.; Schaffer, P.A. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol. 2001, 75, 3240–3249. [Google Scholar] [CrossRef]
- Deschamps, T.; Waisner, H.; Dogrammatzis, C.; Roy, A.; Chacko, S.; Perera, C.; Prisinzano, T.E.; Kalamvoki, M. Discovery of small-molecule inhibitors targeting the E3 ubiquitin ligase activity of the herpes simplex virus 1 ICP0 protein using an in vitro high-throughput screening assay. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Hagglund, R.; Roizman, B. Herpes simplex virus 1 mutant in which the ICP0 HUL-1 E3 ubiquitin ligase site is disrupted stabilizes cdc34 but degrades D-type cyclins and exhibits diminished neurotoxicity. J. Virol. 2003, 77, 13194–13202. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gu, H.; Poon, A.P.; Roizman, B. During its nuclear phase the multifunctional regulatory protein ICP0 undergoes proteolytic cleavage characteristic of polyproteins. Proc. Natl. Acad. Sci. USA 2009, 106, 19132–19137. [Google Scholar] [CrossRef]
- Boutell, C.; Cuchet-Lourenço, D.; Vanni, E.; Orr, A.; Glass, M.; McFarlane, S.; Everett, R.D. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 2011, 7, e1002245. [Google Scholar] [CrossRef]
- Cuchet-Lourenço, D.; Vanni, E.; Glass, M.; Orr, A.; Everett, R.D. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 2012, 86, 11209–11222. [Google Scholar] [CrossRef]
- Sloan, E.; Tatham, M.H.; Groslambert, M.; Glass, M.; Orr, A.; Hay, R.T.; Everett, R.D. Analysis of the SUMO2 proteome during HSV-1 infection. PLoS Pathog. 2015, 11, e1005059. [Google Scholar] [CrossRef]
- Everett, R.D.; Boutell, C.; Pheasant, K.; Cuchet-Lourenço, D.; Orr, A. Sequences related to SUMO interaction motifs in herpes simplex virus 1 protein ICP0 act cooperatively to stimulate virus infection. J. Virol. 2014, 88, 2763–2774. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Parsy, M.-L.; Orr, A. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol. 2009, 83, 4963–4977. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Freemont, P.; Saitoh, H.; Dasso, M.; Orr, A.; Kathoria, M.; Parkinson, J. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 1998, 72, 6581–6591. [Google Scholar] [CrossRef] [PubMed]
- Chelbi-Alix, M.K.; de Thé, H. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 1999, 18, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Blyszcz, T.; González-Prieto, R.; Vertegaal, A.C.O.; Goldberg, A.L. Inhibiting ubiquitination causes an accumulation of SUMOylated newly synthesized nuclear proteins at PML bodies. J. Biol. Chem. 2019, 294, 15218–15234. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Murray, J. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 2005, 79, 5078–5089. [Google Scholar] [CrossRef]
- Maroui, M.A.; Maarifi, G.; McManus, F.P.; Lamoliatte, F.; Thibault, P.; Chelbi-Alix, M.K. Promyelocytic Leukemia Protein (PML) requirement for interferon-induced global cellular SUMOylation. Mol. Cell. Proteom. 2018, 17, 1196–1208. [Google Scholar] [CrossRef] [PubMed]
- Sloan, E.; Orr, A.; Everett, R.D. MORC3, a Component of PML nuclear bodies, has a role in restricting herpes simplex virus 1 and human cytomegalovirus. J. Virol. 2016, 90, 8621–8633. [Google Scholar] [CrossRef] [PubMed]
- Conn, K.L.; Wasson, P.; McFarlane, S.; Tong, L.; Brown, J.R.; Grant, K.G.; Domingues, P.; Boutell, C. Novel role for protein inhibitor of activated STAT 4 (PIAS4) in the restriction of herpes simplex virus 1 by the cellular intrinsic antiviral immune response. J. Virol. 2016, 90, 4807–4826. [Google Scholar] [CrossRef]
- Brown, J.R.; Conn, K.L.; Wasson, P.; Charman, M.; Tong, L.; Grant, K.; McFarlane, S.; Boutell, C. SUMO ligase protein inhibitor of activated STAT1 (PIAS1) is a constituent promyelocytic leukemia nuclear body protein that contributes to the intrinsic antiviral immune response to herpes simplex virus 1. J. Virol. 2016, 90, 5939–5952. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Long, M.C.; Kilvert, M.A.; Lam, V.; Rice, S.A.; Spencer, C.A. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 1996, 70, 7471–7477. [Google Scholar] [CrossRef]
- Parkinson, J.; Lees-Miller, S.P.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol. 1999, 73, 650–657. [Google Scholar] [CrossRef]
- Jackson, S.A.; DeLuca, N.A. Relationship of herpes simplex virus genome configuration to productive and persistent infections. Proc. Natl. Acad. Sci. USA 2003, 100, 7871–7876. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Everett, R.D.; Weitzman, M.D. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 2011, 7, e1002084. [Google Scholar] [CrossRef]
- Chaurushiya, M.S.; Lilley, C.E.; Aslanian, A.; Meisenhelder, J.; Scott, D.C.; Landry, S.; Ticau, S.; Boutell, C.; Yates, J.R., III; Schulman, B.A.; et al. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: Viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol. Cell 2012, 46, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Nagel, C.-H.; Albrecht, N.; Milovic-Holm, K.; Mariyanna, L.; Keyser, B.; Abel, B.; Weseloh, B.; Hofmann, T.G.; Eibl, M.M.; Hauber, J. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. J. Virol. 2011, 85, 7644–7657. [Google Scholar] [CrossRef] [PubMed]
- Conwell, S.E.; White, A.E.; Harper, J.W.; Knipe, D.M. Identification of TRIM27 as a novel degradation target of herpes simplex virus 1 ICP0. J. Virol. 2015, 89, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar] [CrossRef]
- Herrera, F.J.; Triezenberg, S.J. VP16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef]
- Kent, J.R.; Zeng, P.-Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef]
- Huang, J.; Kent, J.R.; Placek, B.; Whelan, K.A.; Hollow, C.M.; Zeng, P.-Y.; Fraser, N.W.; Berger, S.L. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J. Virol. 2006, 80, 5740–5746. [Google Scholar] [CrossRef]
- Cabral, J.M.; Oh, H.S.; Knipe, D.M. ATRX promotes maintenance of herpes simplex virus heterochromatin during chromatin stress. eLife 2018, 7. [Google Scholar] [CrossRef]
- Lee, J.S.; Raja, P.; Knipe, D.M. Herpesviral ICP0 protein promotes two waves of heterochromatin removal on an early viral promoter during lytic infection. mBio 2016, 7. [Google Scholar] [CrossRef]
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 2004, 78, 6744–6757. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.P.W.; Gu, H.; Roizman, B. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 9993–9998. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, W.E., II; DeLuca, N.A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J. Virol. 1999, 73, 8245–8255. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.P.W.; Liang, Y.; Roizman, B. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J. Virol. 2003, 77, 12671–12678. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17721–17726. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. The Histone Acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. J. Virol. 2011, 85, 9472–9477. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. Interwoven roles of cyclin D3 and cdk4 recruited by ICP0 and ICP4 in the expression of herpes simplex virus genes. J. Virol. 2010, 84, 9709–9717. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef]
- Kutluay, S.B.; Triezenberg, S.J. Regulation of histone deposition on the herpes simplex virus type 1 genome during lytic infection. J. Virol. 2009, 83, 5835–5845. [Google Scholar] [CrossRef] [PubMed]
- Placek, B.J.; Huang, J.; Kent, J.R.; Dorsey, J.; Rice, L.; Fraser, N.W.; Berger, S.L. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009, 83, 1416–1421. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 2009, 83, 4376–4385. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liang, Y.; Mandel, G.; Roizman, B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7571–7576. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Roizman, B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. USA 2007, 104, 17134–17139. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Roizman, B. Nuclear retention of ICP0 in cells exposed to HDAC inhibitor or transfected with DNA before infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. USA 2008, 105, 20488–20493. [Google Scholar] [CrossRef]
- Merkl, P.E.; Orzalli, M.H.; Knipe, D.M. Mechanisms of host IFI16, PML, and Daxx protein restriction of herpes simplex virus 1 replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Lukashchuk, V.; Everett, R.D. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 2010, 84, 4026–4040. [Google Scholar] [CrossRef]
- Everett, R.D. Dynamic response of IFI16 and promyelocytic leukemia nuclear body components to herpes simplex virus 1 infection. J. Virol. 2016, 90, 167–179. [Google Scholar] [CrossRef]
- Diner, B.A.; Lum, K.K.; Toettcher, J.E.; Cristea, I.M. Viral DNA sensors IFI16 and cyclic GMP-AMP synthase possess distinct functions in regulating viral gene expression, immune defenses, and apoptotic responses during herpesvirus infection. mBio 2016, 7. [Google Scholar] [CrossRef]
- McFarlane, S.; Orr, A.; Roberts, A.P.E.; Conn, K.L.; Iliev, V.; Loney, C.; Filipe, A.d.S.; Smollett, K.; Gu, Q.; Robertson, N.; et al. The histone chaperone HIRA promotes the induction of host innate immune defences in response to HSV-1 infection. PLoS Pathog. 2019, 15, e1007667. [Google Scholar] [CrossRef]
- Jurak, I.; Silverstein, L.B.; Sharma, M.; Coen, D.M. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J. Virol. 2012, 86, 10093–10102. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative contributions of herpes simplex virus 1 ICP0 and vhs to loss of cellular IFI16 vary in different human cell types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [PubMed]
- Cuchet-Lourenço, D.; Anderson, G.; Sloan, E.; Orr, A.; Everett, R.D. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J. Virol. 2013, 87, 13422–13432. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the antiviral factor interferon gamma-inducible protein 16 (IFI16) mediate immune signaling and herpes simplex virus-1 immunosuppression. Mol. Cell. Proteomics 2015, 14, 2341–2356. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; van Sant, C.; Roizman, B. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 1997, 71, 7328–7336. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. ICP0 enables and monitors the function of D cyclins in herpes simplex virus 1 infected cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14576–14580. [Google Scholar] [CrossRef]
- Li, H.; Baskaran, R.; Krisky, D.M.; Bein, K.; Grandi, P.; Cohen, J.B.; Glorioso, J.C. Chk2 is required for HSV-1 ICP0-mediated G2/M arrest and enhancement of virus growth. Virology 2008, 375, 13–23. [Google Scholar] [CrossRef]
- Lomonte, P.; Sullivan, K.F.; Everett, R.D. Degradation of nucleosome-associated centromeric histone H3-like protein CENP-A induced by herpes simplex virus type 1 protein ICP0. J. Biol. Chem. 2001, 276, 5829–5835. [Google Scholar] [CrossRef]
- Lomonte, P.; Morency, E. Centromeric protein CENP-B proteasomal degradation induced by the viral protein ICP0. FEBS Lett. 2007, 581, 658–662. [Google Scholar] [CrossRef]
- Everett, R.D.; Earnshaw, W.C.; Findlay, J.; Lomonte, P. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 1999, 18, 1526–1538. [Google Scholar] [CrossRef]
- Gross, S.; Catez, F.; Masumoto, H.; Lomonte, P. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS ONE 2012, 7, e44227. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Kim, E.T.; Vladimirova, O.; Dheekollu, J.; Wang, Z.; Newhart, A.; Liu, D.; Myers, J.L.; Hensley, S.E.; Moffat, J.; et al. HSV-1 remodels host telomeres to facilitate viral replication. Cell Rep. 2014, 9, 2263–2278. [Google Scholar] [CrossRef] [PubMed]
- Mossman, K.L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus ICP0 mutants are hypersensitive to interferon. J. Virol. 2000, 74, 2052–2056. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Harrison, T.E.; Laslo, K.M.; Machalek, M.A.; Moorman, N.J.; Virgin, H.W. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J. Exp. Med. 1999, 189, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Burleigh, K.; Maltbaek, J.H.; Cambier, S.; Green, R.; Gale, M.J.; James, R.C.; Stetson, D.B. Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Broekema, N.M.; Diner, B.A.; Hancks, D.C.; Elde, N.C.; Cristea, I.M.; Knipe, D.M. cGAS-mediated stabilization of IFI16 promotes innate signaling during herpes simplex virus infection. Proc. Natl. Acad. Sci. USA 2015, 112, 1773–1781. [Google Scholar] [CrossRef]
- Van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef]
- Lin, R.; Noyce, R.S.; Collins, S.E.; Everett, R.D.; Mossman, K.L. The herpes simplex virus ICP0 RING finger domain inhibits IRF3- and IRF7-mediated activation of interferon-stimulated genes. J. Virol. 2004, 78, 1675–1684. [Google Scholar] [CrossRef]
- Melroe, G.T.; DeLuca, N.A.; Knipe, D.M. Herpes simplex virus 1 has multiple mechanisms for blocking virus-induced interferon production. J. Virol. 2004, 78, 8411–8420. [Google Scholar] [CrossRef]
- Melroe, G.T.; Silva, L.; Schaffer, P.A.; Knipe, D.M. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef]
- Waisner, H.; Kalamvoki, M. The ICP0 protein of herpes simplex virus 1 (HSV-1) downregulates major autophagy adaptor proteins sequestosome 1 and optineurin during the early stages of HSV-1 infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Kalamvoki, M.; Gu, H.; Roizman, B. Overexpression of the ubiquitin-specific protease 7 resulting from transfection or mutations in the ICP0 binding site accelerates rather than depresses herpes simplex virus 1 gene expression. J. Virol. 2012, 86, 12871–12878. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.H.; Thompson, T.W.; Davido, D.J. N-terminal phosphorylation sites of herpes simplex virus 1 ICP0 differentially regulate its activities and enhance viral replication. J. Virol. 2013, 87, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Colleran, A.; Collins, P.E.; O’Carroll, C.; Ahmed, A.; Mao, X.; McManus, B.; Kiely, P.A.; Burstein, E.; Carmody, R.J. Deubiquitination of NF-κB by ubiquitin-specific protease-7 promotes transcription. Proc. Natl. Acad. Sci. USA 2013, 110, 618–623. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Kurakin, A.; Roizman, B. Herpes simplex virus 1 infected cell protein 0 forms a complex with CIN85 and Cbl and mediates the degradation of EGF receptor from cell surfaces. Proc. Natl. Acad. Sci. USA 2005, 102, 5838–5843. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, T.; Dogrammatzis, C.; Mullick, R.; Kalamvoki, M. Cbl E3 ligase mediates the removal of nectin-1 from the surface of herpes simplex virus 1-infected cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Leib, D.A.; Coen, D.M.; Bogard, C.L.; Hicks, K.A.; Yager, D.R.; Knipe, D.M.; Tyler, K.L.; Schaffer, P.A. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 1989, 63, 759–768. [Google Scholar] [CrossRef]
- Halford, W.P.; Kemp, C.D.; Isler, J.A.; Davido, D.J.; Schaffer, P.A. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J. Virol. 2001, 75, 6143–6153. [Google Scholar] [CrossRef]
- Harris, R.A.; Everett, R.D.; Zhu, X.X.; Silverstein, S.; Preston, C.M. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 1989, 63, 3513–3515. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone methyl/phospho switch is required for herpes simplex virus reactivation. Cell Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.L.; Preston, C.M.; Sawtell, N.M. De novo synthesis of VP16 coordinates the exit from HSV latency in vivo. PLoS Pathog. 2009, 5, e1000352. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Dalrymple, M.A.; Davison, A.J.; Dolan, A.; Frame, M.C.; McNab, D.; Perry, L.J.; Scott, J.E.; Taylor, P. The complete DNA sequence of the long unique region in the genome of herpes simplx virus type 1. J. Gen. Virol. 1988, 69, 1531–1574. [Google Scholar] [CrossRef] [PubMed]
- McKnight, J.L.; Pellett, P.E.; Jenkins, F.J.; Roizman, B. Characterization and nucleotide sequence of two herpes simplex virus 1 genes whose products modulate alpha-trans-inducing factor-dependent activation of alpha genes. J. Virol. 1987, 61, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Vittone, V.; Diefenbach, E.; Triffett, D.; Douglas, M.W.; Cunningham, A.L.; Diefenbach, R.J. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 2005, 79, 9566–9571. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.E.; Roizman, B. Identification of three genes nonessential for growth in cell culture near the right terminus of the unique sequences of long component of herpes simplex virus 1. Virology 1990, 177, 684–691. [Google Scholar] [CrossRef]
- Kato, K.; Daikoku, T.; Goshima, F.; Kume, H.; Yamaki, K.; Nishiyama, Y. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 2000, 145, 2149–2162. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, J.; Wang, X.; Yang, T.; Geller, A.I. Enhanced long-term expression from helper virus-free HSV-1 vectors packaged in the presence of deletions in genes that modulate the function of VP16, U L 46 and U L 47. J. Neurosci. Methods 2005, 145, 1–9. [Google Scholar] [CrossRef]
- McLean, G.; Rixon, F.; Langeland, N.; Haarr, L.; Marsden, H. Identification and characterization of the virion protein products of herpes simplex virus type 1 gene UL47. J. Gen. Virol. 1990, 71, 2953–2960. [Google Scholar] [CrossRef]
- Meredith, D.M.; Lindsay, J.A.; Halliburton, I.W.; Whittaker, G.R. Post-translational modification of the tegument proteins (VP13 and VP14) of herpes simplex virus type 1 by glycosylation and phosphorylation. J. Gen. Virol. 1991, 72, 2771–2775. [Google Scholar] [CrossRef]
- Zhang, Y.; McKnight, J.L. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 1993, 67, 1482–1492. [Google Scholar] [CrossRef]
- Kopp, M.; Klupp, B.G.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: Role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 2002, 76, 8820–8833. [Google Scholar] [CrossRef] [PubMed]
- Strunk, U.; Saffran, H.A.; Wu, F.W.; Smiley, J.R. Role of herpes simplex virus VP11/12 tyrosine-based motifs in binding and activation of the Src family kinase Lck and recruitment of p85, Grb2, and Shc. J. Virol. 2013, 87, 11276–11286. [Google Scholar] [CrossRef] [PubMed]
- Yanagida, N.; Yoshida, S.; Nazerian, K.; Lee, L.F. Nucleotide and predicted amino acid sequences of Marek’s disease virus homologues of herpes simplex virus major tegument proteins. J. Gen. Virol. 1993, 74, 1837–1845. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, D.E.; Misra, V. The most abundant protein in bovine herpes 1 virions is a homologue of herpes simplex virus type 1 UL47. J. Gen. Virol. 1991, 72, 3077–3084. [Google Scholar] [CrossRef]
- Whittaker, G.R.; Riggio, M.P.; Halliburton, I.W.; Killington, R.A.; Allen, G.P.; Meredith, D.M. Antigenic and protein sequence homology between VP13/14, a herpes simplex virus type 1 tegument protein, and gp10, a glycoprotein of equine herpesvirus 1 and 4. J. Virol. 1991, 65, 2320–2326. [Google Scholar] [CrossRef]
- Verjans, G.M.; Dings, M.E.; McLauchlan, J.; van der Kooi, A.; Hoogerhout, P.; Brugghe, H.F.; Timmermans, H.A.; Baarsma, G.S.; Osterhaus, A.D. Intraocular T cells of patients with herpes simplex virus (HSV)-induced acute retinal necrosis recognize HSV tegument proteins VP11/12 and VP13/14. J. Infect. Dis. 2000, 182, 923–927. [Google Scholar] [CrossRef]
- Zahariadis, G.; Wagner, M.J.; Doepker, R.C.; Maciejko, J.M.; Crider, C.M.; Jerome, K.R.; Smiley, J.R. Cell-type-specific tyrosine phosphorylation of the herpes simplex virus tegument protein VP11/12 encoded by gene UL46. J. Virol. 2008, 82, 6098–6108. [Google Scholar] [CrossRef][Green Version]
- Wagner, M.J.; Smiley, J.R. Herpes simplex virus requires VP11/12 to induce phosphorylation of the activation loop tyrosine (Y394) of the Src family kinase Lck in T lympocytes. J. Virol. 2009, 83, 12452–12461. [Google Scholar] [CrossRef]
- Benetti, L.; Roizman, B. Protein kinase B/Akt is present in activated form throughout the entire replicative cycle of deltaU(S)3 mutant virus but only at early times after infection with wild-type herpes simplex virus 1. J. Virol. 2006, 80, 3341–3348. [Google Scholar] [CrossRef]
- Wagner, M.J.; Smiley, J.R. Herpes simplex virus requires VP11/12 to activate src family kinase-phosphoinositide 3-kinase-akt signaling. J. Virol. 2011, 85, 2803–2812. [Google Scholar] [CrossRef]
- Strunk, U.; Ramos, D.G.; Saffran, H.A.; Smiley, J.R. Role of Herpes simplex virus 1 VP11/12 tyrosine-based binding motifs for Src family kinases, p85, Grb2 and Shc in activation of the phosphoinositide 3-kinase-Akt pathway. Virology 2016, 498, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Leyton, L.; Hott, M.; Arancibia, Y.; Spichiger, C.; McNiven, M.A.; Court, F.A.; Concha, M.I.; Burgos, P.V.; Otth, C. Herpes simplex virus type 1 neuronal infection perturbs golgi apparatus integrity through activation of src tyrosine kinase and dyn-2 GTPase. Front. Cell. Infect. Microbiol. 2017, 7, 371. [Google Scholar] [CrossRef] [PubMed]
- Bello-Morales, R.; Crespillo, A.J.; Fraile-Ramos, A.; Tabarés, E.; Alcina, A.; López-Guerrero, J.A. Role of the small GTPase Rab27a during herpes simplex virus infection of oligodendrocytic cells. BMC Microbiol. 2012, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Corre, B.; Foulon, E.; Dufour, E.; Veillette, A.; Acuto, O.; Michel, F. T cell receptor for antigen induces linker for activation of T cell-dependent activation of a negative signaling complex involving Dok-2, SHIP-1, and Grb-2. J. Exp. Med. 2006, 203, 2509–2518. [Google Scholar] [CrossRef]
- Némorin, J.-G.; Laporte, P.; Bérubé, G.; Duplay, P. p62 dok negatively regulates CD2 signaling in jurkat cells. J. Immunol. 2001, 166, 4408–4415. [Google Scholar] [CrossRef]
- Lahmidi, S.; Strunk, U.; Smiley, J.R.; Pearson, A.; Duplay, P. Herpes simplex virus 1 infection of T cells causes VP11/12-dependent phosphorylation and degradation of the cellular protein Dok-2. Virology 2017, 511, 66–73. [Google Scholar] [CrossRef]
- Vink, E.I.; Lee, S.; Smiley, J.R.; Mohr, I. Remodeling mTORC1 responsiveness to amino acids by the herpes simplex virus UL46 and Us3 gene products supports replication during nutrient insufficiency. J. Virol. 2018, 92, 1–10. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- You, H.; Zheng, S.; Huang, Z.; Lin, Y.; Shen, Q.; Zheng, C. Herpes simplex virus 1 tegument protein ul46 inhibits tank-binding kinase 1-mediated signaling. mBio 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Xu, J.; Gao, F.; Wu, J.; Zheng, H.; Tong, W.; Cheng, X.; Liu, Y.; Zhu, H.; Fu, X.; Jiang, Y.; et al. Characterization of nucleocytoplasmic shuttling of pseudorabies virus protein UL46. Front. Vet. Sci. 2020, 7, 484. [Google Scholar] [CrossRef]
- Liu, Z.; Kato, A.; Shindo, K.; Noda, T.; Sagara, H.; Kawaoka, Y.; Arii, J.; Kawaguchi, Y. Herpes simplex virus 1 UL47 Interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J. Virol. 2014, 88, 4657–4667. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, M.; Elliott, G. Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J. Virol. 2001, 75, 2566–2574. [Google Scholar] [CrossRef]
- Donnelly, M.; Elliott, G. Fluorescent tagging of herpes simplex virus tegument protein VP13/14 in virus infection. J. Virol. 2001, 75, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Yedowitz, J.C.; Kotsakis, A.; Schlegel, E.F.M.; Blaho, J.A. Nuclear localizations of the herpes simplex virus type 1 tegument proteins VP13/14, vhs, and VP16 precede VP22-dependent microtubule reorganization and VP22 nuclear import. J. Virol. 2005, 79, 4730–4743. [Google Scholar] [CrossRef] [PubMed]
- Dobrikova, E.; Shveygert, M.; Walters, R.; Gromeier, M. Herpes simplex virus proteins ICP27 and UL47 associate with polyadenylate-binding protein and control its subcellular distribution. J. Virol. 2010, 84, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Shu, M.; Taddeo, B.; Zhang, W.; Roizman, B. Selective degradation of mRNAs by the HSV host shutoff RNase is regulated by the UL47 tegument protein. Proc. Natl. Acad. Sci. USA 2013, 110. [Google Scholar] [CrossRef] [PubMed]
- Heine, J.W.; Honess, R.W.; Cassai, E.; Roizman, B. Proteins specified by herpes simplex virus XII. The virion polypeptides of type 1 strains. J. Virol. 1974, 14, 640–651. [Google Scholar] [CrossRef]
- Aints, A.; Güven, H.; Gahrton, G.; Smith, C.I.E.; Dilber, M.S. Mapping of herpes simplex virus-1 VP22 functional domains for inter- and subcellular protein targeting. Gene Ther. 2001, 8, 1051–1056. [Google Scholar] [CrossRef][Green Version]
- Morrison, E.E.; Wang, Y.F.; Meredith, D.M. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J. Virol. 1998, 72, 7108–7114. [Google Scholar] [CrossRef]
- Pomeranz, L.E.; Blaho, J.A. Assembly of infectious herpes simplex virus type 1 virions in the absence of full-length VP22. J. Virol. 2000, 74, 10041–10054. [Google Scholar] [CrossRef]
- Van Leeuwen, H.; Okuwaki, M.; Hong, R.; Chakravarti, D.; Nagata, K.; O’Hare, P. Herpes simplex virus type 1 tegument protein VP22 interacts with TAF-I proteins and inhibits nucleosome assembly but not regulation of histone acetylation by INHAT. J. Gen. Virol. 2003, 84, 2501–2510. [Google Scholar] [CrossRef] [PubMed]
- López, M.R.; Schlegel, E.F.M.; Wintersteller, S.; Blaho, J.A. The major tegument structural protein VP22 targets areas of dispersed nucleolin and marginalized chromatin during productive herpes simplex virus 1 infection. Virus Res. 2008, 136, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liu, L.; Wu, L.; Wang, L.; Dong, C.; Li, W.; Li, Q. Herpes simplex virus type 1 tegument protein VP22 is capable of modulating the transcription of viral TK and gC genes via interaction with viral ICP0. Biochimie 2010, 92, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Kato, A.; Satoh, Y.; Ide, T.; Sagou, K.; Kimura, K.; Hasegawa, H.; Kawaguchi, Y. Herpes simplex virus 1 VP22 regulates translocation of multiple viral and cellular proteins and promotes neurovirulence. J. Virol. 2012, 86, 5264–5277. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, M.T.; Taddeo, B.; Giuffrè-Cuculletto, M.; Medici, M.A.; Mastino, A.; Roizman, B. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J. Virol. 2007, 81, 10924–10932. [Google Scholar] [CrossRef] [PubMed]
- Mbong, E.F.; Woodley, L.; Dunkerley, E.; Schrimpf, J.E.; Morrison, L.A.; Duffy, C. Deletion of the herpes simplex virus 1 UL49 gene results in mRNA and protein translation defects that are complemented by secondary mutations in UL41. J. Virol. 2012, 86, 12351–12361. [Google Scholar] [CrossRef]
- Duffy, C.; Mbong, E.F.; Baines, J.D. VP22 of herpes simplex virus 1 promotes protein synthesis at late times in infection and accumulation of a subset of viral mRNAs at early times in infection. J. Virol. 2009, 83, 1009–1017. [Google Scholar] [CrossRef]
- Brignati, M.J.; Loomis, J.S.; Wills, J.W.; Courtney, R.J. Membrane association of VP22, a herpes simplex virus type 1 tegument protein. J. Virol. 2003, 77, 4888–4898. [Google Scholar] [CrossRef]
- O’Regan, K.J.; Brignati, M.J.; Murphy, M.A.; Bucks, M.A.; Courtney, R.J. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 is facilitated by trans-Golgi network localization and is independent of interaction with glycoprotein E. Virology 2010, 405, 176–192. [Google Scholar] [CrossRef][Green Version]
- Leslie, J.; Rixon, F.J.; Mclauchlan, J. Overexpression of the herpes simplex virus type 1 tegument protein VP22 increases its incorporation into virus particles. Virology 1996, 68, 60–68. [Google Scholar] [CrossRef][Green Version]
- Chi, J.H.I.; Harley, C.A.; Mukhopadhyay, A.; Wilson, D.W. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 2005, 86, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Starkey, J.L.; Han, J.; Chadha, P.; Marsh, J.A.; Wills, J.W. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J. Virol. 2014, 88, 110–119. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, K.J.; Bucks, M.A.; Murphy, M.A.; Wills, J.W.; Courtney, R.J. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 2007, 358, 192–200. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, K.J.; Murphy, M.A.; Bucks, M.A.; Wills, J.W.; Courtney, R.J. Incorporation of the herpes simplex virus type 1 tegument protein VP22 into the virus particle is independent of interaction with VP16. Virology 2007, 369, 263–280. [Google Scholar] [CrossRef]
- Duffy, C.; LaVail, J.H.; Tauscher, A.N.; Wills, E.G.; Blaho, J.A.; Baines, J.D. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 2006, 80, 8664–8675. [Google Scholar] [CrossRef]
- Elliott, G.; Mouzakitis, G.; O’Hare, P. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 1995, 69, 7932–7941. [Google Scholar] [CrossRef]
- Maringer, K.; Stylianou, J.; Elliott, G. A Network of protein interactions around the herpes simplex virus tegument protein VP22. J. Virol. 2012, 86, 12971–12982. [Google Scholar] [CrossRef]
- Kotsakis, A.; Pomeranz, L.E.; Blouin, A.; Blaho, J.A. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 2001, 75, 8697–8711. [Google Scholar] [CrossRef]
- Blouin, A.; Blaho, J.A. Assessment of the subcellular localization of the herpes simplex virus structural protein VP22 in the absence of other viral gene products. Virus Res. 2001, 81, 57–68. [Google Scholar] [CrossRef]
- Van Leeuwen, H.; Elliott, G.; O’Hare, P. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J. Virol. 2002, 76, 3471–3481. [Google Scholar] [CrossRef]
- Liu, J.; Gallo, R.M.; Duffy, C.; Brutkiewicz, R.R. A VP22-null HSV-1 is impaired in inhibiting CD1d-mediated antigen presentation. Viral Immunol. 2016, 29, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Søby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef] [PubMed]
- Maruzuru, Y.; Ichinohe, T.; Sato, R.; Miyake, K.; Okano, T.; Suzuki, T.; Koshiba, T.; Koyanagi, N.; Tsuda, S.; Watanabe, M.; et al. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe 2018, 23, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.; O’Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef]
- Sciortino, M.T.; Taddeo, B.; Poon, A.P.W.; Mastino, A.; Roizman, B. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. USA 2002, 99, 8318–8323. [Google Scholar] [CrossRef]
- Phelan, A.; Elliott, G.; Hare, P.O. Intercellular delivery of functional p53 by the herpesvirus protein VP22. Nat. Biotechnol. 1998, 16, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Xu, B.; Koch, P.; Roth, J.A. Intercellular trafficking of VP22-GFP fusion proteins is not observed in cultured mammalian cells. Gene Ther. 1998, 5, 1420–1424. [Google Scholar] [CrossRef]
- Dilber, M.S.; Phelan, A.; Aints, A.; Mohamed, A.J.; Elliott, G.; Edvard Smith, C.I.; O’Hare, P. Intercellular delivery of thymidine kinase prodrug activating enzyme by the herpes simplex virus protein, VP22. Gene Ther. 1999, 6, 12–21. [Google Scholar] [CrossRef]
- Sheridan, P.J.; Lawrie, A.; Crossman, D.C.; Holt, C.M.; Newman, C.M. VP22-mediated intercellular transport correlates with enhanced biological activity of MybEngrailed but not (HSV-I) thymidine kinase fusion proteins in primary vascular cells following non-viral transfection. J. Gene Med. 2005, 7, 375–385. [Google Scholar] [CrossRef]
- Hakkarainen, T.; Wahlfors, T.; Meriläinen, O.; Loimas, S.; Hemminki, A.; Wahlfors, J. VP22 does not significantly enhance enzyme prodrug cancer gene therapy as a part of a VP22-HSVTk-GFP triple fusion construct. J. Gene Med. 2005, 7, 898–907. [Google Scholar] [CrossRef]
- Beerens, A.M.J.; Rots, M.G.; de Vries, E.F.J.; Haisma, H.J. Fusion of herpes simplex virus thymidine kinase to VP22 does not result in intercellular trafficking of the protein. Int. J. Mol. Med. 2007, 19, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Aints, A.; Dilber, M.S.; Smith, C.I.E. Intercellular spread of GFP-VP22. J. Gene Med. 1999, 1, 275–279. [Google Scholar] [CrossRef]
- Dogrammatzis, C.; Deschamps, T.; Kalamvoki, M. Biogenesis of extracellular vesicles during herpes simplex virus 1 infection: Role of the CD63 tetraspanin. J. Virol. 2018, 93. [Google Scholar] [CrossRef] [PubMed]
- Dogrammatzis, C.; Saleh, S.; Deighan, C.; Kalamvoki, M. Diverse populations of extracellular vesicles with opposite functions during herpes simplex virus 1 infection. J. Virol. 2020, 95, e02357-20. [Google Scholar]
- Greco, O.; Joiner, M.C.; Doleh, A.; Scott, S.D. VP22-mediated intercellular transport for suicide gene therapy under oxic and hypoxic conditions. Gene Ther. 2005, 12, 974–979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, C.S.; Kong, B.H.; Xia, H.H.Q.; Ellem, K.A.O.; Wei, M.Q. VP22 enhanced intercellular trafficking of HSV thymidine kinase reduced the level of ganciclovir needed to cause suicide cell death. J. Gene Med. 2001, 3, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Wybranietz, W.A.; Groß, C.D.; Phelan, A.; O’Hare, P.; Spiegel, M.; Graepler, F.; Bitzer, M.; Stähler, P.; Gregor, M.; Lauer, U.M. Enhanced suicide gene effect by adenoviral transduction of aVP22-cytosine deaminase (CD) fusion gene. Gene Ther. 2001, 8, 1654–1664. [Google Scholar] [CrossRef]
- Lee, K.C.; Hamstra, D.A.; Bullarayasamudram, S.; Bhojani, M.S.; Moffat, B.A.; Dornfeld, K.J.; Ross, B.D.; Rehemtulla, A. Fusion of the HSV-1 tegument protein vp22 to cytosine deaminase confers enhanced bystander effect and increased therapeutic benefit. Gene Ther. 2006, 13, 127–137. [Google Scholar] [CrossRef]
- Cashman, S.M.; Sadowski, S.L.; Morris, D.J.; Frederick, J.; Kumar-Singh, R. Intercellular trafficking of adenovirus-delivered HSV VP22 from the retinal pigment epithelium to the photoreceptors—Implications for gene therapy. Mol. Ther. 2002, 6, 813–823. [Google Scholar] [CrossRef]
- Kretz, A.; Wybranietz, W.A.; Hermening, S.; Lauer, U.M.; Isenmann, S. HSV-1 VP22 augments adenoviral gene transfer to CNS neurons in the retina and striatum in vivo. Mol. Ther. 2003, 7, 659–669. [Google Scholar] [CrossRef]
- Xiong, F.; Xiao, S.; Peng, F.; Zheng, H.; Yu, M.; Ruan, Y.; Li, W.; Shang, Y.; Zhao, C.; Zhou, W.; et al. Herpes simplex virus VP22 enhances adenovirus-mediated microdystrophin gene transfer to skeletal muscles in dystrophin-deficient (mdx) mice. Hum. Gene Ther. 2007, 18, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y.; Xia, Y.; Zhang, L.; Yang, Q.; Lei, J. A DNA vaccine encoding VP22 of herpes simplex virus type I (HSV-1) and OprF confers enhanced protection from Pseudomonas aeruginosa in mice. Vaccine 2016, 34, 4399–4405. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-F.; Cheng, W.-F.; Chai, C.-Y.; Hsu, K.-F.; He, L.; Ling, M.; Wu, T.-C. Improving vaccine potency through intercellular spreading and enhanced MHC class I presentation of antigen. J. Immunol. 2001, 166, 5733–5740. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Proteins specified by herpes simplex virus. XI. Identification and relative molar rates of synthesis of structural and nonstructural herpes virus polypeptides in the infected cell. J. Virol. 1973, 12, 1347–1365. [Google Scholar] [CrossRef] [PubMed]
- Post, L.E.; Roizman, B. A generalized technique for deletion of specific genes in large genomes: A gene 22 of herpes simplex virus 1 is not essential for growth. Cell 1981, 25, 227–232. [Google Scholar] [CrossRef]
- Meignier, B.; Longnecker, R.; Mavromara-Nazos, P.; Sears, A.E.; Roizman, B. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus I. Virology 1988, 162, 251–254. [Google Scholar] [CrossRef]
- Sears, A.E.; Halliburton, I.W.; Meignier, B.; Silver, S.; Roizman, B. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: Growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 1985, 55, 338–346. [Google Scholar] [CrossRef]
- Poffenberger, K.L.; Raichlen, P.E.; Herman, R.C. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes 1993, 7, 171–186. [Google Scholar] [CrossRef]
- Jacquemont, B.; Verrier, B.; Epstein, A.L.; Machuca, I. Expression of immediate-early genes in herpes simplex virus type 1-infected XC cells: Lack of ICP22 (68K) polypeptide. J. Gen. Virol. 1984, 65, 1331–1340. [Google Scholar] [CrossRef]
- Orlando, J.S.; Astor, T.L.; Rundle, S.A.; Schaffer, P.A. The products of the herpes simplex virus type 1 immediate-early US1/US1.5 genes downregulate levels of S-phase-specific cyclins and facilitate virus replication in S-phase Vero cells. J. Virol. 2006, 80, 4005–4016. [Google Scholar] [CrossRef]
- Ogle, W.O.; Roizman, B. Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein. J. Virol. 1999, 73, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Poffenberger, K.L.; Idowu, A.D.; Fraser-Smith, E.B.; Raichlen, P.E.; Herman, R.C. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch. Virol. 1994, 139, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Orlando, J.S.; Balliet, J.W.; Kushnir, A.S.; Astor, T.L.; Kosz-Vnenchak, M.; Rice, S.A.; Knipe, D.M.; Schaffer, P.A. ICP22 is required for wild-type composition and infectivity of herpes simplex virus type 1 virions. J. Virol. 2006, 80, 9381–9390. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.R.; Kolb, A.W. Tyrosine 116 of the herpes simplex virus type 1 IEalpha22 protein is an ocular virulence determinant and potential phosphorylation site. Invest. Ophthalmol. Vis. Sci. 2003, 44, 4601–4607. [Google Scholar] [CrossRef]
- Bowles, R.N.; Blaho, J.A. A truncation mutation of the neurovirulence ICP22 protein produced by a recombinant HSV-1 generated by bacterial artificial chromosome technology targets infected cell nuclei. J. Neurovirol. 2011, 17, 559–569. [Google Scholar] [CrossRef]
- Holden, V.R.; Zhao, Y.; Thompson, Y.; Caughman, G.B.; Smith, R.H.; O’Callaghan, D.J. Characterization of the regulatory function of the ICP22 protein of equine herpesvirus type 1. Virology 1995, 210, 273–282. [Google Scholar] [CrossRef][Green Version]
- Kost, R.G.; Kupinsky, H.; Straus, S.E. Varicella-zoster virus gene 63: Transcript mapping and regulatory activity. Virology 1995, 209, 218–224. [Google Scholar] [CrossRef]
- Heineman, T.C.; Cohen, J.I. The varicella-zoster virus (VZV) open reading frame 47 (ORF47) protein kinase is dispensable for viral replication and is not required for phosphorylation of ORF63 protein, the VZV homolog of herpes simplex virus ICP22. J. Virol. 1995, 69, 7367–7370. [Google Scholar] [CrossRef]
- Gray, W.L.; Gusick, N.J.; Ek-Kommonen, C.; Kempson, S.E.; Fletcher, T.M. 3rd The inverted repeat regions of the simian varicella virus and varicella-zoster virus genomes have a similar genetic organization. Virus Res. 1995, 39, 181–193. [Google Scholar] [CrossRef]
- Cai, M.; Jiang, S.; Zeng, Z.; Li, X.; Mo, C.; Yang, Y.; Chen, C.; Xie, P.; Bian, Y.; Wang, J.; et al. Probing the nuclear import signal and nuclear transport molecular determinants of PRV ICP22. Cell Biosci. 2016, 6, 3. [Google Scholar] [CrossRef]
- Blaho, J.A.; Mitchell, C.; Roizman, B. Guanylylation and adenylylation of the alpha regulatory proteins of herpes simplex virus require a viral beta or gamma function. J. Virol. 1993, 67, 3891–3900. [Google Scholar] [CrossRef] [PubMed]
- Purves, F.C.; Roizman, B. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc. Natl. Acad. Sci. USA 1992, 89, 7310–7314. [Google Scholar] [CrossRef] [PubMed]
- Purves, F.C.; Ogle, W.O.; Roizman, B. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. USA 1993, 90, 6701–6705. [Google Scholar] [CrossRef] [PubMed]
- Blaho, J.A.; Zong, C.S.; Mortimer, K.A. Tyrosine phosphorylation of the herpes simplex virus type 1 regulatory protein ICP22 and a cellular protein which shares antigenic determinants with ICP22. J. Virol. 1997, 71, 9828–9832. [Google Scholar] [CrossRef]
- O’Toole, J.M.; Aubert, M.; Kotsakis, A.; Blaho, J.A. Mutation of the protein tyrosine kinase consensus site in the herpes simplex virus 1 alpha22 gene alters ICP22 posttranslational modification. Virology 2003, 305, 153–167. [Google Scholar] [CrossRef]
- Stelz, G.; Rücker, E.; Rosorius, O.; Meyer, G.; Stauber, R.H.; Spatz, M.; Eibl, M.M.; Hauber, J. Identification of two nuclear import signals in the alpha-gene product ICP22 of herpes simplex virus 1. Virology 2002, 295, 360–370. [Google Scholar] [CrossRef][Green Version]
- Ng, T.I.; Chang, Y.E.; Roizman, B. Infected cell protein 22 of herpes simplex virus 1 regulates the expression of virion host shutoff gene U(L)41. Virology 1997, 234, 226–234. [Google Scholar] [CrossRef]
- Advani, S.J.; Brandimarti, R.; Weichselbaum, R.R.; Roizman, B. The disappearance of cyclins A and B and the increase in activity of the G(2)/M-phase cellular kinase cdc2 in herpes simplex virus 1-infected cells require expression of the alpha22/U(S)1.5 and U(L)13 viral genes. J. Virol. 2000, 74, 8–15. [Google Scholar] [CrossRef]
- Advani, S.J.; Weichselbaum, R.R.; Roizman, B. The role of cdc2 in the expression of herpes simplex virus genes. Proc. Natl. Acad. Sci. USA 2000, 97, 10996–11001. [Google Scholar] [CrossRef]
- Advani, S.J.; Weichselbaum, R.R.; Roizman, B. Herpes simplex virus 1 activates cdc2 to recruit topoisomerase II alpha for post-DNA synthesis expression of late genes. Proc. Natl. Acad. Sci. USA 2003, 100, 4825–4830. [Google Scholar] [CrossRef]
- Smith-Donald, B.A.; Roizman, B. The interaction of herpes simplex virus 1 regulatory protein ICP22 with the cdc25C phosphatase is enabled in vitro by viral protein kinases US3 and UL13. J. Virol. 2008, 82, 4533–4543. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.A.; Long, M.C.; Lam, V.; Schaffer, P.A.; Spencer, C.A. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol. 1995, 69, 5550–5559. [Google Scholar] [CrossRef] [PubMed]
- Rice, S.A.; Long, M.C.; Lam, V.; Spencer, C.A. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 1994, 68, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Dahmus, M.E.; Rice, S.A. Repression of host RNA polymerase II transcription by herpes simplex virus type 1. J. Virol. 1997, 71, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, H.L.; Spencer, C.A. RNA polymerase II holoenzyme modifications accompany transcription reprogramming in herpes simplex virus type 1-infected cells. J. Virol. 2001, 75, 9872–9884. [Google Scholar] [CrossRef] [PubMed]
- Long, M.C.; Leong, V.; Schaffer, P.A.; Spencer, C.A.; Rice, S.A. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol. 1999, 73, 5593–5604. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; Rice, S.A. Identification of sequences in herpes simplex virus type 1 ICP22 that influence RNA polymerase II modification and viral late gene expression. J. Virol. 2009, 83, 128–139. [Google Scholar] [CrossRef]
- Bowman, J.J.; Schaffer, P.A. Origin of expression of the herpes simplex virus type 1 protein U(S)1.5. J. Virol. 2009, 83, 9183–9194. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, R.; Ward, P.L.; Ogle, W.O.; Roizman, B. Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the U(L)13 proteinkinase. J. Virol. 1997, 71, 1133–1139. [Google Scholar] [CrossRef]
- Jahedi, S.; Markovitz, N.S.; Filatov, F.; Roizman, B. Colocalization of the herpes simplex virus 1 UL4 protein with infected cell protein 22 in small, dense nuclear structures formed prior to onset of DNA synthesis. J. Virol. 1999, 73, 5132–5138. [Google Scholar] [CrossRef]
- Wu, N.; Watkins, S.C.; Schaffer, P.A.; DeLuca, N.A. Prolonged gene expression and cell survival after infection by a herpes simplex virus mutant defective in the immediate-early genes encoding ICP4, ICP27, and ICP22. J. Virol. 1996, 70, 6358–6369. [Google Scholar] [CrossRef] [PubMed]
- Cun, W.; Chen, J.; Zhang, Y.; Liu, L.; Li, Q. Analysis of the cellular localization of herpes simplex virus 1 immediate-early protein ICP22. Virol. Sin. 2010, 25, 158–167. [Google Scholar] [CrossRef]
- Durand, L.O.; Roizman, B. Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J. Virol. 2008, 82, 10591–10599. [Google Scholar] [CrossRef] [PubMed]
- Ou, M.; Sandri-Goldin, R.M. Inhibition of cdk9 during herpes simplex virus 1 infection impedes viral transcription. PLoS ONE 2013, 8, e79007. [Google Scholar] [CrossRef] [PubMed]
- Fraser, K.A.; Rice, S.A. Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J. Virol. 2007, 81, 5091–5101. [Google Scholar] [CrossRef]
- Fraser, K.A.; Rice, S.A. Herpes simplex virus type 1 infection leads to loss of serine-2 phosphorylation on the carboxyl-terminal domain of RNA polymerase II. J. Virol. 2005, 79, 11323–11334. [Google Scholar] [CrossRef]
- Guo, L.; Wu, W.; Liu, L.; Wang, L.; Zhang, Y.; Wu, L.; Guan, Y.; Li, Q. Herpes simplex virus 1 ICP22 inhibits the transcription of viral gene promoters by binding to and blocking the recruitment of P-TEFb. PLoS ONE 2012, 7, e45749. [Google Scholar] [CrossRef]
- Zaborowska, J.; Baumli, S.; Laitem, C.; O’Reilly, D.; Thomas, P.H.; O’Hare, P.; Murphy, S. Herpes simplex virus 1 (HSV-1) ICP22 protein directly interacts with cyclin-dependent kinase (CDK)9 to inhibit RNA polymerase II transcription elongation. PLoS ONE 2014, 9, e107654. [Google Scholar] [CrossRef]
- Prod’hon, C.; Machuca, I.; Berthomme, H.; Epstein, A.; Jacquemont, B. Characterization of regulatory functions of the HSV-1 immediate-early protein ICP22. Virology 1996, 226, 393–402. [Google Scholar] [CrossRef][Green Version]
- Kwun, H.J.; Yim, S.W.; Lee, D.H.; Jang, K.L. Activation of the thymidine kinase promoter by herpes simplex virus type 1 immediate early proteins. Mol. Cells 1999, 9, 277–280. [Google Scholar]
- Poon, A.P.W.; Roizman, B. Herpes simplex virus 1 ICP22 regulates the accumulation of a shorter mRNA and of a truncated US3 protein kinase that exhibits altered functions. J. Virol. 2005, 79, 8470–8479. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Y.; Yang, Z.; Cao, Y.; Zhang, S.; Li, H.; Huang, Y.; Ding, Y.-Q.; Liu, X. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem. J. 2008, 413, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.S.; Pipas, J.M. The virus-chaperone connection. Virology 2001, 287, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Adlakha, M.; Livingston, C.M.; Bezsonova, I.; Weller, S.K. The herpes simplex virus 1 immediate early protein ICP22 is a functional mimic of a cellular J protein. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Bastian, T.W.; Livingston, C.M.; Weller, S.K.; Rice, S.A. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J. Virol. 2010, 84, 2384–2394. [Google Scholar] [CrossRef] [PubMed]
- Burch, A.D.; Weller, S.K. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J. Virol. 2005, 79, 10740–10749. [Google Scholar] [CrossRef]
- Burch, A.D.; Weller, S.K. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol. 2004, 78, 7175–7185. [Google Scholar] [CrossRef]
- Livingston, C.M.; Ifrim, M.F.; Cowan, A.E.; Weller, S.K. Virus-Induced Chaperone-Enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog. 2009, 5, e1000619. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Davido, D.J. Herpes simplex virus 1 ICP22 but not US 1.5 is required for efficient acute replication in mice and VICE domain formation. J. Virol. 2013, 87, 13510–13519. [Google Scholar] [CrossRef]
- Maruzuru, Y.; Shindo, K.; Liu, Z.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kato, A.; Kawaguchi, Y. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J. Virol. 2014, 88, 7445–7454. [Google Scholar] [CrossRef]
- Matundan, H.; Ghiasi, H. Herpes simplex virus 1 ICP22 suppresses CD80 Expression by murine dendritic cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Tormanen, K.; Wang, S.; Ghiasi, H. CD80 plays a critical role in increased inflammatory responses in herpes simplex virus 1-infected mouse corneas. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Matundan, H.H.; Jaggi, U.; Wang, S.; Ghiasi, H. Loss of ICP22 in HSV-1 elicits immune infiltration and maintains stromal keratitis despite reduced primary and latent virus infectivity. Invest. Ophthalmol. Vis. Sci. 2019, 60, 3398–3406. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Fu, M.; Li, M.; Hu, H.; Gong, S.; Hu, Q. Herpes simplex virus type 2 inhibits type I IFN signaling mediated by the novel E3 ubiquitin protein ligase activity of viral protein ICP22. J. Immunol. 2020, 205, 1281–1292. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. The terminal a sequence of the herpes simplex virus genome contains the promoter of a gene located in the repeat sequences of the L component. J. Virol. 1986, 57, 629–637. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Chou, J.; Sarmiento, M.; Lerner, R.A.; Roizman, B. Identification by antibody to a synthetic peptide of a protein specified by a diploid gene located in the terminal repeats of the L component of herpes simplex virus genome. J. Virol. 1986, 58, 843–850. [Google Scholar] [CrossRef]
- Taha, M.; Clements, G.; Brown, S. A variant of herpes simplex virus type 2 strain HG52 with a 1.5 kb deletion in RL between 0 to 0.02 and 0.81 to 0.83 map units is non-neurovirulent for mice. J. Gen. Virol. 1989, 70, 705–716. [Google Scholar] [CrossRef]
- Taha, M.Y.; Clements, G.B.; Brown, S.M. The herpes simplex virus type 2 (HG52) variant JH2604 has a 1488 bp deletion which eliminates neurovirulence in mice. J. Gen. Virol. 1989, 70, 3073–3078. [Google Scholar] [CrossRef]
- Whitley, R.J.; Kern, E.R.; Chatterjee, S.; Chou, J.; Roizman, B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J. Clin. Investig. 1993, 91, 2837–2843. [Google Scholar] [CrossRef]
- MacLean, A.R.; Ul-Fareed, M.; Robertson, L.; Harland, J.; Brown, S.M. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the “a” sequence. J. Gen. Virol. 1991, 72, 631–639. [Google Scholar] [CrossRef]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef] [PubMed]
- Robertson, L.M.; MacLean, A.R.; Brown, S.M. Peripheral replication and latency reactivation kinetics of the non-neurovirulent herpes simplex virus type 1 variant 1716. J. Gen. Virol. 1992, 73, 967–970. [Google Scholar] [CrossRef]
- Dolan, A.; McKie, E.; MacLean, A.R.; McGeoch, D.J. Status of the ICP34.5 gene in herpes simplex virus type 1 strain 17. J. Gen. Virol. 1992, 73, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Perng, G.C.; Thompson, R.L.; Sawtell, N.M.; Taylor, W.E.; Slanina, S.M.; Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol. 1995, 69, 3033–3041. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.L.; Stevens, J.G. Biological characterization of a herpes simplex virus intertypic recombinant which is completely and specifically non-neurovirulent. Virology 1983, 131, 171–179. [Google Scholar] [CrossRef]
- Bolovan, C.A.; Sawtell, N.M.; Thompson, R.L. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J. Virol. 1994, 68, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Perng, G.C.; Ghiasi, H.; Slanina, S.M.; Nesburn, A.B.; Wechsler, S.L. High-dose ocular infection with a herpes simplex virus type 1 ICP34.5 deletion mutant produces no corneal disease or neurovirulence yet results in wild-type levels of spontaneous reactivation. J. Virol. 1996, 70, 2883–2893. [Google Scholar] [CrossRef]
- Mattila, R.K.; Harila, K.; Kangas, S.M.; Paavilainen, H.; Heape, A.M.; Mohr, I.J.; Hukkanen, V. An investigation of herpes simplex virus type 1 latency in a novel mouse dorsal root ganglion model suggests a role for ICP34.5 in reactivation. J. Gen. Virol. 2015, 96, 2304–2313. [Google Scholar] [CrossRef]
- Brown, S.M.; Harland, J.; MacLean, A.R.; Podlech, J.; Clements, J.B. Cell type and cell state determine differential in vitro growth of non-neurovirulent ICP34.5-negative herpes simplex virus types 1 and 2. J. Gen. Virol. 1994, 75, 2367–2377. [Google Scholar] [CrossRef]
- Kesari, S.; Lasner, T.M.; Balsara, K.R.; Randazzo, B.P.; Lee, V.M.; Trojanowski, J.Q.; Fraser, N.W. A neuroattenuated ICP34.5-deficient herpes simplex virus type 1 replicates in ependymal cells of the murine central nervous system. J. Gen. Virol. 1998, 79, 525–536. [Google Scholar] [CrossRef]
- Mao, H.; Rosenthal, K.S. Strain-dependent structural variants of herpes simplex virus type 1 ICP34.5 determine viral plaque size, efficiency of glycoprotein processing, and viral release and neuroinvasive disease potential. J. Virol. 2003, 77, 3409–3417. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; MacLean, A.R.; McKie, E.A.; Harland, J. The herpes simplex virus virulence factor ICP34.5 and the cellular protein MyD116 complex with proliferating cell nuclear antigen through the 63-amino-acid domain conserved in ICP34.5, MyD116, and GADD34. J. Virol. 1997, 71, 9442–9449. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. Herpes simplex virus 1 gamma(1)34.5 gene function, which blocks the host response to infection, maps in the homologous domain of the genes expressed during growth arrest and DNA damage. Proc. Natl. Acad. Sci. USA 1994, 91, 5247–5251. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Barnett, B.C. Neurovirulence factor. Nature 1991, 353, 609. [Google Scholar] [CrossRef]
- Chou, J.; Chen, J.J.; Gross, M.; Roizman, B. Association of a M(r) 90,000 phosphoprotein with protein kinase PKR in cells exhibiting enhanced phosphorylation of translation initiation factor eIF-2 alpha and premature shutoff of protein synthesis after infection with gamma 134.5- mutants of herpes si. Proc. Natl. Acad. Sci. USA 1995, 92, 10516–10520. [Google Scholar] [CrossRef] [PubMed]
- Chou, J.; Roizman, B. The gamma 1(34.5) gene of herpes simplex virus 1 precludes neuroblastoma cells from triggering total shutoff of protein synthesis characteristic of programed cell death in neuronal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3266–3270. [Google Scholar] [CrossRef]
- Cassady, K.A.; Gross, M.; Roizman, B. The second-site mutation in the herpes simplex virus recombinants lacking the gamma134.5 genes precludes shutoff of protein synthesis by blocking the phosphorylation of eIF-2alpha. J. Virol. 1998, 72, 7005–7011. [Google Scholar] [CrossRef]
- Ward, S.L.; Scheuner, D.; Poppers, J.; Kaufman, R.J.; Mohr, I.; Leib, D.A. In vivo replication of an ICP34.5 second-site suppressor mutant following corneal infection correlates with in vitro regulation of eIF2 alpha phosphorylation. J. Virol. 2003, 77, 4626–4634. [Google Scholar] [CrossRef]
- Tallóczy, Z.; Virgin, H.W., IV; Levine, B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2006, 2, 24–29. [Google Scholar] [CrossRef]
- Wylie, K.M.; Schrimpf, J.E.; Morrison, L.A. Increased eIF2alpha phosphorylation attenuates replication of herpes simplex virus 2 vhs mutants in mouse embryonic fibroblasts and correlates with reduced accumulation of the PKR antagonist ICP34.5. J. Virol. 2009, 83, 9151–9162. [Google Scholar] [CrossRef]
- He, B.; Gross, M.; Roizman, B. The gamma(1)34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activ. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Brett, M.-E.; He, B. Val193 and Phe195 of the γ134.5 protein of herpes simplex virus 1 are required for viral resistance to interferon-α/β. Virology 2001, 290, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Leib, D.A.; Machalek, M.A.; Williams, B.R.G.; Silverman, R.H.; Virgin, H.W. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl. Acad. Sci. USA 2000, 97, 6097–6101. [Google Scholar] [CrossRef] [PubMed]
- Verpooten, D.; Ma, Y.; Hou, S.; Yan, Z.; He, B. Control of TANK-binding kinase 1-mediated signaling by the gamma(1)34.5 protein of herpes simplex virus 1. J. Biol. Chem. 2009, 284, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Manivanh, R.; Mehrbach, J.; Knipe, D.M.; Leib, D.A. Role of herpes simplex virus 1 γ34.5 in the regulation of IRF3 signaling. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef]
- Tallóczy, Z.; Jiang, W.; Virgin, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.-L.; Levine, B. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef]
- Leib, D.A.; Alexander, D.E.; Cox, D.; Yin, J.; Ferguson, T.A. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J. Virol. 2009, 83, 12164–12171. [Google Scholar] [CrossRef]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippé, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef]
- Gobeil, P.A.M.; Leib, D.A. Herpes simplex virus γ34.5 interferes with autophagosome maturation and antigen presentation in dendritic cells. mBio 2012, 3. [Google Scholar] [CrossRef]
- Alexander, D.E.; Ward, S.L.; Mizushima, N.; Levine, B.; Leib, D.A. Analysis of the role of autophagy in replication of herpes simplex virus in cell culture. J. Virol. 2007, 81, 12128–12134. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.E.; Leib, D.A. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy 2008, 4, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Santana, S.; Bullido, M.J.; Recuero, M.; Valdivieso, F.; Aldudo, J. Herpes simplex virus type I induces an incomplete autophagic response in human neuroblastoma cells. J. Alzheimers Dis. 2012, 30, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.B.; Horan, K.A.; Holm, C.K.; Stranks, A.J.; Mettenleiter, T.C.; Simon, A.K.; Jensen, S.B.; Rixon, F.J.; He, B.; Paludan, S.R. Activation of autophagy by α-herpesviruses in myeloid cells is mediated by cytoplasmic viral DNA through a mechanism dependent on stimulator of IFN genes. J. Immunol. 2011, 187, 5268–5276. [Google Scholar] [CrossRef]
- Budida, R.; Stankov, M.V.; Döhner, K.; Buch, A.; Panayotova-Dimitrova, D.; Tappe, K.A.; Pohlmann, A.; Sodeik, B.; Behrens, G.M.N. Herpes simplex virus 1 interferes with autophagy of murine dendritic cells and impairs their ability to stimulate CD8(+) T lymphocytes. Eur. J. Immunol. 2017, 47, 1819–1834. [Google Scholar] [CrossRef]
- York, I.A.; Roop, C.; Andrews, D.W.; Riddell, S.R.; Graham, F.L.; Johnson, D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994, 77, 525–535. [Google Scholar] [CrossRef]
- Banks, T.A.; Nair, S.; Rouse, B.T. Recognition by and in vitro induction of cytotoxic T lymphocytes against predicted epitopes of the immediate-early protein ICP27 of herpes simplex virus. J. Virol. 1993, 67, 613–616. [Google Scholar] [CrossRef]
- Fruh, K.; Ahn, K.; Djaballah, H.; Sempe, P.; van Endert, P.M.; Tampé, R.; Peterson, P.A.; Yang, Y. A viral inhibitor of peptide transporters for antigen presentation. Nature 1995, 375, 415–418. [Google Scholar] [CrossRef]
- Tomazin, R.; Hill, A.B.; Jugovic, P.; York, I.; van Endert, P.; Ploegh, H.L.; Andrews, D.W.; Johnson, D.C. Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J. 1996, 15, 3256–3266. [Google Scholar] [CrossRef]
- Galocha, B.; Hill, A.; Barnett, B.C.; Dolan, A.; Raimondi, A.; Cook, R.F.; Brunner, J.; Mcgeoch, D.J.; Ploegh, H.L. The active site of ICP47, a herpes simplex virus-encoded inhibitor of the major histocompatibility complex (MHC)-encoded peptide transported associated with antigen processing (TAP), maps to the NH2-terminal 35 residues. J. Exp. Med. 1997, 185, 1565–1572. [Google Scholar] [CrossRef]
- Ahn, K.; Meyer, T.H.; Uebel, S.; Sempé, P.; Djaballah, H.; Yang, Y.; Peterson, P.A.; Früh, K.; Tampé, R. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus protein ICP47. EMBO J. 1996, 15, 3247–3255. [Google Scholar] [CrossRef]
- Tomazin, R.; van Schoot, N.E.G.; Goldsmith, K.; Jugovic, P.; Sempé, P.; Früh, K.; Johnson, D.C. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J. Virol. 1998, 72, 2560–2563. [Google Scholar] [CrossRef]
- Jugovic, P.; Hill, A.M.; Tomazin, R.; Ploegh, H.; Johnson, D.C. Inhibition of major histocompatibility complex class I antigen presentation in pig and primate cells by herpes simplex virus type 1 and 2 ICP47. J. Virol. 1998, 72, 5076–5084. [Google Scholar] [CrossRef] [PubMed]
- Orr, M.T.; Edelmann, K.H.; Vieira, J.; Corey, L.; Raulet, D.H.; Wilson, C.B. Inhibition of MHC class I is a virulence factor in herpes simplex virus infection of mice. PLoS Pathog. 2005, 1, e7. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Kuncheria, L.; Roulstone, V.; Kyula, J.N.; Mansfield, D.; Bommareddy, P.K.; Smith, H.; Kaufman, H.L.; Harrington, K.J.; Coffin, R.S. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J. Immunother. Cancer 2019, 7, 1–17. [Google Scholar] [CrossRef]
- Saric, T.; Chang, S.C.; Hattori, A.; York, I.A.; Markant, S.; Rock, K.L.; Tsujimoto, M.; Goldberg, A.L. An IFN-γ-induced aminopeptidase in the ER, ERAP I, trims precursors to MHC class I-presented peptides. Nat. Immunol. 2002, 3, 1169–1176. [Google Scholar] [CrossRef]
- Marijt, K.A.; van Hall, T. To TAP or not to TAP: Alternative peptides for immunotherapy of cancer. Curr. Opin. Immunol. 2020, 64, 15–19. [Google Scholar] [CrossRef]
- Raafat, N.; Sadowski-Cron, C.; Mengus, C.; Heberer, M.; Spagnoli, G.C.; Zajac, P. Preventing vaccinia virus class-I epitopes presentation by HSV-ICP47 enhances the immunogenicity of a TAP-independent cancer vaccine epitope. Int. J. Cancer 2012, 131, 659–669. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Kurachi, R.; Daikoku, T.; Umene, K. The US 9, 10, 11, and 12 genes of herpes simplex virus type 1 are of no importance for its neurovirulence and latency in mice. Virology 1993, 194, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, B.K.; Chen, W.; Johnson, D.C.; Hendricks, R.L. Infected Cell Protein (ICP)47 enhances herpes simpex virus neurovirulence by blocking the CD8 T cell response. J. Exp. Med. 1998, 187, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Burgos, J.S.; Serrano-Saiz, E.; Sastre, I.; Valdivieso, F. ICP47 mediates viral neuroinvasiveness by induction of TAP protein following intravenous inoculation of herpes simplex virus type 1 in mice. J. Neurovirol. 2006, 12, 420–427. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. DNA repair enzymes. Annu. Rev. Biochem. 1982, 51, 61–87. [Google Scholar] [CrossRef]
- Worrad, D.M.; Caradonna, S. Identification of the coding sequence for herpes simplex virus uracil-DNA glycosylase. J. Virol. 1988, 62, 4774–4777. [Google Scholar] [CrossRef]
- Savva, R.; Pearl, L.H. Crystallization and preliminary X-ray analysis of the uracil-DNA glycosylase DNA repair enzyme from herpes simplex virus type 1. J. Mol. Biol. 1993, 234, 910–912. [Google Scholar] [CrossRef]
- Caradonna, S.J.; Cheng, Y.C. Induction of uracil-DNA glycosylase and dUTP nucleotidohydrolase activity in herpes simplex virus-infected human cells. J. Biol. Chem. 1981, 256, 9834–9837. [Google Scholar]
- Caradonna, S.; Worrad, D.; Lirette, R. Isolation of a herpes simplex virus cDNA encoding the DNA repair enzyme uracil-DNA glycosylase. J. Virol. 1987, 61, 3040–3047. [Google Scholar] [CrossRef]
- Focher, F.; Verri, A.; Verzeletti, S.; Mazzarello, P.; Spadari, S. Uracil in Oris of herpes simplex 1 alters its specific recognition by origin binding protein (OBP): Does virus induced uracil-DNA glycosylase play a key role in viral reactivation and replication? Chromosoma 1992, 102, 67–71. [Google Scholar] [CrossRef]
- Pyles, R.B.; Thompson, R.L. Evidence that the herpes simplex virus type 1 uracil DNA glycosylase is required for efficient viral replication and latency in the murine nervous system. J. Virol. 1994, 68, 4963–4972. [Google Scholar] [CrossRef]
- Bogani, F.; Chua, C.N.; Boehmer, P.E. Reconstitution of uracil DNA glycosylase-initiated base excision repair in herpes simplex virus-1. J. Biol. Chem. 2009, 284, 16784–16790. [Google Scholar] [CrossRef] [PubMed]
- Bogani, F.; Corredeira, I.; Fernandez, V.; Sattler, U.; Rutvisuttinunt, W.; Defais, M.; Boehmer, P.E. Association between the herpes simplex virus-1 DNA polymerase and uracil DNA glycosylase. J. Biol. Chem. 2010, 285, 27664–27672. [Google Scholar] [CrossRef] [PubMed]
- Prichard, M.N.; Lawlor, H.; Duke, G.M.; Mo, C.; Wang, Z.; Dixon, M.; Kemble, G.; Kern, E.R. Human cytomegalovirus uracil DNA glycosylase associates with ppUL44 and accelerates the accumulation of viral DNA. Virol. J. 2005, 2, 55. [Google Scholar] [CrossRef] [PubMed]
- Courcelle, C.T.; Courcelle, J.; Prichard, M.N.; Mocarski, E.S. Requirement for uracil-DNA glycosylase during the transition to late-phase cytomegalovirus DNA replication. J. Virol. 2001, 75, 7592–7601. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zou, X.; Wang, Y.; Xu, Z.; Ou, X.; Li, Y.; Liu, D.; Guo, Y.; Deng, Y.; Jiang, S.; et al. The nuclear localization signal-mediated nuclear targeting of herpes simplex virus 1 early protein UL2 is important for efficient viral production. Aging 2020, 12, 2921–2938. [Google Scholar] [CrossRef] [PubMed]
- Mullaney, J.; Moss, H.W.; McGeoch, D.J. Gene UL2 of herpes simplex virus type 1 encodes a uracil-DNA glycosylase. J. Gen. Virol. 1989, 70, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Dean, H.J.; Cheung, A.K. A 3’ coterminal gene cluster in pseudorabies virus contains herpes simplex virus UL1, UL2, and UL3 gene homologs and a unique UL3.5 open reading frame. J. Virol. 1993, 67, 5955–5961. [Google Scholar] [CrossRef]
- Khattar, S.K.; van Drunen Littel-van den Hurk, S.; Babiuk, L.A.; Tikoo, S.K. Identification and transcriptional analysis of a 3’-coterminal gene cluster containing UL1, UL2, UL3, and UL3.5 open reading frames of bovine herpesvirus-1. Virology 1995, 213, 28–37. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Han, Z.; Shao, Y.; Chen, S.; Kong, X. Comparative analysis of the genes UL1 through UL7 of the duck enteritis virus and other herpesviruses of the subfamily Alphaherpesvirinae. Genet. Mol. Biol. 2009, 32, 121–128. [Google Scholar] [CrossRef]
- Prichard, M.N.; Duke, G.M.; Mocarski, E.S. Human cytomegalovirus uracil DNA glycosylase is required for the normal temporal regulation of both DNA synthesis and viral replication. J. Virol. 1996, 70, 3018–3025. [Google Scholar] [CrossRef]
- Puvion-Dutilleul, F.; Pichard, E. Viral alkaline nuclease in intranuclear dense bodies induced by herpes simplex infection. Biol. Cell 1986, 58, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M. The role of viral and cellular nuclear proteins in herpes simplex virus replication. Adv. Virus Res. 1989, 37, 85–123. [Google Scholar] [PubMed]
- Banks, L.M.; Halliburton, I.W.; Purifoy, D.J.; Killington, R.A.; Powell, K.L. Studies on the herpes simplex virus alkaline nuclease: Detection of type-common and type-specific epitopes on the enzyme. J. Gen. Virol. 1985, 66, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Daikoku, T.; Yamashita, Y.; Tsurumi, T.; Nishiyama, Y. The US3 protein kinase of herpes simplex virus type 2 is associated with phosphorylation of the UL12 alkaline nuclease in vitro. Arch. Virol. 1995, 140, 1637–1644. [Google Scholar] [CrossRef]
- Costa, R.H.; Draper, K.G.; Banks, L.; Powell, K.L.; Cohen, G.; Eisenberg, R.; Wagner, E.K. High-resolution characterization of herpes simplex virus type 1 transcripts encoding alkaline exonuclease and a 50,000-dalton protein tentatively identified as a capsid protein. J. Virol. 1983, 48, 591–603. [Google Scholar] [CrossRef]
- Preston, C.M.; Cordingley, M.G. mRNA- and DNA-directed synthesis of herpes simplex virus-coded exonuclease in Xenopus laevis oocytes. J. Virol. 1982, 43, 386–394. [Google Scholar] [CrossRef]
- Wathen, M.W.; Hay, J. Physical mapping of the herpes simplex virus type 2 nuc- lesion affecting alkaline exonuclease activity by using herpes simplex virus type 1 deletion clones. J. Virol. 1984, 51, 237–241. [Google Scholar] [CrossRef]
- Draper, K.G.; Devi-Rao, G.; Costa, R.H.; Blair, E.D.; Thompson, R.L.; Wagner, E.K. Characterization of the genes encoding herpes simplex virus type 1 and type 2 alkaline exonucleases and overlapping proteins. J. Virol. 1986, 57, 1023–1036. [Google Scholar] [CrossRef]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Séguin, C. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 1984, 310, 207–211. [Google Scholar] [CrossRef]
- Grose, C.; Tyler, S.; Peters, G.; Hiebert, J.; Stephens, G.M.; Ruyechan, W.T.; Jackson, W.; Storlie, J.; Tipples, G.A. Complete DNA sequence analyses of the first two varicella-zoster virus glycoprotein E (D150N) mutant viruses found in North America: Evolution of genotypes with an accelerated cell spread phenotype. J. Virol. 2004, 78, 6799–6807. [Google Scholar] [CrossRef][Green Version]
- De Wind, N.; Peeters, B.P.; Zuderveld, A.; Gielkens, A.L.; Berns, A.J.; Kimman, T.G. Mutagenesis and characterization of a 41-kilobase-pair region of the pseudorabies virus genome: Transcription map, search for virulence genes, and comparison with homologs of herpes simplex virus type 1. Virology 1994, 200, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Robertson, B.J.; McCann, P.J.; O’Boyle, D.R.; Weller, S.K.; Newcomb, W.W.; Brown, J.C.; Weinheimer, S.P. Functional conservations of the alkaline nuclease of herpes simplex type 1 and human cytomegalovirus. Virology 1998, 249, 460–470. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weller, S.K.; Seghatoleslami, M.R.; Shao, L.; Rowse, D.; Carmichael, E.P. The herpes simplex virus type 1 alkaline nuclease is not essential for viral DNA synthesis: Isolation and characterization of a lacZ insertion mutant. J. Gen. Virol. 1990, 71, 2941–2952. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Rapp, L.M.; Weller, S.K. Herpes simplex virus 1 alkaline nuclease is required for efficient egress of capsids from the nucleus. Virology 1993, 196, 146–162. [Google Scholar] [CrossRef]
- Sagou, K.; Uema, M.; Kawaguchi, Y. Nucleolin is required for efficient nuclear egress of herpes simplex virus type 1 nucleocapsids. J. Virol. 2010, 84, 2110–2121. [Google Scholar] [CrossRef][Green Version]
- Porter, I.M.; Stow, N.D. Virus particles produced by the herpes simplex virus type 1 alkaline nuclease null mutant ambUL12 contain abnormal genomes. J. Gen. Virol. 2004, 85, 583–591. [Google Scholar] [CrossRef]
- Grady, L.M.; Szczepaniak, R.; Murelli, R.P.; Masaoka, T.; Le Grice, S.F.J.; Wright, D.L.; Weller, S.K. The exonuclease activity of herpes simplex virus 1 UL12 is required for production of viral DNA that can be packaged to produce infectious virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Martinez, R.; Sarisky, R.T.; Weber, P.C.; Weller, S.K. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J. Virol. 1996, 70, 2075–2085. [Google Scholar] [CrossRef]
- Goldstein, J.N.; Weller, S.K. In vitro processing of herpes simplex virus type 1 DNA replication intermediates by the viral alkaline nuclease, UL12. J. Virol. 1998, 72, 8772–8781. [Google Scholar] [CrossRef]
- Henderson, J.O.; Ball-Goodrich, L.J.; Parris, D.S. Structure–function analysis of the herpes simplex virus type 1 UL12 gene: Correlation of deoxyribonuclease activityin vitrowith replication function. Virology 1998, 243, 247–259. [Google Scholar] [CrossRef][Green Version]
- Reuven, N.B.; Staire, A.E.; Myers, R.S.; Weller, S.K. The herpes simplex virus type 1 alkaline nuclease and single-stranded DNA binding protein mediate strand exchange in vitro. J. Virol. 2003, 77, 7425–7433. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Willcox, S.; Griffith, J.D.; Weller, S.K. Catalysis of strand exchange by the HSV-1 UL12 and ICP8 proteins: Potent ICP8 recombinase activity is revealed upon resection of dsDNA substrate by nuclease. J. Mol. Biol. 2004, 342, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Reuven, N.B.; Weller, S.K. Herpes simplex virus type 1 single-strand DNA binding protein ICP8 enhances the nuclease activity of the UL12 alkaline nuclease by increasing its processivity. J. Virol. 2005, 79, 9356–9358. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef] [PubMed]
- Tolun, G.; Makhov, A.M.; Ludtke, S.J.; Griffith, J.D. Details of ssDNA annealing revealed by an HSV-1 ICP8-ssDNA binary complex. Nucleic Acids Res. 2013, 41, 5927–5937. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, N.; Bai, P.; Buchek, G.; Korza, G.; Weller, S.K. Physical interaction between the herpes simplex virus type 1 exonuclease, UL12, and the DNA double-strand break-sensing MRN complex. J. Virol. 2010, 84, 12504–12514. [Google Scholar] [CrossRef] [PubMed]
- Bronstein, J.C.; Weller, S.K.; Weber, P.C. The product of the UL12.5 gene of herpes simplex virus type 1 is a capsid-associated nuclease. J. Virol. 1997, 71, 3039–3047. [Google Scholar] [CrossRef]
- Reuven, N.B.; Antoku, S.; Weller, S.K. The UL12.5 gene product of herpes simplex virus type 1 exhibits nuclease and strand exchange activities but does not localize to the nucleus. J. Virol. 2004, 78, 4599–4608. [Google Scholar] [CrossRef]
- Martinez, R.; Goldstein, J.N.; Weller, S.K. The product of the UL12.5 gene of herpes simplex virus type 1 is not essential for lytic viral growth and is not specifically associated with capsids. Virology 2002, 298, 248–257. [Google Scholar] [CrossRef]
- Saffran, H.A.; Pare, J.M.; Corcoran, J.A.; Weller, S.K.; Smiley, J.R. Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep. 2007, 8, 188–193. [Google Scholar] [CrossRef]
- Duguay, B.A.; Smiley, J.R. Mitochondrial nucleases ENDOG and EXOG participate in mitochondrial DNA depletion initiated by herpes simplex virus 1 UL12.5. J. Virol. 2013, 87, 11787–11797. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.A.; Saffran, H.A.; Duguay, B.A.; Smiley, J.R. Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol. 2009, 83, 2601–2610. [Google Scholar] [CrossRef] [PubMed]
- Duguay, B.A.; Saffran, H.A.; Ponomarev, A.; Duley, S.A.; Eaton, H.E.; Smiley, J.R. Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J. Virol. 2014, 88, 2967–2976. [Google Scholar] [CrossRef] [PubMed]
- Björnberg, O.; Bergman, A.C.; Rosengren, A.M.; Persson, R.; Lehman, I.R.; Nyman, P.O. dUTPase from herpes simplex virus type 1; purification from infected green monkey kidney (Vero) cells and from an overproducing Escherichia coli strain. Protein Expr. Purif. 1993, 4, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Preston, V.G.; Fisher, F.B. Identification of the herpes simplex virus type 1 gene encoding the dUTPase. Virology 1984, 138, 58–68. [Google Scholar] [CrossRef]
- Caradonna, S.J.; Adamkiewicz, D.M. Purification and properties of the deoxyuridine triphosphate nucleotidohydrolase enzyme derived from HeLa S3 cells. Comparison to a distinct dUTP nucleotidohydrolase induced in herpes simplex virus-infected HeLa S3 cells. J. Biol. Chem. 1984, 259, 5459–5464. [Google Scholar]
- Williams, M. V Deoxyuridine triphosphate nucleotidohydrolase induced by herpes simplex virus type 1. Purification and characterization of induced enzyme. J. Biol. Chem. 1984, 259, 10080–10084. [Google Scholar]
- Pyles, R.B.; Sawtell, N.M.; Thompson, R.L. Herpes simplex virus type 1 dUTPase mutants are attenuated for neurovirulence, neuroinvasiveness, and reactivation from latency. J. Virol. 1992, 66, 6706–6713. [Google Scholar] [CrossRef]
- Lirette, R.; Caradonna, S. Inhibition of phosphorylation of cellular dUTP nucleotidohydrolase as a consequence of herpes simplex virus infection. J. Cell. Biochem. 1990, 43, 339–353. [Google Scholar] [CrossRef]
- Kato, A.; Tsuda, S.; Liu, Z.; Kozuka-Hata, H.; Oyama, M.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J. Virol. 2014, 88, 655–666. [Google Scholar] [CrossRef]
- Kato, A.; Shindo, K.; Maruzuru, Y.; Kawaguchi, Y. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase, Us3, dictates viral pathogenicity in the central nervous system but not at the periphery. J. Virol. 2014, 88, 2775–2785. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Hirohata, Y.; Arii, J.; Kawaguchi, Y. Phosphorylation of herpes simplex virus 1 dUTPase upregulated viral dUTPase activity to compensate for low cellular dUTPase activity for efficient viral replication. J. Virol. 2014, 88, 7776–7785. [Google Scholar] [CrossRef] [PubMed]
- Riggio, M.P.; Onions, D.E. DNA sequence of a gene cluster in the equine herpesvirus-4 genome which contains a newly identified herpesvirus gene encoding a membrane protein. Arch. Virol. 1993, 133, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, J.; Klupp, B.G.; Mettenleiter, T.C. Pseudorabies virus and equine herpesvirus 1 share a nonessential gene which is absent in other herpesviruses and located adjacent to a highly conserved gene cluster. J. Virol. 1995, 69, 5560–5567. [Google Scholar] [CrossRef]
- Williams, M.V.J. Demonstration of a herpes simplex virus type 2-induced deoxyuridine triphosphate nucleotidohydrolase in infected KB cells and in biochemically transformed HeLa cells. J. Gen. Virol. 1984, 65, 209–213. [Google Scholar] [CrossRef]
- Liang, X.; Tang, M.; Manns, B.; Babiuk, L.A.; Zamb, T.J. Identification and deletion mutagenesis of the bovine herpesvirus 1 dUTPase gene and a gene homologous to herpes simplex virus UL49.5. Virology 1993, 195, 42–50. [Google Scholar] [CrossRef]
- Ariza, M.-E.; Glaser, R.; Kaumaya, P.T.P.; Jones, C.; Williams, M. V The EBV-encoded dUTPase activates NF-κB through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef]
- Glaser, R.; Litsky, M.L.; Padgett, D.A.; Baiocchi, R.A.; Yang, E.V.; Chen, M.; Yeh, P.-E.; Green-Church, K.B.; Caligiuri, M.A.; Williams, M. V EBV-encoded dUTPase induces immune dysregulation: Implications for the pathophysiology of EBV-associated disease. Virology 2006, 346, 205–218. [Google Scholar] [CrossRef]
- Ariza, M.E.; Glaser, R.; Williams, M. V Human herpesviruses-encoded dUTPases: A family of proteins that modulate dendritic cell function and innate immunity. Front. Microbiol. 2014, 5, 504. [Google Scholar] [CrossRef]
- Goldstein, D.J.; Weller, S.K. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: Characterization of an ICP6 deletion mutant. Virology 1988, 166, 41–51. [Google Scholar] [CrossRef]
- Bacchetti, S.; Evelegh, M.J.; Muirhead, B. Identification and separation of the two subunits of the herpes simplex virus ribonucleotide reductase. J. Virol. 1986, 57, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y. Glycoprotein M and ESCRT in Herpes Simplex Virus Type 1 Assembly. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2011. [Google Scholar]
- Paradis, H.; Gaudreau, P.; Brazeau, P.; Langelier, Y. Mechanism of inhibition of herpes simplex virus (HSV) ribonucleotide reductase by a nonapeptide corresponding to the carboxyl terminus of its subunit 2. Specific binding of a photoaffinity analog, [4’-azido-Phe6]HSV H2-(6-15), to subunit 1. J. Biol. Chem. 1988, 263, 16045–16050. [Google Scholar] [PubMed]
- Spector, T.; Stonehuerner, J.G.; Biron, K.K.; Averett, D.R. Ribonucleotide reductase induced by varicella zoster virus. Characterization, and potentiation of acyclovir by its inhibition. Biochem. Pharmacol. 1987, 36, 4341–4346. [Google Scholar] [CrossRef]
- Coen, D.M.; Kosz-Vnenchak, M.; Jacobson, J.G.; Leib, D.A.; Bogard, C.L.; Schaffer, P.A.; Tyler, K.L.; Knipe, D.M. Thymidine kinase-negative herpes simplex virus mutants establish latency in mouse trigeminal ganglia but do not reactivate. Proc. Natl. Acad. Sci. USA 1989, 86, 4736–4740. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; De Giovanni, C.; Gatta, V.; Nanni, P.; Lollini, P.L.; Menotti, L. Rethinking herpes simplex virus: The way to oncolytic agents. Rev. Med. Virol. 2011, 21, 213–226. [Google Scholar] [CrossRef]
- Gober, M.D.; Wales, S.Q.; Hunter, J.C.; Sharma, B.K.; Aurelian, L. Stress up-regulates neuronal expression of the herpes simplex virus type 2 large subunit of ribonucleotide reductase (R1; ICP10) by activating activator protein 1. J. Neurovirol. 2005, 11, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Langelier, Y.; Bergeron, S.; Chabaud, S.; Lippens, J.; Guilbault, C.; Sasseville, A.M.J.; Denis, S.; Mosser, D.D.; Massie, B. The R1 subunit of herpes simplex virus ribonucleotide reductase protects cells against apoptosis at, or upstream of, caspase-8 activation. J. Gen. Virol. 2002, 83, 2779–2789. [Google Scholar] [CrossRef] [PubMed]
- Dufour, F.; Sasseville, A.M.J.; Chabaud, S.; Massie, B.; Siegel, R.M.; Langelier, Y. The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFα- and FasL-induced apoptosis by interacting with caspase-8. Apoptosis 2011, 16, 256–271. [Google Scholar] [CrossRef] [PubMed]
- Brandt, C.R.; Kintner, R.L.; Pumfery, A.M.; Visalli, R.J.; Grau, D.R. The herpes simplex virus ribonucleotide reductase is required for ocular virulence. J. Gen. Virol. 1991, 72, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Turk, S.R.; Kik, N.A.; Birch, G.M.; Chiego, D.J., Jr.; Shipman, C., Jr. Herpes simplex virus type 1 ribonucleotide reductase null mutants induce lesions in guinea pigs. Virology 1989, 173, 733–735. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Thompson, T.W.; Konen, A.J.; Haenchen, S.D.; Hilliard, J.G.; Macdonald, S.J.; Morrison, L.A.; Davido, D.J. Herpes simplex virus 1 mutant with point mutations in UL39 Is impaired for acute viral replication in mice, establishment of latency, and explant-induced reactivation. J. Virol. 2018, 92, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Davido, D.J.; Tu, E.M.; Wang, H.; Korom, M.; Gazquez Casals, A.; Reddy, P.J.; Mostafa, H.H.; Combs, B.; Haenchen, S.D.; Morrison, L.A. Attenuated herpes simplex virus 1 (HSV-1) expressing a mutant form of ICP6 stimulates a strong immune response that protects mice against HSV-1-induced corneal disease. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, M.L.; Clark, J. Early and delayed shut-off of host protein synthesis in cells infected with herpes simplex virus. J. Gen. Virol. 1982, 61, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Fenwick, M.L.; McMenamin, M.M. Early virion-associated suppression of cellular protein synthesis by herpes simplex virus is accompanied by inactivation of mRNA. J. Gen. Virol. 1984, 65, 1225–1228. [Google Scholar] [CrossRef]
- Fenwick, M.L.; Walker, M.J. Suppression of the synthesis of cellular macromolecules by herpes simplex virus. J. Gen. Virol. 1978, 41, 37–51. [Google Scholar] [CrossRef]
- Kwong, A.D.; Frenkel, N. Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 1926–1930. [Google Scholar] [CrossRef]
- Kwong, A.D.; Kruper, J.A.; Frenkel, N. Herpes simplex virus virion host shutoff function. J. Virol. 1988, 62, 912–921. [Google Scholar] [CrossRef]
- Oroskar, A.A.; Read, G.S. Control of mRNA stability by the virion host shutoff function of herpes simplex virus. J. Virol. 1989, 63, 1897–1906. [Google Scholar] [CrossRef]
- Read, G.S.; Frenkel, N. Herpes simplex virus mutants defective in the virion-associated shutoff of host polypeptide synthesis and exhibiting abnormal synthesis of alpha (immediate early) viral polypeptides. J. Virol. 1983, 46, 498–512. [Google Scholar] [CrossRef]
- Schek, N.; Bachenheimer, S.L. Degradation of cellular mRNAs induced by a virion-associated factor during herpes simplex virus infection of Vero cells. J. Virol. 1985, 55, 601–610. [Google Scholar] [CrossRef]
- Berthomme, H.; Jacquemont, B.; Epstein, A. The pseudorabies virus host-shutoff homolog gene: Nucleotide sequence and comparison with alphaherpesvirus protein counterparts. Virology 1993, 193, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Jones, F.E.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus virion host shutoff protein requires a mammalian factor for efficient in vitro endoribonuclease activity. J. Virol. 2001, 75, 1172–1185. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, T.J.; Morrison, L.A.; Leib, D.A. Pathogenesis of herpes simplex virus type 2 virion host shutoff (vhs) mutants. J. Virol. 2002, 76, 2054–2061. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Strelow, L.I.; Leib, D.A. Role of the virion host shutoff (vhs) of herpes simplex virus type 1 in latency and pathogenesis. J. Virol. 1995, 69, 6779–6786. [Google Scholar] [CrossRef]
- Smibert, C.A.; Smiley, J.R. Differential regulation of endogenous and transduced beta-globin genes during infection of erythroid cells with a herpes simplex virus type 1 recombinant. J. Virol. 1990, 64, 3882–3894. [Google Scholar] [CrossRef]
- Strelow, L.; Smith, T.; Leib, D. The virion host shutoff function of herpes simplex virus type 1 plays a role in corneal invasion and functions independently of the cell cycle. Virology 1997, 231, 28–34. [Google Scholar] [CrossRef]
- Strelow, L.I.; Leib, D.A. Analysis of conserved domains of UL41 of herpes simplex virus type 1 in virion host shutoff and pathogenesis. J. Virol. 1996, 70, 5665–5667. [Google Scholar] [CrossRef]
- Fenwick, M.L.; Everett, R.D. Inactivation of the shutoff gene (UL41) of herpes simplex virus types 1 and 2. J. Gen. Virol. 1990, 71, 2961–2967. [Google Scholar] [CrossRef]
- Strom, T.; Frenkel, N. Effects of herpes simplex virus on mRNA stability. J. Virol. 1987, 61, 2198–2207. [Google Scholar] [CrossRef]
- Krikorian, C.R.; Read, G.S. In vitro mRNA degradation system to study the virion host shutoff function of herpes simplex virus. J. Virol. 1991, 65, 112–122. [Google Scholar] [CrossRef]
- Oroskar, A.A.; Read, G.S. A mutant of herpes simplex virus type 1 exhibits increased stability of immediate-early (alpha) mRNAs. J. Virol. 1987, 61, 604–606. [Google Scholar] [CrossRef] [PubMed]
- Lam, Q.; Smibert, C.A.; Koop, K.E.; Lavery, C.; Capone, J.P.; Weinheimer, S.P.; Smiley, J.R. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 1996, 15, 2575–2581. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Sciortino, M.T.; Zhang, W.; Roizman, B. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. USA 2007, 104, 12163–12168. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Zhang, W.; Roizman, B. The herpes simplex virus host shutoff RNase degrades cellular and viral mRNAs made before infection but not viral mRNA made after infection. J. Virol. 2013, 87, 4516–4522. [Google Scholar] [CrossRef] [PubMed]
- Esclatine, A.; Taddeo, B.; Evans, L.; Roizman, B. The herpes simplex virus 1 UL41 gene-dependent destabilization of cellular RNAs is selective and may be sequence-specific. Proc. Natl. Acad. Sci. USA 2004, 101, 3603–3608. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, C.M.; Hart, P.A.; Ross, J. Analysis of herpes simplex virus-induced mRNA destabilizing activity using an in vitro mRNA decay system. Nucleic Acids Res. 1991, 19, 4459–4465. [Google Scholar] [CrossRef] [PubMed]
- Zelus, B.D.; Stewart, R.S.; Ross, J. The virion host shutoff protein of herpes simplex virus type 1: Messenger ribonucleolytic activity in vitro. J. Virol. 1996, 70, 2411–2419. [Google Scholar] [CrossRef]
- David, N.E.; Feng, P.; Mian, I.S.; Read, G.S. mRNA Degradation by the virion host shutoff (vhs) protein of herpes simplex virus: Genetic and Biochemical evidence that vhs is a nuclease. J. Virol. 2002, 76, 8560–8571. [Google Scholar] [CrossRef]
- Elgadi, M.M.; Hayes, C.E.; Smiley, J.R. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 1999, 73, 7153–7164. [Google Scholar] [CrossRef]
- Doherty, A.J.; Serpell, L.C.; Ponting, C.P. The helix-hairpin-helix DNA-binding motif: A structural basis for non-sequence-specific recognition of DNA. Nucleic Acids Res. 1996, 24, 2488–2497. [Google Scholar] [CrossRef]
- Taddeo, B.; Roizman, B. The virion host shutoff protein (UL41) of herpes simplex virus 1 is an endoribonuclease with a substrate specificity similar to that of RNase A. J. Virol. 2006, 80, 9341–9345. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Esclatine, A.; Zhang, W.; Roizman, B. The stress-inducible immediate-early responsive gene IEX-1 is activated in cells infected with herpes simplex virus 1, but several viral mechanisms, including 3′ degradation of its RNA, preclude expression of the gene. J. Virol. 2003, 77, 6178–6187. [Google Scholar] [CrossRef] [PubMed]
- Esclatine, A.; Taddeo, B.; Roizman, B. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J. Virol. 2004, 78, 8582–8592. [Google Scholar] [CrossRef] [PubMed]
- Karr, B.M.; Read, G.S. The virion host shutoff function of herpes simplex virus degrades the 5′ end of a target mRNA before the 3′ end. Virology 1999, 264, 195–204. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perez-Parada, J.; Saffran, H.A.; Smiley, J.R. RNA degradation induced by the herpes simplex virus vhs protein proceeds 5′ to 3′ in vitro. J. Virol. 2004, 78, 13391–13394. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Feng, P.; Everly, D.N., Jr.; Read, G.S. mRNA decay during herpesvirus infections: Interaction between a putative viral nuclease and a cellular translation factor. J. Virol. 2001, 75, 10272–10280. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Everly, D.N.J.; Read, G.S. mRNA decay during herpes simplex virus (HSV) infections: Protein-protein interactions involving the HSV virion host shutoff protein and translation factors eIF4H and eIF4A. J. Virol. 2005, 79, 9651–9664. [Google Scholar] [CrossRef]
- Benetti, L.; Munger, J.; Roizman, B. The herpes simplex virus 1 US3 protein kinase blocks caspase-dependent double cleavage and activation of the proapoptotic protein BAD. J. Virol. 2003, 77, 6567–6573. [Google Scholar] [CrossRef]
- Doepker, R.C.; Hsu, W.-L.; Saffran, H.A.; Smiley, J.R. Herpes simplex virus virion host shutoff protein is stimulated by translation initiation factors eIF4B and eIF4H. J. Virol. 2004, 78, 4684–4699. [Google Scholar] [CrossRef]
- Barzilai, A.; Zivony-Elbom, I.; Sarid, R.; Noah, E.; Frenkel, N. The herpes simplex virus type 1 vhs-UL41 gene secures viral replication by temporarily evading apoptotic cellular response to infection: Vhs-UL41 activity might require interactions with elements of cellular mRNA degradation machinery. J. Virol. 2006, 80, 505–513. [Google Scholar] [CrossRef]
- Shiflett, L.A.; Read, G.S. mRNA decay during herpes simplex virus (HSV) infections: Mutations that affect translation of an mRNA influence the sites at which it is cleaved by the HSV virion host shutoff (Vhs) protein. J. Virol. 2013, 87, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Suzutani, T.; Nagamine, M.; Shibaki, T.; Ogasawara, M.; Yoshida, I.; Daikoku, T.; Nishiyama, Y.; Azuma, M. The role of the UL41 gene of herpes simplex virus type 1 in evasion of non-specific host defence mechanisms during primary infection. J. Gen. Virol. 2000, 81, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Samady, L.; Costigliola, E.; MacCormac, L.; McGrath, Y.; Cleverley, S.; Lilley, C.E.; Smith, J.; Latchman, D.S.; Chain, B.; Coffin, R.S. Deletion of the virion host shutoff protein (vhs) from herpes simplex virus (hsv) relieves the viral block to dendritic cell activation: Potential of vhs- HSV vectors for dendritic cell-mediated immunotherapy. J. Virol. 2003, 77, 3768–3776. [Google Scholar] [CrossRef] [PubMed]
- Cotter, C.R.; Nguyen, M.L.; Yount, J.S.; López, C.B.; Blaho, J.A.; Moran, T.M. The virion host shut-off (vhs) protein blocks a TLR-independent pathway of herpes simplex virus type 1 recognition in human and mouse dendritic cells. PLoS ONE 2010, 5, e8684. [Google Scholar] [CrossRef]
- Eisemann, J.; Mühl-Zürbes, P.; Steinkasserer, A.; Kummer, M. Infection of mature dendritic cells with herpes simplex virus type 1 interferes with the interferon signaling pathway. Immunobiology 2007, 212, 877–886. [Google Scholar] [CrossRef]
- Pasieka, T.J.; Cilloniz, C.; Lu, B.; Teal, T.H.; Proll, S.C.; Katze, M.G.; Leib, D.A. Host responses to wild-type and attenuated herpes simplex virus infection in the absence of Stat1. J. Virol. 2009, 83, 2075–2087. [Google Scholar] [CrossRef]
- Burgess, H.M.; Mohr, I. Defining the role of stress granules in innate immune suppression by the herpes simplex virus 1 endoribonuclease VHS. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Dauber, B.; Poon, D.; dos Santos, T.; Duguay, B.A.; Mehta, N.; Saffran, H.A.; Smiley, J.R. The herpes simplex virus virion host shutoff protein enhances translation of viral true late mRNAs independently of suppressing protein kinase R and stress granule formation. J. Virol. 2016, 90, 6049–6057. [Google Scholar] [CrossRef]
- Sciortino, M.T.; Parisi, T.; Siracusano, G.; Mastino, A.; Taddeo, B.; Roizman, B. The virion host shutoff RNase plays a key role in blocking the activation of protein kinase R in cells infected with herpes simplex virus 1. J. Virol. 2013, 87, 3271–3276. [Google Scholar] [CrossRef]
- Dauber, B.; Saffran, H.A.; Smiley, J.R. The herpes simplex virus 1 virion host shutoff protein enhances translation of viral late mRNAs by preventing mRNA overload. J. Virol. 2014, 88, 9624–9632. [Google Scholar] [CrossRef]
- Yao, X.-D.; Rosenthal, K.L. Herpes simplex virus type 2 virion host shutoff protein suppresses innate dsRNA antiviral pathways in human vaginal epithelial cells. J. Gen. Virol. 2011, 92, 1981–1993. [Google Scholar] [CrossRef] [PubMed]
- Kit, S.; Dubbs, D.R.; Piekarski, L.J.; Hsu, T.C. Deletion of thymidine kinase activity from L cells resistant to bromodeoxyuridine. Exp. Cell Res. 1963, 31, 297–312. [Google Scholar] [CrossRef]
- Deville-Bonne, D.; el Amri, C.; Meyer, P.; Chen, Y.; Agrofoglio, L.A.; Janin, J. Human and viral nucleoside/nucleotide kinases involved in antiviral drug activation: Structural and catalytic properties. Antiviral Res. 2010, 86, 101–120. [Google Scholar] [CrossRef] [PubMed]
- McKnight, S.L. The nucleotide sequence and transcript map of the herpes simplex virus thymidine kinase gene. Nucleic Acids Res. 1980, 8, 5949–5964. [Google Scholar] [CrossRef][Green Version]
- Liu, Q.; Summers, W.C. Site-directed mutagenesis of a nucleotide-binding domain in HSV-1 thymidine kinase: Effects on catalytic activity. Virology 1988, 163, 638–642. [Google Scholar] [CrossRef]
- Gentry, G.A. Viral thymidine kinases and their relatives. Pharmacol. Ther. 1992, 54, 319–355. [Google Scholar] [CrossRef]
- Wild, K.; Bohner, T.; Folkers, G.; Schulz, G.E. The structures of thymidine kinase from Herpes simplex virus type 1 in complex with substrates and a substrate analogue. Protein Sci. 1997, 6, 2097–2106. [Google Scholar] [CrossRef]
- Elion, G.B.; Furman, P.A.; Fyfe, J.A.; de Miranda, P.; Beauchamp, L.; Schaeffer, H.J. Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl) guanine. Proc. Natl. Acad. Sci. USA 1977, 74, 5716–5720. [Google Scholar] [CrossRef]
- Fyfe, J.A.; Keller, P.M.; Furman, P.A.; Miller, R.L.; Elion, G.B. Thymidine kinase from herpes simplex virus phosphorylates the new antiviral compound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem. 1978, 253, 8721–8727. [Google Scholar]
- Gustafson, E.A.; Chillemi, A.C.; Sage, D.R.; Fingeroth, J.D. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 1998, 42, 2923–2931. [Google Scholar] [CrossRef]
- Gustafson, E.A.; Schinazi, R.F.; Fingeroth, J.D. Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J. Virol. 2000, 74, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Lock, M.J.; Thorley, N.; Teo, J.; Emery, V.C. Azidodeoxythymidine and didehydrodeoxythymidine as inhibitors and substrates of the human herpesvirus 8 thymidine kinase. J. Antimicrob. Chemother. 2002, 49, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Solaroli, N.; Johansson, M.; Balzarini, J.; Karlsson, A. Substrate specificity of three viral thymidine kinases (TK): Vaccinia virus TK, feline herpesvirus TK, and canine herpesvirus TK. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, B.; Dural, G.; Marschall, M.; Schregel, V.; Goltz, M.; Hentschke, J. Endotheliotropic elephant herpesvirus, the first betaherpesvirus with a thymidine kinase gene. J. Gen. Virol. 2006, 87, 2781–2789. [Google Scholar] [CrossRef]
- Mittal, S.K.; Field, H.J. Analysis of the bovine herpesvirus type 1 thymidine kinase (TK) gene from wild-type virus and TK-deficient mutants. J. Gen. Virol. 1989, 70, 901–918. [Google Scholar] [CrossRef]
- Dong, J.; Bai, J.; Sun, T.; Gu, Z.; Wang, J.; Sun, H.; Jiang, P. Comparative pathogenicity and immunogenicity of triple and double gene-deletion pseudorabies virus vaccine candidates. Res. Vet. Sci. 2017, 115, 17–23. [Google Scholar] [CrossRef]
- Pellicer, A.; Wigler, M.; Axel, R.; Silverstein, S. The transfer and stable integration of the HSV thymidine kinase gene into mouse cells. Cell 1978, 14, 133–141. [Google Scholar] [CrossRef]
- Field, H.J.; Darby, G. Pathogenicity in mice of strains of herpes simplex virus which are resistant to acyclovir in vitro and in vivo. Antimicrob. Agents Chemother. 1980, 17, 209–216. [Google Scholar] [CrossRef]
- Field, H.J.; Wildy, P. The pathogenicity of thymidine kinase-deficient mutants of herpes simplex virus in mice. J. Hyg. 1978, 81, 267–277. [Google Scholar] [CrossRef]
- Gordon, Y.J.; Gilden, D.M.; Becker, Y. HSV-1 thymidine kinase promotes virulence and latency in the mouse. Investig. Ophthalmol. Vis. Sci. 1983, 24, 599–602. [Google Scholar]
- Price, R.W.; Khan, A. Resistance of peripheral autonomic neurons to in vivo productive infection by herpes simplex virus mutants deficient in thymidine kinase activity. Infect. Immun. 1981, 34, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Cook, W.J.; Grove, K.L.; Coen, D.M. Human thymidine kinase can functionally replace herpes simplex virus type 1 thymidine kinase for viral replication in mouse sensory ganglia and reactivation from latency upon explant. J. Virol. 1998, 72, 6710–6715. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Lin, Y.-W.; Griffiths, A.; Huang, W.-Y.; Chen, S.-H. Competition and complementation between thymidine kinase-negative and wild-type herpes simplex virus during co-infection of mouse trigeminal ganglia. J. Gen. Virol. 2006, 87, 3495–3502. [Google Scholar] [CrossRef] [PubMed]
- Leist, T.P.; Sandri-Goldin, R.M.; Stevens, J.G. Latent infections in spinal ganglia with thymidine kinase-deficient herpes simplex virus. J. Virol. 1989, 63, 4976–4978. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-H.; Pearson, A.; Coen, D.M.; Chen, S.-H. Failure of thymidine kinase-negative herpes simplex virus to reactivate from latency following efficient establishment. J. Virol. 2004, 78, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-Y.; Yao, H.-W.; Wang, L.-C.; Shen, F.-H.; Hsu, S.-M.; Chen, S.-H. Thymidine kinase-negative herpes simplex virus 1 can efficiently establish persistent infection in neural tissues of nude mice. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Stroop, W.G.; Banks, M.C.; Qavi, H.; Chodosh, J.; Brown, S.M. A thymidine kinase deficient HSV-2 strain causes acute keratitis and establishes trigeminal ganglionic latency, but poorly reactivates in vivo. J. Med. Virol. 1994, 43, 297–309. [Google Scholar] [CrossRef]
- Jacobson, J.G.; Ruffner, K.L.; Kosz-Vnenchak, M.; Hwang, C.B.; Wobbe, K.K.; Knipe, D.M.; Coen, D.M. Herpes simplex virus thymidine kinase and specific stages of latency in murine trigeminal ganglia. J. Virol. 1993, 67, 6903–6908. [Google Scholar] [CrossRef]
- Tenser, R.B.; Hay, K.A.; Edris, W.A. Latency-associated transcript but not reactivatable virus is present in sensory ganglion neurons after inoculation of thymidine kinase-negative mutants of herpes simplex virus type 1. J. Virol. 1989, 63, 2861–2865. [Google Scholar] [CrossRef]
- Field, H.J.; Bell, S.E.; Elion, G.B.; Nash, A.A.; Wildy, P. Effect of acycloguanosine treatment of acute and latent herpes simplex infections in mice. Antimicrob. Agents Chemother. 1979, 15, 554–561. [Google Scholar] [CrossRef]
- Wilcox, C.L.; Crnic, L.S.; Pizer, L.I. Replication, latent infection, and reactivation in neuronal culture with a herpes simplex virus thymidine kinase-negative mutant. Virology 1992, 187, 348–352. [Google Scholar] [CrossRef]
- Morfin, F.; Thouvenot, D. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 2003, 26, 29–37. [Google Scholar] [CrossRef]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Coen, D.M.; Schaffer, P.A. Antiherpesvirus drugs: A promising spectrum of new drugs and drug targets. Nat. Rev. Drug Discov. 2003, 2, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Safrin, S. Treatment of acyclovir-resistant herpes simplex virus infections in patients with AIDS. J. Acquir. Immune Defic. Syndr. 1992, 5, 29–32. [Google Scholar]
- Chen, H.; Beardsley, G.P.; Coen, D.M. Mechanism of ganciclovir-induced chain termination revealed by resistant viral polymerase mutants with reduced exonuclease activity. Proc. Natl. Acad. Sci. USA 2014, 111, 17462–17467. [Google Scholar] [CrossRef]
- Zeng, Z.-J.; Xiang, S.-G.; Xue, W.-W.; Li, H.-D.; Ma, N.; Ren, Z.-J.; Xu, Z.-J.; Jiao, C.-H.; Wang, C.-Y.; Hu, W.-X. The cell death and DNA damages caused by the Tet-On regulating HSV-tk/GCV suicide gene system in MCF-7 cells. Biomed. Pharmacother. 2014, 68, 887–892. [Google Scholar] [CrossRef]
- Osaki, T.; Tanio, Y.; Tachibana, I.; Hosoe, S.; Kumagai, T.; Kawase, I.; Oikawa, S.; Kishimoto, T. Gene therapy for carcinoembryonic antigen-producing human lung cancer cells by cell type-specific expression of herpes simplex virus thymidine kinase gene. Cancer Res. 1994, 54, 5258–5261. [Google Scholar]
- Wei, S.-J.; Chao, Y.; Hung, Y.-M.; Lin, W.; Yang, D.-M.; Shih, Y.-L.; Ch’ang, L.-Y.; Whang-Peng, J.; Yang, W.K. S- and G2-phase cell cycle arrests and apoptosis induced by ganciclovir in murine melanoma cells transduced with herpes simplex virus thymidine kinase. Exp. Cell Res. 1998, 241, 66–75. [Google Scholar] [CrossRef]
- Tomicic, M.T.; Thust, R.; Kaina, B. Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: Critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene 2002, 21, 2141–2153. [Google Scholar] [CrossRef]
- Yin, X.; Yu, B.; Tang, Z.; He, B.; Ren, J.; Xiao, X.; Tang, W. Bifidobacterium infantis-mediated HSV-TK/GCV suicide gene therapy induces both extrinsic and intrinsic apoptosis in a rat model of bladder cancer. Cancer Gene Ther. 2013, 20, 77–81. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qiu, Y.; Peng, G.-L.; Liu, Q.-C.; Li, F.-L.; Zou, X.-S.; He, J.-X. Selective killing of lung cancer cells using carcinoembryonic antigen promoter and double suicide genes, thymidine kinase and cytosine deaminase (pCEA-TK/CD). Cancer Lett. 2012, 316, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Beltinger, C.; Fulda, S.; Kammertoens, T.; Meyer, E.; Uckert, W.; Debatin, K.-M. Herpes simplex virus thymidine kinase/ganciclovir-induced apoptosis involves ligand-independent death receptor aggregation and activation of caspases. Proc. Natl. Acad. Sci. USA 1999, 96, 8699–8704. [Google Scholar] [CrossRef] [PubMed]
- Beltinger, C.; Fulda, S.; Kammertoens, T.; Uckert, W.; Debatin, K.-M. Mitochondrial amplification of death signals determines thymidine kinase/ganciclovir-triggered activation of apoptosis. Cancer Res. 2000, 60, 3212–3217. [Google Scholar]
- Liu, X.; Wang, S.; Guo, X.; Wei, F.; Yin, J.; Zang, Y.; Li, N.; Chen, D. Exogenous p53 and ASPP2 expression enhances rAdV-TK/ GCV-induced death in hepatocellular carcinoma cells lacking functional p53. Oncotarget 2016, 7, 18896–18905. [Google Scholar] [CrossRef][Green Version]
- Moolten, F.L.; Wells, J.M. Curability of tumors bearing herpes thymidine kinase genes transfered by retroviral vectors. J. Natl. Cancer Inst. 1990, 82, 297–300. [Google Scholar] [CrossRef]
- Ram, Z.; Culver, K.W.; Walbridge, S.; Blaese, R.M.; Oldfield, E.H. In situ retroviral-mediated gene transfer for the treatment of brain tumors in rats. Cancer Res. 1993, 53, 83–88. [Google Scholar]
- O’Malley, B.W.; Chen, S.-H.; Schwartz, M.R.; Woo, S.L.C. Adenovirus-mediated gene therapy for human head and neck squamous cell cancer in a nude mouse model. Cancer Res. 1995, 55, 1080–1085. [Google Scholar]
- Hall, S.J.; Sanford, M.A.; Atkinson, G.; Chen, S.-H. Induction of potent antitumor natural killer cell activity by herpes simplex virus-thymidine kinase and ganciclovir therapy in an orthotopic mouse model of prostate cancer. Cancer Res. 1998, 58, 3221–3225. [Google Scholar]
- Ram, Z.; Walbridge, S.; Shawker, T.; Culver, K.W.; Blaese, R.M.; Oldfield, E.H. The effect of thymidine kinase transduction and ganciclovir therapy on tumor vasculature and growth of 9L gliomas in rats. J. Neurosurg. 1994, 81, 256–260. [Google Scholar] [CrossRef]
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The “Bystander Effect”: Tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993, 53, 5274–5283. [Google Scholar] [PubMed]
- Hamel, W.; Magnelli, L.; Chiarugi, V.P.; Israel, M.A. Herpes simplex virus thymidine kinase/ganciclovir-mediated apoptotic death of bystander cells. Cancer Res. 1996, 56, 2697–2702. [Google Scholar] [PubMed]
- Vile, R.G.; Nelson, J.A.; Castleden, S.; Chong, H.; Hart, I.R. Systemic gene therapy of murine melanoma using tissue specific expression of the HSVtk gene involves an immune component. Cancer Res. 1994, 54, 6228–6234. [Google Scholar] [PubMed]
- Pastorakova, A.; Jakubechova, J.; Altanerova, U.; Altaner, C. Suicide gene therapy mediated with exosomes produced by mesenchymal stem/stromal cells stably transduced with HSV thymidine kinase. Cancers 2020, 12, 1096. [Google Scholar] [CrossRef]
- Tamura, R.; Miyoshi, H.; Morimoto, Y.; Oishi, Y.; Sampetrean, O.; Iwasawa, C.; Mine, Y.; Saya, H.; Yoshida, K.; Okano, H.; et al. Gene therapy using neural stem/progenitor cells derived from human induced pluripotent stem cells: Visualization of migration and bystander killing effect. Hum. Gene Ther. 2020, 31, 352–366. [Google Scholar] [CrossRef]
- Voges, J.; Reszka, R.; Gossmann, A.; Dittmar, C.; Richter, R.; Garlip, G.; Kracht, L.; Coenen, H.H.; Sturm, V.; Wienhard, K.; et al. Imaging-guided convection-enhanced delivery and gene therapy of glioblastoma. Ann. Neurol. 2003, 54, 479–487. [Google Scholar] [CrossRef]
- Li, N.; Zhou, J.; Weng, D.; Zhang, C.; Li, L.; Wang, B.; Song, Y.; He, Q.; Lin, D.; Chen, D.; et al. Adjuvant adenovirus-mediated delivery of herpes simplex virus thymidine kinase administration improves outcome of liver transplantation in patients with advanced hepatocellular carcinoma. Clin. Cancer Res. 2007, 13, 5847–5854. [Google Scholar] [CrossRef]
- Nasu, Y.; Saika, T.; Ebara, S.; Kusaka, N.; Kaku, H.; Abarzua, F.; Manabe, D.; Thompson, T.C.; Kumon, H. Suicide gene therapy with adenoviral delivery of HSV-tK gene for patients with local recurrence of prostate cancer after hormonal therapy. Mol. Ther. 2007, 15, 834–840. [Google Scholar] [CrossRef]
- Van Putten, E.H.P.; Dirven, C.M.F.; van den Bent, M.J.; Lamfers, M.L.M. Sitimagene ceradenovec: A gene-based drug for the treatment of operable high-grade glioma. Futur. Oncol. 2010, 6, 1691–1710. [Google Scholar] [CrossRef]
- Sangro, B.; Mazzolini, G.; Ruiz, M.; Ruiz, J.; Quiroga, J.; Herrero, I.; Qian, C.; Benito, A.; Larrache, J.; Olagüe, C.; et al. A phase I clinical trial of thymidine kinase-based gene therapy in advanced hepatocellular carcinoma. Cancer Gene Ther. 2010, 17, 837–843. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.M.B.U.; Hong, Y.M.; Ornella, M.S.C.; Ngabire, D.; Jang, H.; Cho, E.; Kim, E.-K.; Hale, J.J.J.; Kim, C.H.; Ahn, S.C.; et al. Engineering and preclinical evaluation of western reserve oncolytic vaccinia virus expressing A167Y mutant herpes simplex virus thymidine kinase. Biomedicines 2020, 8, 426. [Google Scholar] [CrossRef] [PubMed]
- Hossain, J.A.; Ystaas, L.R.; Mrdalj, J.; Välk, K.; Riecken, K.; Fehse, B.; Bjerkvig, R.; Grønli, J.; Miletic, H. Lentiviral HSV-Tk.007-mediated suicide gene therapy is not toxic for normal brain cells. J. Gene Med. 2016, 18, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Kenarkoohi, A.; Bamdad, T.; Soleimani, M.; Soleimanjahi, H.; Fallah, A.; Falahi, S. HSV-TK Expressing mesenchymal stem cells exert inhibitory effect on cervical cancer model. Int. J. Mol. Cell. Med. 2020, 9, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.B.; Lee, H.; Lee, K.W.; Kim, S.J.; Hong, D.; Park, J.B. Complete regression of metastatic de-differentiated liposarcoma with engineered mesenchymal stromal cells with dTRAIL and HSV-TK. Am. J. Transl. Res. 2020, 12, 3993–4000. [Google Scholar] [PubMed]
- Islam, S.M.B.U.; Lee, B.; Jiang, F.; Kim, E.-K.; Ahn, S.C.; Hwang, T.-H. Engineering and characterization of oncolytic vaccinia virus expressing truncated herpes simplex virus thymidine kinase. Cancers 2020, 12, 228. [Google Scholar] [CrossRef] [PubMed]
- Dührsen, L.; Hartfuß, S.; Hirsch, D.; Geiger, S.; Maire, C.L.; Sedlacik, J.; Guenther, C.; Westphal, M.; Lamszus, K.; Hermann, F.G.; et al. Preclinical analysis of human mesenchymal stem cells: Tumor tropism and therapeutic efficiency of local HSV-TK suicide gene therapy in glioblastoma. Oncotarget 2019, 10, 6049–6061. [Google Scholar] [CrossRef]
- Li, H.; Du, H.; Zhang, G.; Wu, Y.; Qiu, P.; Liu, J.; Guo, J.; Liu, X.; Sun, L.; Du, B.; et al. Curcumin plays a synergistic role in combination with HSV-TK/GCV in inhibiting growth of murine B16 melanoma cells and melanoma xenografts. PeerJ 2019, 7, e7760. [Google Scholar] [CrossRef]
- Sagou, K.; Imai, T.; Sagara, H.; Uema, M.; Kawaguchi, Y. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by Autophosphorylation and its role in pathogenesis. J. Virol. 2009, 83, 5773–5783. [Google Scholar] [CrossRef]
- Morimoto, T.; Arii, J.; Tanaka, M.; Sata, T.; Akashi, H.; Yamada, M.; Nishiyama, Y.; Uema, M.; Kawaguchi, Y. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J. Virol. 2009, 83, 11624–11634. [Google Scholar] [CrossRef]
- Wisner, T.W.; Wright, C.C.; Kato, A.; Kawaguchi, Y.; Mou, F.; Baines, J.D.; Roller, R.J.; Johnson, D.C. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 2009, 83, 3115–3126. [Google Scholar] [CrossRef] [PubMed]
- Wild, P.; Leisinger, S.; de Oliveira, A.P.; Doehner, J.; Schraner, E.M.; Fraevel, C.; Ackermann, M.; Kaech, A. Nuclear envelope impairment is facilitated by the herpes simplex virus 1 Us3 kinase. F1000Research 2019, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Tobler, K.; Senn, C.; Schraner, E.M.; Ackermann, M.; Fraefel, C.; Wild, P. The herpes simplex virus 1 Us3 kinase is involved in assembly of membranes needed for viral envelopment and in distribution of glycoprotein K. F1000Research 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef] [PubMed]
- Ryckman, B.J.; Roller, R.J. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 2004, 78, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Mou, F.; Forest, T.; Baines, J.D. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 2007, 81, 6459–6470. [Google Scholar] [CrossRef] [PubMed]
- Bjerke, S.L.; Roller, R.J. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 2006, 347, 261–276. [Google Scholar] [CrossRef]
- Mou, F.; Wills, E.; Baines, J.D. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 2009, 83, 5181–5191. [Google Scholar] [CrossRef]
- Purves, F.C.; Spector, D.; Roizman, B. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 1991, 65, 5757–5764. [Google Scholar] [CrossRef]
- Reynolds, A.E.; Ryckman, B.J.; Baines, J.D.; Zhou, Y.; Liang, L.; Roller, R.J. UL31 and UL34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001, 75, 8803–8817. [Google Scholar] [CrossRef]
- Kato, A.; Liu, Z.; Minowa, A.; Imai, T.; Tanaka, M.; Sugimoto, K.; Nishiyama, Y.; Arii, J.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J. Virol. 2011, 85, 9599–9613. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, R.; van Sant, C.; Roizman, B. The herpes simplex virus 1 protein kinase Us3 is required for protection from apoptosis induced by the virus. Proc. Natl. Acad. Sci. USA 1997, 94, 7891–7896. [Google Scholar] [CrossRef] [PubMed]
- Galvan, V.; Roizman, B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell-type-dependent manner. Proc. Natl. Acad. Sci. USA 1998, 95, 3931–3936. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Roizman, B. The Us3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 10410–10415. [Google Scholar] [CrossRef]
- Hagglund, R.; Munger, J.; Poon, A.P.W.; Roizman, B. U(S)3 protein kinase of herpes simplex virus 1 blocks caspase 3 activation induced by the products of U(S)1.5 and U(L)13 genes and modulates expression of transduced U(S)1.5 open reading frame in a cell type-specific manner. J. Virol. 2002, 76, 743–754. [Google Scholar] [CrossRef]
- Leopardi, R.; Roizman, B. The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc. Natl. Acad. Sci. USA 1996, 93, 9583–9587. [Google Scholar] [CrossRef]
- Benetti, L.; Roizman, B. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 9411–9416. [Google Scholar] [CrossRef]
- Walters, M.S.; Kinchington, P.R.; Banfield, B.W.; Silverstein, S. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 2010, 84, 9666–9676. [Google Scholar] [CrossRef]
- Chuluunbaatar, U.; Roller, R.; Feldman, M.E.; Brown, S.; Shokat, K.M.; Mohr, I. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev. 2010, 24, 2627–2639. [Google Scholar] [CrossRef]
- Chuluunbaatar, U.; Mohr, I. A herpesvirus kinase that masquerades as Akt: You don’t have to look like Akt, to act like it. Cell Cycle 2011, 10, 2064–2068. [Google Scholar] [CrossRef]
- Sloan, D.D.; Zahariadis, G.; Posavad, C.M.; Pate, N.T.; Kussick, S.J.; Jerome, K.R. CTL Are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J. Immunol. 2003, 171, 6733–6741. [Google Scholar] [CrossRef] [PubMed]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes simplex virus US3 tegument protein inhibits Toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J. Virol. 2013, 87, 12814–12827. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhang, R.; Lang, Y.; Shao, A.; Xu, A.; Feng, W.; Han, J.; Wang, M.; He, W.; Yu, C.; et al. Bclaf1 critically regulates the type I interferon response and is degraded by alphaherpesvirus US3. PLoS Pathog. 2019, 15, e1007559. [Google Scholar] [CrossRef]
- Rubio, R.M.; Mohr, I. Inhibition of ULK1 and Beclin1 by an α-herpesvirus Akt-like Ser/Thr kinase limits autophagy to stimulate virus replication. Proc. Natl. Acad. Sci. USA 2019, 116, 26941–26950. [Google Scholar] [CrossRef]
- Kato, A.; Arii, J.; Shiratori, I.; Akashi, H.; Arase, H.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and Regulates its expression on the cell surface. J. Virol. 2009, 83, 250–261. [Google Scholar] [CrossRef]
- Rao, P.; Pham, H.T.; Kulkarni, A.; Yang, Y.; Liu, X.; Knipe, D.M.; Cresswell, P.; Yuan, W. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J. Virol. 2011, 85, 8093–8104. [Google Scholar] [CrossRef]
- Cunningham, C.; Davison, A.J.; Dolan, A.; Frame, M.C.; McGeoch, D.J.; Meredith, D.M.; Moss, H.W.; Orr, A.C. The UL13 virion protein of herpes simplex virus type 1 is phosphorylated by a novel virus-induced protein kinase. J. Gen. Virol. 1992, 73, 303–311. [Google Scholar] [CrossRef]
- Smith, R.F.; Smith, T.F. Identification of new protein kinase-related genes in three herpesviruses, herpes simplex virus, varicella-zoster virus, and Epstein-Barr virus. J. Virol. 1989, 63, 450–455. [Google Scholar] [CrossRef]
- Chee, M.S.; Lawrence, G.L.; Barrell, B.G. Alpha-, beta- and gammaherpesviruses encode a putative phosphotransferase. J. Gen. Virol. 1989, 70, 1151–1160. [Google Scholar] [CrossRef]
- Overton, H.A.; McMillan, D.J.; Klavinskis, L.S.; Hope, L.; Ritchie, A.J.; Wong-kai-in, P. Herpes simplex virus type 1 gene UL13 encodes a phosphoprotein that is a component of the virion. Virology 1992, 190, 184–192. [Google Scholar] [CrossRef]
- Lemaster, S.; Roizman, B. Herpes simplex virus phosphoproteins. II. Characterization of the virion protein kinase and of the polypeptides phosphorylated in the virion. J. Virol. 1980, 35, 798–811. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, T.K.; Lynch, J.; Hay, J.; Ruyechan, W.; Grose, C. Varicella-zoster virus ORF47 protein serine kinase: Characterization of a cloned, biologically active phosphotransferase and two viral substrates, ORF62 and ORF63. J. Virol. 2001, 75, 8854–8858. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chee, M.S.; Bankier, A.T.; Beck, S.; Bohni, R.; Brown, C.M.; Cerny, R.; Horsnell, T.; Hutchison, C.A., III; Kouzarides, T.; Martignetti, J.A. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 1990, 154, 125–169. [Google Scholar] [CrossRef]
- Lawrence, G.L.; Chee, M.; Craxton, M.A.; Gompels, U.A.; Honess, R.W.; Barrell, B.G. Human herpesvirus 6 is closely related to human cytomegalovirus. J. Virol. 1990, 64, 287–299. [Google Scholar] [CrossRef]
- Ng, T.I.; Talarico, C.; Burnette, T.C.; Biron, K.; Roizman, B. Partial substitution of the functions of the herpes simplex virus 1 U(L)13 gene by the human cytomegalovirus U(L)97 gene. Virology 1996, 225, 347–358. [Google Scholar] [CrossRef][Green Version]
- Daikoku, T.; Shibata, S.; Goshima, F.; Oshima, S.; Tsurumi, T.; Yamada, H.; Yamashita, Y.; Nishiyama, Y. Purification and characterization of the protein kinase encoded by the UL13 gene of herpes simplex virus type 2. Virology 1997, 235, 82–93. [Google Scholar] [CrossRef]
- Coulter, L.J.; Moss, H.W.; Lang, J.; McGeoch, D.J. A mutant of herpes simplex virus type 1 in which the UL13 protein kinase gene is disrupted. J. Gen. Virol. 1993, 74, 387–395. [Google Scholar] [CrossRef]
- Overton, H.; McMillan, D.; Hope, L.; Wong-Kai-In, P. Production of host shutoff-defective mutants of herpes simplex virus type 1 by inactivation of the UL13 gene. Virology 1994, 202, 97–106. [Google Scholar] [CrossRef]
- Tanaka, M.; Nishiyama, Y.; Sata, T.; Kawaguchi, Y. The role of protein kinase activity expressed by the UL13 gene of herpes simplex virus 1: The activity is not essential for optimal expression of UL41 and ICP0. Virology 2005, 341, 301–312. [Google Scholar] [CrossRef]
- Shibaki, T.; Suzutani, T.; Yoshida, I.; Ogasawara, M.; Azuma, M. Participation of type I interferon in the decreased virulence of the UL13 gene-deleted mutant of herpes simplex virus type 1. J. Interf. Cytokine Res. 2001, 21, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Ogle, W.O.; Ng, T.I.; Carter, K.L.; Roizman, B. The UL13 protein kinase and the infected cell type are determinants of posttranslational modification of ICP0. Virology 1997, 235, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Asai, R.; Ohno, T.; Kato, A.; Kawaguchi, Y. Identification of proteins directly phosphorylated by UL13 protein kinase from herpes simplex virus 1. Microbes Infect. 2007, 9, 1434–1438. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Du, T.; Zhou, G.; Roizman, B. The stability of herpes simplex virus 1 ICP0 early after infection is defined by the RING finger and the UL13 protein kinase. J. Virol. 2014, 88, 5437–5443. [Google Scholar] [CrossRef] [PubMed]
- Eaton, H.E.; Saffran, H.A.; Wu, F.W.; Quach, K.; Smiley, J.R. Herpes simplex virus protein kinases US3 and UL13 modulate VP11/12 phosphorylation, virion packaging, and phosphatidylinositol 3-kinase/Akt signaling activity. J. Virol. 2014, 88, 7379–7388. [Google Scholar] [CrossRef]
- Geiss, B.J.; Cano, G.L.; Tavis, J.E.; Morrison, L.A. Herpes simplex virus 2 VP22 phosphorylation induced by cellular and viral kinases does not influence intracellular localization. Virology 2004, 330, 74–81. [Google Scholar] [CrossRef][Green Version]
- Ng, T.I.; Ogle, W.O.; Roizman, B. UL13 protein kinase of herpes simplex virus 1 complexes with glycoprotein E and mediates the phosphorylation of the viral Fc receptor: Glycoproteins E and I. Virology 1998, 241, 37–48. [Google Scholar] [CrossRef][Green Version]
- Attrill, H.L.; Cumming, S.A.; Clements, J.B.; Graham, S.V. The herpes simplex virus type 1 US11 protein binds the coterminal UL12, UL13, and UL14 RNAs and regulates UL13 expression in vivo. J. Virol. 2002, 76, 8090–8100. [Google Scholar] [CrossRef][Green Version]
- Gershburg, S.; Geltz, J.; Peterson, K.E.; Halford, W.P.; Gershburg, E. The UL13 and US3 protein kinases of herpes simplex virus 1 cooperate to promote the assembly and release of mature, infectious virions. PLoS ONE 2015, 10, e0131420. [Google Scholar] [CrossRef]
- Sato, Y.; Koshizuka, T.; Ishibashi, K.; Hashimoto, K.; Ishioka, K.; Ikuta, K.; Yokota, S.-I.; Fujii, N.; Suzutani, T. Involvement of herpes simplex virus type 1 UL13 protein kinase in induction of SOCS genes, the negative regulators of cytokine signaling. Microbiol. Immunol. 2017, 61, 159–167. [Google Scholar] [CrossRef]
- Yokota, S.; Yokosawa, N.; Okabayashi, T.; Suzutani, T.; Miura, S.; Jimbow, K.; Fujii, N. Induction of suppressor of cytokine signaling-3 by herpes simplex virus type 1 contributes to inhibition of the interferon signaling pathway. J. Virol. 2004, 78, 6282–6286. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.G.; Ahmed, C.M.I.; Dabelic, R.; Jager, L.D.; Noon-Song, E.N.; Haider, S.M.; Johnson, H.M.; Bigley, N.J. HSV-1-induced SOCS-1 expression in keratinocytes: Use of a SOCS-1 antagonist to block a novel mechanism of viral immune evasion. J. Immunol. 2009, 183, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Reichard, A.C.; Cheemarla, N.R.; Bigley, N.J. SOCS1/3 expression levels in HSV-1-infected, cytokine-polarized and -unpolarized macrophages. J. Interf. Cytokine Res. 2015, 35, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, R.; Musarra-Pizzo, M.; Lei, Z.; Zhou, G.G.; Sciortino, M.T. VHS, US3 and UL13 viral tegument proteins are required for herpes simplex virus-induced modification of protein kinase R. Sci. Rep. 2020, 10, 5580. [Google Scholar] [CrossRef]
- Merrick, W.C. Mechanism and regulation of eukaryotic protein synthesis. Microbiol. Rev. 1992, 56, 291–315. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Van Sant, C.; Roizman, B. Eukaryotic elongation factor 1delta is hyperphosphorylated by the protein kinase encoded by the U(L)13 gene of herpes simplex virus 1. J. Virol. 1998, 72, 1731–1736. [Google Scholar] [CrossRef]
- Perkins, D.; Pereira, E.F.R.; Gober, M.; Yarowsky, P.J.; Aurelian, L. The herpes simplex virus type 2 R1 protein kinase (ICP10 PK) blocks apoptosis in hippocampal neurons, involving activation of the MEK/MAPK survival pathway. J. Virol. 2002, 76, 1435–1449. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kato, K.; Tanaka, M.; Kanamori, M.; Nishiyama, Y.; Yamanashi, Y. Conserved protein kinases encoded by herpesviruses and cellular protein kinase cdc2 target the same phosphorylation site in eukaryotic elongation factor 1delta. J. Virol. 2003, 77, 2359–2368. [Google Scholar] [CrossRef]
- Baines, J.D.; Roizman, B. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 1991, 65, 938–944. [Google Scholar] [CrossRef]
- Jun, P.; Strelow, L.; Herman, R.; Marsden, H.; Eide, T.; Haarr, L.; Leib, D. The UL4 gene of herpes simplex virus type 1 is dispensable for latency, reactivation and pathogenesis in mice. J. Gen. Virol. 1998, 79, 1603–1611. [Google Scholar] [CrossRef][Green Version]
- Worrad, D.M.; Caradonna, S. The herpes simplex virus type 2 UL3 open reading frame encodes a nuclear localizing phosphoprotein. Virology 1993, 195, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Lee, L.F.; Yanagida, N.; Nazerian, K. Identification and characterization of a Marek’s disease virus gene homologous to glycoprotein L of herpes simplex virus. Virology 1994, 204, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, H.; Perng, G.C.; Cai, S.; Nesburn, A.B.; Wechsler, S.L. The UL3 open reading frame of herpes simplex virus type 1 codes for a phosphoprotein. Virus Res. 1996, 44, 137–142. [Google Scholar] [CrossRef]
- McGeoch, D.J.; Cunningham, C.; McIntyre, G.; Dolan, A. Comparative sequence analysis of the long repeat regions and adjoining parts of the long unique regions in the genomes of herpes simplex viruses types 1 and 2. J. Gen. Virol. 1991, 72, 3057–3075. [Google Scholar] [CrossRef]
- Davison, A.J. DNA sequence of the US component of the varicella-zoster virus genome. EMBO J. 1983, 2, 2203–2209. [Google Scholar] [CrossRef]
- Telford, E.A.R.; Watson, M.S.; McBride, K.; Davison, A.J. The DNA sequence of equine herpesvirus-1. Virology 1992, 189, 304–316. [Google Scholar] [CrossRef]
- Dean, H.J.; Cheung, A.K. Identification of the pseudorabies virus UL4 and UL5 (helicase) genes. Virology 1994, 202, 962–967. [Google Scholar] [CrossRef]
- Vlcek, C.; Benes, V.; Lu, Z.; Kutish, G.F.; Paces, V.; Rock, D.; Letchworth, G.J.; Schwyzer, M. Nucleotide sequence analysis of a 30-kb region of the bovine herpesvirus 1 genome which exhibits a colinear gene arrangement with the UL21 to UL4 genes of herpes simplex virus. Virology 1995, 210, 100–108. [Google Scholar] [CrossRef]
- Yamada, H.; Jiang, Y.-M.; Zhu, H.-Y.; Inagaki-Ohara, K.; Nishiyama, Y. Nucleolar localization of the UL3 protein of herpes simplex virus type 2. J. Gen. Virol. 1999, 80, 2157–2164. [Google Scholar] [CrossRef]
- Zheng, C.; Lin, F.; Wang, S.; Xing, J. A novel virus-encoded nucleocytoplasmic shuttling protein: The UL3 protein of herpes simplex virus type 1. J. Virol. Methods 2011, 177, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Eide, T.; Marsden, H.S.; Leib, D.A.; Cunningham, C.; Davison, A.J.; Langeland, N.; Haarr, L. Identification of the UL4 protein of herpes simplex virus type 1. J. Gen. Virol. 1998, 79, 3033–3038. [Google Scholar] [CrossRef] [PubMed]
- Markovitz, N.S.; Roizman, B. Small dense nuclear bodies are the site of localization of herpes simplex virus 1 U(L)3 and U(L)4 proteins and of ICP22 only when the latter protein is present. J. Virol. 2000, 74, 523–528. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ward, P.L.; Taddeo, B.; Markovitz, N.S.; Roizman, B. Identification of a novel expressed open reading frame situated between genes U(L)20 and U(L)21 of the herpes simplex virus 1 genome. Virology 2000, 266, 275–285. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Loret, S.; Lippé, R. Biochemical analysis of infected cell polypeptide (ICP)0, ICP4, ul7 and ul23 incorporated into extracellular herpes simplex virus type 1 virions. J. Gen. Virol. 2012, 93, 624–634. [Google Scholar] [CrossRef]
- Roller, R.J.; Fetters, R. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J. Virol. 2015, 89, 3112–3122. [Google Scholar] [CrossRef]
- Albecka, A.; Owen, D.J.; Ivanova, L.; Brun, J.; Liman, R.; Davies, L.; Ahmed, M.F.; Colaco, S.; Hollinshead, M.; Graham, S.C.; et al. Dual function of the pUL7-pUL51 tegument protein complex in herpes simplex virus 1 infection. J. Virol. 2017, 91, 1–19. [Google Scholar] [CrossRef]
- Xu, X.; Fan, S.; Zhou, J.; Zhang, Y.; Che, Y.; Cai, H.; Wang, L.; Guo, L.; Liu, L.; Li, Q. The mutated tegument protein UL7 attenuates the virulence of herpes simplex virus 1 by reducing the modulation of α-4 gene transcription. Virol. J. 2016, 13, 1–12. [Google Scholar] [CrossRef]
- Nozawa, N.; Daikoku, T.; Yamauchi, Y.; Takakuwa, H.; Goshima, F.; Yoshikawa, T.; Nishiyama, Y. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genes 2002, 24, 257–266. [Google Scholar] [CrossRef]
- Feutz, E.; McLeland-Wieser, H.; Ma, J.; Roller, R.J. Functional interactions between herpes simplex virus pUL51, pUL7 and gE reveal cell-specific mechanisms for epithelial cell-to-cell spread. Virology 2019, 537, 84–96. [Google Scholar] [CrossRef]
- Tanaka, M.; Sata, T.; Kawaguchi, Y. The product of the Herpes simplex virus 1 UL7 gene interacts with a mitochondrial protein, adenine nucleotide translocator 2. Virol. J. 2008, 5, 1–13. [Google Scholar] [CrossRef]
- Nozawa, N.; Daikoku, T.; Koshizuka, T.; Yamauchi, Y.; Yoshikawa, T.; Nishiyama, Y. Subcellular Localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in golgi apparatus targeting. J. Virol. 2003, 77, 3204–3216. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Oda, S.; Watanabe, M.; Oyama, M.; Kozuka-Hata, H.; Koyanagi, N.; Maruzuru, Y.; Arii, J.; Kawaguchi, Y. Roles of the phosphorylation of herpes simplex virus 1 UL51 at a specific site in viral replication and pathogenicity. J. Virol. 2018, 92, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, N.; Kawaguchi, Y.; Tanaka, M.; Kato, A.; Kato, A.; Kimura, H.; Nishiyama, Y. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 2005, 79, 6947–6956. [Google Scholar] [CrossRef] [PubMed]
- Roller, R.J.; Haugo, A.C.; Yang, K.; Baines, J.D. The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J. Virol. 2014, 88, 4058–4068. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.; Arii, J.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. The interaction between herpes simplex virus 1 tegument proteins UL51 and UL14 and its role in virion morphogenesis. J. Virol. 2016, 90, 8754–8767. [Google Scholar] [CrossRef] [PubMed]
- Maclean, C.A.; Robertson, L.M.; Jamieson, F.E. Characterization of the UL10 gene product of herpes simplex virus type and investigation of its role in vivo. J. Gen. Virol. 1993, 74, 975–983. [Google Scholar] [CrossRef] [PubMed]
- MacLean, C.A.; Efstathiou, S.; Elliott, M.L.; Jamieson, F.E.; McGeoch, D.J. Investigation of herpes simplex virus type 1 genes encoding multiply inserted membrane proteins. J. Gen. Virol. 1991, 72, 897–906. [Google Scholar] [CrossRef]
- Baines, J.D.; Wills, E.; Jacob, R.J.; Pennington, J.; Roizman, B. Glycoprotein M of herpes simplex virus 1 is incorporated into virions during budding at the inner nuclear membrane. J. Virol. 2007, 81, 800–812. [Google Scholar] [CrossRef]
- Zhang, J.; Nagel, C.-H.; Sodeik, B.; Lippé, R. Early, active, and specific localization of herpes simplex virus type 1 gM to nuclear membranes. J. Virol. 2009, 83, 12984–12997. [Google Scholar] [CrossRef]
- El Kasmi, I.; Lippé, R. Herpes simplex virus 1 gN partners with gM To modulate the viral fusion machinery. J. Virol. 2015, 89, 2313–2323. [Google Scholar] [CrossRef]
- Crump, C.M.; Bruun, B.; Bell, S.; Pomeranz, L.E.; Minson, T.; Browne, H.M. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 2004, 85, 3517–3527. [Google Scholar] [CrossRef] [PubMed]
- Striebinger, H.; Funk, C.; Raschbichler, V.; Bailer, S.M. Subcellular trafficking and functional relationship of the HSV-1 glycoproteins N and M. Viruses 2016, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Schnee, M.; Ruzsics, Z.; Bubeck, A.; Koszinowski, U.H. Common and specific properties of Herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J. Virol. 2006, 80, 11658–11666. [Google Scholar] [CrossRef]
- Striebinger, H.; Zhang, J.; Ott, M.; Funk, C.; Radtke, K.; Duron, J.; Ruzsics, Z.; Haas, J.; Lippé, R.; Bailer, S.M. Subcellular trafficking and functional importance of herpes simplex virus type 1 glycoprotein M domains. J. Gen. Virol. 2015, 96, 3313–3325. [Google Scholar] [CrossRef]
- Leege, T.; Fuchs, W.; Granzow, H.; Kopp, M.; Klupp, B.G.; Mettenleiter, T.C. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 2009, 83, 896–907. [Google Scholar] [CrossRef]
- Ren, Y.; Bell, S.; Zenner, H.L.; Kathy Lau, S.Y.; Crump, C.M. Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J. Gen. Virol. 2012, 93, 319–329. [Google Scholar] [CrossRef]
- Koyano, S.; Mar, E.C.; Stamey, F.R.; Inoue, N. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J. Gen. Virol. 2003, 84, 1485–1491. [Google Scholar] [CrossRef]
- El Kasmi, I.; Khadivjam, B.; Lackman, M.; Duron, J.; Bonneil, E.; Thibault, P.; Lippé, R. Extended synaptotagmin 1 interacts with herpes simplex virus 1 glycoprotein M and Negatively modulates virus-induced membrane fusion. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Cheshenko, N.; del Rosario, B.; Woda, C.; Marcellino, D.; Satlin, L.M.; Herold, B.C. Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 2003, 163, 283–293. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. Bcl-2 blocks accretion or depletion of stored calcium but has no effect on the redistribution of IP3 receptor I mediated by glycoprotein E of herpes simplex virus 1. J. Virol. 2007, 81, 6316–6325. [Google Scholar] [CrossRef][Green Version]
- Boruchowicz, H.; Hawkins, J.; Cruz-Palomar, K.; Lippé, R. The XPO6 exportin mediates herpes simplex virus 1 gM nuclear release late in infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, C.; Pelchen-Matthews, A.; Mlcochova, P.; Marsh, M.; Milne, R.S.B.; Towers, G.J. Tetherin restricts herpes simplex virus 1 and is antagonized by glycoprotein M. J. Virol. 2013, 87, 13124–13133. [Google Scholar] [CrossRef] [PubMed]
- MacLean, C.A.; Clark, B.; McGeoch, D.J. Gene UL11 of herpes simplex virus type 1 encodes a virion protein which is myristylated. J. Gen. Virol. 1989, 70, 3147–3157. [Google Scholar] [CrossRef] [PubMed]
- Loomis, J.S.; Bowzard, J.B.; Courtney, R.J.; Wills, J.W. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2001, 75, 12209–12219. [Google Scholar] [CrossRef] [PubMed]
- Loomis, J.S.; Courtney, R.J.; Wills, J.W. Packaging determinants in the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2006, 80, 10534–10541. [Google Scholar] [CrossRef]
- MacLean, C.A.; Dolan, A.; Jamieson, F.E.; McGeoch, D.J. The myristylated virion proteins of herpes simplex virus type 1: Investigation of their role in the virus life cycle. J. Gen. Virol. 1992, 73, 539–547. [Google Scholar] [CrossRef]
- Baird, N.L.; Starkey, J.L.; Hughes, D.J.; Wills, J.W. Myristylation and palmitylation of HSV-1 UL11 are not essential for its function. Virology 2010, 397, 80–88. [Google Scholar] [CrossRef]
- Loomis, J.S.; Courtney, R.J.; Wills, J.W. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2003, 77, 11417–11424. [Google Scholar] [CrossRef]
- Yeh, P.-C.; Meckes, D.G.; Wills, J.W. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 2008, 82, 10693–10700. [Google Scholar] [CrossRef] [PubMed]
- Farnsworth, A.; Wisner, T.W.; Johnson, D.C. Cytoplasmic Residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 2007, 81, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chadha, P.; Meckes, D.G.; Baird, N.L.; Wills, J.W. Interaction and Interdependent packaging of tegument protein UL11 and glycoprotein E of herpes simplex virus. J. Virol. 2011, 85, 9437–9446. [Google Scholar] [CrossRef] [PubMed]
- Fulmer, P.A.; Melancon, J.M.; Baines, J.D.; Kousoulas, K.G. UL20 protein functions precede and are required for the UL11 functions of herpes simplex virus type 1 cytoplasmic virion envelopment. J. Virol. 2007, 81, 3097–3108. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. [Google Scholar] [CrossRef]
- Metrick, C.M.; Koenigsberg, A.L.; Heldwein, E.E. Conserved outer tegument component UL11 from herpes simplex virus 1 is an intrinsically disordered, RNA-binding protein. mBio 2020, 11, 1–22. [Google Scholar] [CrossRef]
- Baird, N.L.; Yeh, P.C.; Courtney, R.J.; Wills, J.W. Sequences in the UL11 tegument protein of herpes simplex virus that control association with detergent-resistant membranes. Virology 2008, 374, 315–321. [Google Scholar] [CrossRef]
- Chadha, P.; Sarfo, A.; Zhang, D.; Abraham, T.; Carmichael, J.; Han, J.; Wills, J.W. Domain interaction studies of herpes simplex virus 1 tegument protein UL16 reveal its interaction with mitochondria. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Nalwanga, D.; Rempel, S.; Roizman, B.; Baines, J.D. The U(L)16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 1996, 226, 236–242. [Google Scholar] [CrossRef]
- Carmichael, J.C.; Wills, J.W. Differential requirements for gE, gI, and UL16 among herpes simplex virus 1 syncytial variants suggest unique modes of dysregulating the mechanism of cell-to-cell spread. J. Virol. 2019, 93, 1–20. [Google Scholar] [CrossRef]
- Harper, A.L.; Meckes, D.G.; Marsh, J.A.; Ward, M.D.; Yeh, P.-C.; Baird, N.L.; Wilson, C.B.; Semmes, O.J.; Wills, J.W. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J. Virol. 2010, 84, 2963–2971. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Wills, J.W. Structural rearrangement within an enveloped virus upon binding to the host cell. J. Virol. 2008, 82, 10429–10435. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.-C.; Han, J.; Chadha, P.; Meckes, D.G.; Ward, M.D.; Semmes, O.J.; Wills, J.W. Direct and specific binding of the UL16 tegument protein of herpes simplex virus to the cytoplasmic tail of glycoprotein E. J. Virol. 2011, 85, 9425–9436. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chadha, P.; Starkey, J.L.; Wills, J.W. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc. Natl. Acad. Sci. USA 2012, 109, 19798–19803. [Google Scholar] [CrossRef]
- Gao, J.; Yan, X.; Banfield, B.W. Comparative analysis of UL16 mutants derived from multiple strains of herpes simplex virus 2 (HSV-2) and HSV-1 reveals species-specific requirements for the UL16 protein. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Carmichael, J.C.; Starkey, J.; Zhang, D.; Sarfo, A.; Chadha, P.; Wills, J.W.; Han, J. Glycoprotein D of HSV-1 is dependent on tegument protein UL16 for packaging and contains a motif that is differentially required for syncytia formation. Virology 2019, 527, 64–76. [Google Scholar] [CrossRef]
- Hutchinson, L.; Goldsmith, K.; Snoddy, D.; Ghosh, H.; Graham, F.L.; Johnson, D.C. Identification and characterization of a novel herpes simplex virus glycoprotein, gK, involved in cell fusion. J. Virol. 1992, 66, 5603–5609. [Google Scholar] [CrossRef]
- Foster, T.P.; Alvarez, X.; Kousoulas, K.G. Plasma membrane topology of syncytial domains of herpes simplex virus type 1 glycoprotein K (gK): The UL20 protein enables cell surface localization of gK but not gK-mediated cell-to-cell fusion. J. Virol. 2003, 77, 499–510. [Google Scholar] [CrossRef]
- Hutchinson, L.; Roop-Beauchamp, C.; Johnson, D.C. Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J. Virol. 1995, 69, 4556–4563. [Google Scholar] [CrossRef]
- Jayachandra, S.; Baghian, A.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 1997, 71, 5012–5024. [Google Scholar] [CrossRef]
- Hutchinson, L.; Graham, F.L.; Cai, W.; Debroy, C.; Person, S.; Johnson, D.C. Herpes simplex virus (HSV) glycoproteins B and K inhibit cell fusion induced by HSV syncytial mutants. Virology 1993, 196, 514–531. [Google Scholar] [CrossRef] [PubMed]
- Avitabile, E.; Lombardi, G.; Campadelli-Fiume, G. Herpes simplex virus glycoprotein K, but not its syncytial allele, inhibits cell-cell fusion mediated by the four fusogenic glycoproteins, gD, gB, gH, and gL. J. Virol. 2003, 77, 6836–6844. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Foster, T.P.; Melancon, J.M.; Baines, J.D.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 2004, 78, 5347–5357. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.P.; Rybachuk, G.V.; Kousoulas, K.G. Glycoprotein K specified by herpes simplex virus type 1 is expressed on virions as a golgi complex-dependent glycosylated species and functions in virion entry. J. Virol. 2001, 75, 12431–12438. [Google Scholar] [CrossRef]
- Foster, T.P.; Chouljenko, V.N.; Kousoulas, K.G. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 2008, 82, 6310–6323. [Google Scholar] [CrossRef]
- Chouljenko, V.N.; Iyer, A.V.; Chowdhury, S.; Kim, J.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 2010, 84, 8596–8606. [Google Scholar] [CrossRef][Green Version]
- Chouljenko, V.N.; Iyer, A.V.; Chowdhury, S.; Chouljenko, D.V.; Kousoulas, K.G. The amino terminus of herpes simplex virus type 1 glycoprotein K (gK) modulates gB-mediated virus-induced cell fusion and virion egress. J. Virol. 2009, 83, 12301–12313. [Google Scholar] [CrossRef][Green Version]
- Jambunathan, N.; Chowdhury, S.; Subramanian, R.; Chouljenko, V.N.; Walker, J.D.; Kousoulas, K.G. Site-specific proteolytic cleavage of the amino terminus of herpes simplex virus glycoprotein K on virion particles inhibits virus entry. J. Virol. 2011, 85, 12910–12918. [Google Scholar] [CrossRef]
- Lau, S.Y.K.; Crump, C.M. HSV-1 gm and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 2015, 7, 915–938. [Google Scholar] [CrossRef]
- Musarrat, F.; Jambunathan, N.; Rider, P.J.F.; Chouljenko, V.N.; Kousoulas, K.G. The amino terminus of herpes simplex virus 1 glycoprotein K (gK) is required for gb binding to akt, release of intracellular calcium, and fusion of the viral envelope with plasma membranes. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Saied, A.A.; Chouljenko, V.N.; Subramanian, R.; Kousoulas, K.G. A replication competent HSV-1(McKrae) with a mutation in the amino-terminus of Glycoprotein K (gK) is unable to infect mouse trigeminal ganglia after cornea infection. Curr. Eye Res. 2014, 39, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Matundan, H.H.; Mott, K.R.; Akhtar, A.A.; Breunig, J.J.; Ghiasi, H. Mutations within the pathogenic region of herpes simplex virus 1 gK signal sequences alter cell surface expression and neurovirulence. J. Virol. 2015, 89, 2530–2542. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takakuwa, H.; Goshima, F.; Koshizuka, T.; Murata, T.; Daikoku, T.; Nishiyama, Y. Herpes simplex virus encodes a virion-associated protein which promotes long cellular processes in over-expressing cells. Genes Cells 2001, 6, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Muto, Y.; Goshima, F.; Ushijima, Y.; Kimura, H.; Nishiyama, Y. Generation and characterization of UL21-null herpes simplex virus type 1. Front. Microbiol. 2012, 3, 1–6. [Google Scholar] [CrossRef]
- Le Sage, V.; Jung, M.; Alter, J.D.; Wills, E.G.; Johnston, S.M.; Kawaguchi, Y.; Baines, J.D.; Banfield, B.W. The herpes simplex virus 2 UL21 protein is essential for virus propagation. J. Virol. 2013, 87, 5904–5915. [Google Scholar] [CrossRef]
- Metrick, C.M.; Heldwein, E.E. Novel structure and unexpected RNA-binding ability of the C-terminal domain of herpes simplex virus 1 tegument protein UL21. J. Virol. 2016, 90, 7007–7008. [Google Scholar] [CrossRef]
- Sarfo, A.; Starkey, J.; Mellinger, E.; Zhang, D.; Chadha, P.; Carmichael, J.; Wills, J.W. The UL21 Tegument protein of herpes simplex virus 1 is differentially required for the syncytial phenotype. J. Virol. 2017, 91, 1–18. [Google Scholar] [CrossRef]
- Jacobson, J.G.; Chen, S.H.; Cook, W.J.; Kramer, M.F.; Coen, D.M. Importance of the herpes simplex virus UL24 gene for productive ganglionic infection in mice. Virology 1998, 242, 161–169. [Google Scholar] [CrossRef]
- Abdeljelil, N.B.; Rochette, P.A.; Pearson, A. The UL24 protein of herpes simplex virus 1 affects the sub-cellular distribution of viral glycoproteins involved in fusion. Virology 2013, 444, 263–273. [Google Scholar] [CrossRef]
- Pearson, A.; Coen, D.M. Identification, localization, and regulation of expression of the UL24 protein of herpes simplex virus type 1. J. Virol. 2002, 76, 10821–10828. [Google Scholar] [CrossRef]
- Dridi, S.; Richerioux, N.; Suarez, C.E.G.; Vanharen, M.; Sanabria-Solano, C.; Pearson, A. A Mutation in the UL24 gene abolishes expression of the newly identified UL24.5 protein of herpes simplex virus 1 and leads to an increase in pathogenicity in mice. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Rochette, P.A.; Bourget, A.; Sanabria-Solano, C.; Lahmidi, S.; Lavalleé, G.O.; Pearson, A. Mutation of UL24 impedes the dissemination of acute herpes simplex virus 1 infection from the cornea to neurons of trigeminal ganglia. J. Gen. Virol. 2015, 96, 2794–2805. [Google Scholar] [CrossRef]
- Blakeney, S.; Kowalski, J.; Tummolo, D.; DeStefano, J.; Cooper, D.; Guo, M.; Gangolli, S.; Long, D.; Zamb, T.; Natuk, R.J.; et al. Herpes simplex virus type 2 UL24 gene is a virulence determinant in murine and guinea pig disease models. J. Virol. 2005, 79, 10498–10506. [Google Scholar] [CrossRef] [PubMed]
- Kniżewski, Ł.; Kinch, L.; Grishin, N.V.; Rychlewski, L.; Ginalski, K. Human herpesvirus 1 UL24 gene encodes a potential PD-(D/E)XK endonuclease. J. Virol. 2006, 80, 2575–2577. [Google Scholar] [CrossRef] [PubMed]
- Lymberopoulos, M.H.; Pearson, A. Involvement of UL24 in herpes-simplex-virus-1-induced dispersal of nucleolin. Virology 2007, 363, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Leiva-Torres, G.A.; Hyjazie, H.; Pearson, A. Conserved residues in the UL24 protein of herpes simplex virus 1 are important for dispersal of the nucleolar protein nucleolin. J. Virol. 2010, 84, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Leiva-Torres, G.A.; Rochette, P.A.; Pearson, A. Differential importance of highly conserved residues in UL24 for herpes simplex virus 1 replication in vivo and reactivation. J. Gen. Virol. 2010, 91, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Callé, A.; Ugrinova, I.; Epstein, A.L.; Bouvet, P.; Diaz, J.-J.; Greco, A. Nucleolin is required for an efficient herpes simplex virus type 1 infection. J. Virol. 2008, 82, 4762–4773. [Google Scholar] [CrossRef]
- Box, J.K.; Paquet, N.; Adams, M.N.; Boucher, D.; Bolderson, E.; O’Byrne, K.J.; Richard, D.J. Nucleophosmin: From structure and function to disease development. BMC Mol. Biol. 2016, 17, 1–12. [Google Scholar] [CrossRef]
- Lymberopoulos, M.H.; Bourget, A.; Abdeljelil, N.B.; Pearson, A. Involvement of the UL24 protein in herpes simplex virus 1-induced dispersal of B23 and in nuclear egress. Virology 2011, 412, 341–348. [Google Scholar] [CrossRef]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J. Virol. 2017, 91, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shiba, C.; Daikoku, T.; Goshima, F.; Takakuwa, H.; Yamauchi, Y.; Koiwai, O.; Nishiyama, Y. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J. Gen. Virol. 2000, 81, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.-J.; Roizman, B. The essential protein encoded by the UL31 gene of herpes simplex virus 1 depends for its stability on the presence of UL34 protein. Proc. Natl. Acad. Sci. USA 2000, 97, 11002–11007. [Google Scholar] [CrossRef] [PubMed]
- Funk, C.; Ott, M.; Raschbichler, V.; Nagel, C.H.; Binz, A.; Sodeik, B.; Bauerfeind, R.; Bailer, S.M. The herpes simplex virus protein pUL31 escorts nucleocapsids to sites of nuclear egress, a process coordinated by Its N-terminal domain. PLoS Pathog. 2015, 11, e1004957. [Google Scholar] [CrossRef]
- Chang, Y.E.; van Sant, C.; Krug, P.W.; Sears, A.E.; Roizman, B. The null mutant of the U(L)31 gene of herpes simplex virus 1: Construction and phenotype in infected cells. J. Virol. 1997, 71, 8307–8315. [Google Scholar] [CrossRef]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 UL34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef]
- Monier, K.; Armas, J.C.G.; Etteldorf, S.; Ghazal, P.; Sullivan, K.F. Annexation of the interchromosomal space during viral infection. Nat. Cell Biol. 2000, 2, 661–665. [Google Scholar] [CrossRef]
- Simpson-Holley, M.; Baines, J.; Roller, R.; Knipe, D.M. Herpes simplex virus 1 UL31 and UL34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 2004, 78, 5591–5600. [Google Scholar] [CrossRef]
- Reynolds, A.E.; Liang, L.; Baines, J.D. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes UL31 and UL34. J. Virol. 2004, 78, 5564–5575. [Google Scholar] [CrossRef]
- Morrison, A.L.; DeLassus, G.S. Breach of the nuclear lamina during assembly of herpes simplex viruses. Nucleus 2011, 2, 271–276. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Fuchs, W.; Keil, G.M.; Finke, S.; Mettenleiter, T.C. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 7241–7246. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.J.; Pryce, E.N.; Henson, B.W.; Luitweiler, E.M.; Cothran, J. Reconstitution of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J. Virol. 2012, 86, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Luitweiler, E.M.; Henson, B.W.; Pryce, E.N.; Patel, V.; Coombs, G.; McCaffery, J.M.; Desai, P.J. Interactions of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J. Virol. 2013, 87, 3915–3929. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef] [PubMed]
- Zeev-Ben-Mordehai, T.; Weberruß, M.; Lorenz, M.; Cheleski, J.; Hellberg, T.; Whittle, C.; el Omari, K.; Vasishtan, D.; Dent, K.C.; Harlos, K.; et al. Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep. 2015, 13, 2645–2652. [Google Scholar] [CrossRef]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef]
- Tandon, R.; Mocarski, E.S.; Conway, J.F. The A, B, Cs of herpesvirus capsids. Viruses 2015, 7, 899–914. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Homa, F.L.; Brown, J.C. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 2006, 80, 6286–6294. [Google Scholar] [CrossRef]
- Yang, K.; Baines, J.D. Selection of HSV capsids for envelopment involves interaction between capsid surface components pU L31, pU L17, and pU L25. Proc. Natl. Acad. Sci. USA 2011, 108, 14276–14281. [Google Scholar] [CrossRef]
- Chen, D.-H.; Jakana, J.; McNab, D.; Mitchell, J.; Zhou, Z.H.; Dougherty, M.; Chiu, W.; Rixon, F.J. The Pattern of tegument-capsid interaction in the herpes simplex virus type 1 virion is not influenced by the small hexon-associated protein VP26. J. Virol. 2001, 75, 11863–11867. [Google Scholar] [CrossRef][Green Version]
- Zhou, Z.H.; He, J.; Jakana, J.; Tatman, J.D.; Rixon, F.J.; Chiu, W. Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nature 1995, 2, 1026–1030. [Google Scholar] [CrossRef] [PubMed]
- Bowman, B.R.; Baker, M.L.; Rixon, F.J.; Chiu, W.; Quiocho, F.A. Structure of the herpesvirus major capsid protein. EMBO J. 2003, 22, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Vittone, V.; Diefenbach, E.; Cunningham, A.L.; Diefenbach, R.J. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 2008, 378, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Rixon, F.J.; Addison, C.; McGregor, A.; Macnab, S.J.; Nicholson, P.; Preston, V.G.; Tatman, J.D. Multiple interactions control the intracellular localization of the herpes simplex virus type 1 capsid proteins. J. Gen. Virol. 1996, 77, 2251–2260. [Google Scholar] [CrossRef]
- Thomsen, D.R.; Roof, L.L.; Homa, F.L. Assembly of herpes simplex virus (HSV) intermediate capsids in insect cells infected with recombinant baculoviruses expressing HSV capsid proteins. J. Virol. 1994, 68, 2442–2457. [Google Scholar] [CrossRef]
- Desai, P.; DeLuca, N.A.; Person, S. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 1998, 247, 115–124. [Google Scholar] [CrossRef]
- Kobayashi, R.; Kato, A.; Oda, S.; Koyanagi, N.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kawaguchi, Y. Function of the herpes simplex virus 1 small capsid protein VP26 is regulated by phosphorylation at a specific site. J. Virol. 2015, 89, 6141–6147. [Google Scholar] [CrossRef]
- Kobayashi, R.; Kato, A.; Sagara, H.; Watanabe, M.; Maruzuru, Y.; Koyanagi, N.; Arii, J.; Kawaguchi, Y. Herpes simplex virus 1 small capsomere-interacting protein VP26 regulates nucleocapsid maturation. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Douglas, M.W.; Diefenbach, R.J.; Homa, F.L.; Miranda-Saksena, M.; Rixon, F.J.; Vittone, V.; Byth, K.; Cunningham, A.L. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 2004, 279, 28522–28530. [Google Scholar] [CrossRef]
- Apcarian, A.; Cunningham, A.L.; Diefenbach, R.J. Identification of binding domains in the herpes simplex virus type 1 small capsid protein pUL35 (VP26). J. Gen. Virol. 2010, 91, 2659–2663. [Google Scholar] [CrossRef]
- Carter, K.L.; Ward, P.L.; Roizman, B. Characterization of the products of the U(L)43 gene of herpes simplex virus 1: Potential implications for regulation of gene expression by antisense transcription. J. Virol. 1996, 70, 7663–7668. [Google Scholar] [CrossRef] [PubMed]
- Loret, S.; Guay, G.; Lippé, R. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 2008, 82, 8605–8618. [Google Scholar] [CrossRef] [PubMed]
- Herold, B.C.; WuDunn, D.; Soltys, N.; Spear, P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [CrossRef]
- Trybala, E.; Bergstrom, T.; Svennerholm, B.; Jeansson, S.; Glorioso, J.C.; Olofsson, S. Localization of a functional site on herpes simplex virus type 1 glycoprotein C involved in binding to cell surface heparan sulphate. J. Gen. Virol. 1994, 75, 743–752. [Google Scholar] [CrossRef]
- Komala Sari, T.; Gianopulos, K.A.; Weed, D.J.; Schneider, S.M.; Pritchard, S.M.; Nicola, A.V. Herpes simplex virus glycoprotein c regulates low-pH Entry. mSphere 2020, 5, 1–16. [Google Scholar] [CrossRef]
- Visalli, R.J.; Brandt, C.R. The HSV-1 UL45 18 kDa gene product is a true late protein and a component of the virion. Virus Res. 1993, 29, 167–178. [Google Scholar] [CrossRef]
- Visalli, R.J.; Brandt, C.R. The HSV-1 UL45 gene product is not required for growth in vero cells. Virology 1991, 185, 419–423. [Google Scholar] [CrossRef]
- Visalli, R.J.; Brandt, C.R. Mutation of the herpes simplex virus 1 KOS UL45 gene reveals dose dependent effects on central nervous system growth. Arch. Virol. 2002, 147, 519–532. [Google Scholar] [CrossRef]
- Dollery, S.J.; Lane, K.D.; Delboy, M.G.; Roller, D.G.; Nicola, A.V. Role of the UL45 protein in herpes simplex virus entry via low pH-dependent endocytosis and its relationship to the conformation and function of glycoprotein B. Virus Res. 2010, 149, 115–118. [Google Scholar] [CrossRef]
- Haanes, E.J.; Nelson, C.M.; Soule, C.L.; Goodman, J.L. The UL45 gene product is required for herpes simplex virus type 1 glycoprotein B-induced fusion. J. Virol. 1994, 68, 5825–5834. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.V.; Hou, J.; Major, E.O.; Straus, S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, C.; Moyal, M.; Rossen-Wolff, A.; Darai, G.; Becker, Y. Herpes simplex virus type 1 (HSV-1) UL56 gene is involved in viral intraperitoneal pathogenicity to immunocompetent mice. Arch. Virol. 1994, 134, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Kolb, A.W.; Lee, K.; Larsen, I.; Craven, M.; Brandt, C.R. Quantitative trait locus based virulence determinant mapping of the HSV-1 genome in murine ocular infection: Genes involved in viral regulatory and innate immune networks contribute to virulence. PLoS Pathog. 2016, 12, e1005499. [Google Scholar] [CrossRef] [PubMed]
- Nash, T.C.; Spivack, J.G. The UL55 and UL56 genes of herpes simplex virus type 1 are not required for viral replication, intraperitoneal virulence, or establishment of latency in mice. Virology 1994, 204, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Umene, K.; Fukumaki, Y. DNA genome of spontaneously occurring deletion mutants of herpes simplex virus type 1 lacking one copy of the inverted repeat sequences of the L component. Arch. Virol. 2011, 156, 1305–1315. [Google Scholar] [CrossRef] [PubMed]
- Kehm, R.; Gelderblom, H.R.; Darai, G. Identification of the UL56 protein of herpes simplex virus type 1 within the virion by immune electron microscopy. Virus Genes 1998, 17, 49–53. [Google Scholar] [CrossRef]
- Koshizuka, T.; Kawaguchi, Y.; Goshima, F.; Mori, I.; Nishiyama, Y. Association of two membrane proteins encoded by herpes simplex virus type 2, UL11 and UL56. Virus Genes 2006, 32, 153–163. [Google Scholar] [CrossRef]
- Ushijima, Y.; Koshizuka, T.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes simplex virus type 2 UL56 interacts with the ubiquitin ligase Nedd4 and Increases its ubiquitination. J. Virol. 2008, 82, 5220–5233. [Google Scholar] [CrossRef]
- Ushijima, Y.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes simplex virus type 2 tegument protein UL56 relocalizes ubiquitin ligase Nedd4 and has a role in transport and/or release of virions. Virol. J. 2009, 6, 1–13. [Google Scholar] [CrossRef]
- Ushijima, Y.; Luo, C.; Kamakura, M.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes simplex virus UL56 interacts with and regulates the Nedd4-family ubiquitin ligase Itch. Virol. J. 2010, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J. On the predictive recognition of signal peptide sequences. Virus Res. 1985, 3, 271–286. [Google Scholar] [CrossRef]
- Weber, P.C.; Levine, M.; Glorioso, J.C. Rapid identification of nonessential genes of herpes simplex virus type 1 by Tn5 mutagenesis. Science 1987, 236, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Glück, B.; Möbius, S.; Pfaff, F.; Zell, R.; Sauerbrei, A. Novel method for genotyping clinical herpes simplex virus type 1 isolates. Arch. Virol. 2015, 160, 2807–2811. [Google Scholar] [CrossRef]
- Jiang, Y.-M.; Yamada, H.; Goshima, F.; Daikoku, T.; Oshima, S.; Wada, K.; Nishiyama, Y. Characterization of the herpes simplex virus type 2 (HSV-2) US2 gene product and a US2-deficient HSV-2 mutant. J. Gen. Virol. 1998, 79, 1989–1995. [Google Scholar] [CrossRef]
- Goshima, F.; Watanabe, D.; Suzuki, H.; Takakuwa, H.; Yamada, H.; Nishiyama, Y. The US2 gene product of herpes simplex virus type 2 interacts with cytokeratin 18. Arch. Virol. 2001, 146, 2201–2209. [Google Scholar] [CrossRef]
- Norrild, B.; Lehto, V.P.; Virtanen, I. Organization of cytoskeleton elements during herpes simplex virus type I infection of human fibroblasts: An immunofluorescence study. J. Gen. Virol. 1986, 67, 97–105. [Google Scholar] [CrossRef]
- Wu, Y.; Wei, F.; Tang, L.; Liao, Q.; Wang, H.; Shi, L.; Gong, Z.; Zhang, W.; Zhou, M.; Xiang, B.; et al. Herpesvirus acts with the cytoskeleton and promotes cancer progression. J. Cancer 2019, 10, 2185–2193. [Google Scholar] [CrossRef]
- Mingo, R.M.; Han, J.; Newcomb, W.W.; Brown, J.C. Replication of herpes simplex virus: Egress of progeny virus at specialized cell membrane sites. J. Virol. 2012, 86, 7084–7097. [Google Scholar] [CrossRef]
- Kang, M.-H.; Roy, B.B.; Finnen, R.L.; le Sage, V.; Johnston, S.M.; Zhang, H.; Banfield, B.W. The Us2 gene product of herpes simplex virus 2 is a membrane-associated ubiquitin-interacting protein. J. Virol. 2013, 87, 9590–9603. [Google Scholar] [CrossRef]
- Lu, X.; Huang, C.; Zhang, Y.; Lin, Y.; Wang, X.; Li, Q.; Liu, S.; Tang, J.; Zhou, L. The Us2 gene product of herpes simplex virus 2 modulates NF-κB activation by targeting TAK1. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Balan, P.; Davis-Poynter, N.; Bell, S.; Atkinson, H.; Browne, H.; Minson, T. An analysis of the in vitro and in vivo phenotypes of mutants of herpes simplex virus type 1 lacking glycoproteins gG, gE, gI or the putative gJ. J. Gen. Virol. 1994, 75, 1245–1258. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.C.; Kissner, J.M.; Westerman, L.E.; Sears, A.E. A herpes simplex virus 1 recombinant lacking the glycoprotein G coding sequences is defective in entry through apical surfaces of polarized epithelial cells in culture and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. Baculovirus-expressed glycoprotein g of herpes simplex virus type 1 partially protects vaccinated mice against lethal HSV-1 challenge. Virology 1992, 190, 233–239. [Google Scholar] [CrossRef]
- Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Slanina, S.; Wechsler, S.L. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): Comparative protection against lethal challenge in mice. J. Virol. 1994, 68, 2118–2126. [Google Scholar] [CrossRef]
- Blacklaws, B.A.; Krishna, S.; Minson, A.C.; Nash, A.A. Immunogenicity of herpes simplex virus type 1 glycoproteins expressed in vaccinia virus recombinants. Virology 1990, 177, 727–736. [Google Scholar] [CrossRef]
- Ghiasi, H.; Hofman, F.M.; Cai, S.; Perng, G.C.; Nesburn, A.B.; Wechsler, S.L. Vaccination with different HSV-1 glycoproteins induces different patterns of ocular cytokine responses following HSV-1 challenge of vaccinated mice. Vaccine 1999, 17, 2576–2582. [Google Scholar] [CrossRef]
- Baggiolini, M. Chemokines and leukocyte traffic. Nat. Immunol. 1998, 392, 949–952. [Google Scholar] [CrossRef]
- Thapa, M.; Carr, D.J.J. CXCR3 deficiency increases susceptibility to genital herpes simplex virus type 2 infection: Uncoupling of CD8+ T-cell effector function but not migration. J. Virol. 2009, 83, 9486–9501. [Google Scholar] [CrossRef]
- Richman, D.D.; Buckmaster, A.; Bell, S.; Hodgman, C.; Minson, A.C. Identification of a new glycoprotein of herpes simplex virus type 1 and genetic mapping of the gene that codes for it. J. Virol. 1986, 57, 647–655. [Google Scholar] [CrossRef]
- Viejo-Borbolla, A.; Martinez-Martín, N.; Nel, H.J.; Rueda, P.; Martín, R.; Blanco, S.; Arenzana-Seisdedos, F.; Thelen, M.; Fallon, P.G.; Alcamí, A. Enhancement of chemokine function as an immunomodulatory strategy employed by human herpesviruses. PLoS Pathog. 2012, 8, e1002497. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Martin, N.; Viejo-Borbolla, A.; Martín, R.; Blanco, S.; Benovic, J.L.; Thelen, M.; Alcamí, A. Herpes simplex virus enhances chemokine function through modulation of receptor trafficking and oligomerization. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- McLean, T.I.; Bachenheimer, S.L. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 1999, 73, 8415–8426. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Martín, N.; Viejo-Borbolla, A.; Alcami, A. Herpes simplex virus particles interact with chemokines and enhance cell migration. J. Gen. Virol. 2016, 97, 3007–3016. [Google Scholar] [CrossRef]
- Aubert, M.; Chen, Z.; Lang, R.; Dang, C.H.; Fowler, C.; Sloan, D.D.; Jerome, K.R. The antiapoptotic herpes simplex virus glycoprotein J localizes to multiple cellular organelles and induces reactive oxygen species formation. J. Virol. 2008, 82, 617–629. [Google Scholar] [CrossRef]
- Jerome, K.R.; Chen, Z.; Lang, R.; Torres, M.R.; Hofmeister, J.; Smith, S.; Fox, R.; Froelich, C.J.; Corey, L. HSV and glycoprotein J inhibit caspase activation and apoptosis induced by granzyme B or fas. J. Immunol. 2001, 167, 3928–3935. [Google Scholar] [CrossRef]
- Johnson, D.C.; Frame, M.C.; Ligas, M.W.; Cross, A.M.; Stow, N.D. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 1988, 62, 1347–1354. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Johnson, D.C. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 1998, 72, 8933–8942. [Google Scholar] [CrossRef]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef]
- Frank, I.; Friedman, H.M. A novel function of the herpes simplex virus type 1 Fc receptor: Participation in bipolar bridging of antiviral immunoglobulin G. J. Virol. 1989, 63, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Dubin, G.; Socolof, E.; Frank, I.; Friedman, H.M. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J. Virol. 1991, 65, 7046–7050. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, K.E.; de Graaf-Miltenburg, L.A.; Verhoef, J.; Van Strijp, J.A. Direct evidence for antibody bipolar bridging on herpes simplex virus-infected cells. Immunology 1992, 77, 109–115. [Google Scholar] [PubMed]
- Neidhardt, H.; Schröder, C.H.; Kaerner, H.C. Herpes simplex virus type 1 glycoprotein E is not indispensable for viral infectivity. J. Virol. 1987, 61, 600–603. [Google Scholar] [CrossRef]
- Saldanha, C.E.; Lubinski, J.; Martin, C.; Nagashunmugam, T.; Wang, L.; van der Keyl, H.; Tal-Singer, R.; Friedman, H.M. Herpes simplex virus type 1 glycoprotein E domains involved in virus spread and disease. J. Virol. 2000, 74, 6712–6719. [Google Scholar] [CrossRef]
- Polcicova, K.; Biswas, P.S.; Banerjee, K.; Wisner, T.W.; Rouse, B.T.; Johnson, D.C. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA 2005, 102, 11462–11467. [Google Scholar] [CrossRef]
- Wang, F.; Tang, W.; McGraw, H.M.; Bennett, J.; Enquist, L.W.; Friedman, H.M. Herpes simplex virus type 1 glycoprotein E is required for axonal localization of capsid, tegument, and membrane glycoproteins. J. Virol. 2005, 79, 13362–13372. [Google Scholar] [CrossRef]
- Snyder, A.; Polcicova, K.; Johnson, D.C. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 2008, 82, 10613–10624. [Google Scholar] [CrossRef]
- Howard, P.W.; Wright, C.C.; Howard, T.; Johnson, D.C. Herpes simplex virus gE/gI extracellular domains promote axonal transport and spread from neurons to epithelial cells. J. Virol. 2014, 88, 11178–11186. [Google Scholar] [CrossRef][Green Version]
- LaVail, J.H.; Tauscher, A.N.; Sucher, A.; Harrabi, O.; Brandimarti, R. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 2007, 146, 974–985. [Google Scholar] [CrossRef]
- Frame, M.C.; McGeoch, D.J.; Rixon, F.J.; Orr, A.C.; Marsden, H.S. The 10K virion phosphoprotein encoded by gene US9 from herpes simplex virus type 1. Virology 1986, 150, 321–332. [Google Scholar] [CrossRef]
- Brideau, A.D.; Banfield, B.W.; Enquist, L.W. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J. Virol. 1998, 72, 4560–4570. [Google Scholar] [CrossRef] [PubMed]
- Brideau, A.D.; Eldridge, M.G.; Enquist, L.W. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J. Virol. 2000, 74, 4549–4561. [Google Scholar] [CrossRef] [PubMed][Green Version]
- DuRaine, G.; Wisner, T.W.; Howard, P.; Williams, M.; Johnson, D.C. Erratum for DuRaine et al., “Herpes simplex virus gE/gI and US9 promote both envelopment and sorting of virus particles in the cytoplasm of neurons, two processes that precede anterograde transport in axons”. J. Virol. 2017, 91, 1–20. [Google Scholar] [CrossRef]
- Zhang, W.; Gao, P.; Gui, X.; Zhou, L.; Ge, X.; Guo, X.; Wills, J.W.; Han, J.; Yang, H. Induction of rod-shaped structures by herpes simplex virus glycoprotein I. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Georgopoulou, U.; Michaelidou, A.; Roizman, B.; Mavromara-Nazos, P. Identification of a new transcriptional unit that yields a gene product within the unique sequences of the short component of the herpes simplex virus 1 genome. J. Virol. 1993, 67, 3961–3968. [Google Scholar] [CrossRef]
- Willemse, M.J.; Strijdveen, I.G.L.; van Schooneveld, S.H.B.; van den Berg, M.C.; Sondermeijer, P.J.A. Transcriptional analysis of the short segment of the feline herpesvirus type 1 genome and insertional mutagenesis of a unique reading frame. Virology 1995, 208, 704–711. [Google Scholar] [CrossRef][Green Version]
- Yamada, H.; Daikoku, T.; Yamashita, Y.; Jiang, Y.M.; Tsurumi, T.; Nishiyama, Y. The product of the US10 gene of herpes simplex virus type 1 is a capsid/tegument-associated phosphoprotein which copurifies with the nuclear matrix. J. Gen. Virol. 1997, 78, 2923–2931. [Google Scholar] [CrossRef]
- Johnson, P.A.; MacLean, C.; Marsden, H.S.; Dalziel, R.G.; Everett, R.D. The product of gene US11 of herpes simplex type 1 is expressed as a true late gene. J. Gen. Virol. 1986, 67, 871–883. [Google Scholar] [CrossRef]
- Roller, R.J.; Roizman, B. The herpes simplex virus Us11 open reading frame encodes a sequence-specific RNA-binding protein. J. Virol. 1990, 64, 3463–3470. [Google Scholar] [CrossRef]
- Roller, R.J.; Roizman, B. Herpes simplex virus 1 RNA-binding protein US11 negatively regulates the accumulation of a truncated viral mRNA. J. Virol. 1991, 65, 5873–5879. [Google Scholar] [CrossRef] [PubMed]
- Roller, R.J.; Roizman, B. A herpes simplex virus 1 US11-expressing cell line is resistant to herpes simplex virus infection at a step in viral entry mediated by glycoprotein D. J. Virol. 1994, 68, 2830–2839. [Google Scholar] [CrossRef] [PubMed]
- Simonin, D.; Diaz, J.-J.; Kindbeiter, K.; Pernas, P.; Madjar, J.-J. Phosphorylation of herpes simplex virus type 1 Us11 protein is independent of viral genome expression. Electrophoresis 1995, 16, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Cassady, K.A.; Gross, M. The herpes simplex virus type 1 US11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J. Virol. 2002, 76, 2029–2035. [Google Scholar] [CrossRef] [PubMed]
- Greco, A.; Arata, L.; Soler, E.; Gaume, X.; Coute, Y.; Hacot, S.; Calle, A.; Monier, K.; Epstein, A.L.; Sanchez, J.-C.; et al. Nucleolin interacts with US11 protein of herpes simplex virus 1 and is involved in its trafficking. J. Virol. 2012, 86, 1449–1457. [Google Scholar] [CrossRef]
- Diaz, J.-J.; Dodon, M.D.; Schaerer-Uthurralt, N.; Simonin, D.; Kindbeiter, K.; Gazzolo, L.; Madjar, J.-J. Post-transcriptional transctivation of human retroviral envelope glycoprotein expression by herpes simplex virus Us11 protein. Nature 1996, 379, 273–277. [Google Scholar] [CrossRef]
- Schaerer-Uthurralt, N.; Erard, M.; Kindbeiter, K.; Madjar, J.J.; Diaz, J.J. Distinct domains in herpes simplex virus type 1 US11 protein mediate post-transcriptional transactivation of human T-lymphotropic virus type I envelope glycoprotein gene expression and specific binding to the Rex responsive element. J. Gen. Virol. 1998, 79, 1593–1602. [Google Scholar] [CrossRef]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef]
- Lussignol, M.; Queval, C.; Bernet-Camard, M.-F.; Cotte-Laffitte, J.; Beau, I.; Codogno, P.; Esclatine, A. The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR. J. Virol. 2013, 87, 859–871. [Google Scholar] [CrossRef]
- Sadler, A.J.; Latchoumanin, O.; Hawkes, D.; Mak, J.; Williams, B.R.G. An antiviral response directed by PKR phosphorylation of the RNA helicase A. PLoS Pathog. 2009, 5, e1000311. [Google Scholar] [CrossRef]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef] [PubMed]
- Kew, C.; Lui, P.-Y.; Chan, C.-P.; Liu, X.; Au, S.W.N.; Mohr, I.; Jin, D.-Y.; Kok, K.-H. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J. Virol. 2013, 87, 13141–13149. [Google Scholar] [CrossRef] [PubMed]
- Sànchez, R.; Mohr, I. Inhibition of cellular 2′-5′ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 2007, 81, 3455–3464. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Main, D.; Ma, Y.; He, B. Herpes simplex virus 1 inhibits TANK-binding kinase 1 through formation of the Us11-Hsp90 complex. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Liyana, A.; Vanessa, S.S. The emerging role of human TBK1 in virus-induced autophagy. Autophagy 2019, 15, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Miranda-Saksena, M.; Diefenbach, E.; Holland, D.J.; Boadle, R.A.; Armati, P.J.; Cunningham, A.L. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 2002, 76, 3282–3291. [Google Scholar] [CrossRef]
- Roller, R.J.; Monk, L.L.; Stuart, D.; Roizman, B. Structure and function in the herpes simplex virus 1 RNA-binding protein U(s)11: Mapping of the domain required for ribosomal and nucleolar association and RNA binding in vitro. J. Virol. 1996, 70, 2842–2851. [Google Scholar] [CrossRef]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus Type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef]
- Charron, A.J.; Ward, S.L.; North, B.J.; Ceron, S.; Leib, D.A. The US11 gene of herpes simplex virus 1 promotes neuroinvasion and periocular replication following corneal infection. J. Virol. 2019, 93, 1–19. [Google Scholar] [CrossRef]
- Melancon, J.M.; Luna, R.E.; Foster, T.P.; Kousoulas, K.G. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 2005, 79, 299–313. [Google Scholar] [CrossRef]
- Croft, C.L.; Noble, W. Preparation of organotypic brain slice cultures for the study of Alzheimer’s disease [version 1; peer review: 3 approved]. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Visalli, R.J.; Courtney, R.J.; Meyers, C. Infection and replication of herpes simplex virus type 1 in an organotypic epithelial culture system. Virology 1997, 230, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Conlon, J.; Burdette, D.L.; Sharma, S.; Bhat, N.; Thompson, M.; Jiang, Z.; Rathinam, V.A.K.; Monks, B.; Jin, T.; Xiao, T.S.; et al. Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-Dimethylxanthenone-4-Acetic acid. J. Immunol. 2013, 190, 5216–5225. [Google Scholar] [CrossRef] [PubMed]
- Cavlar, T.; Deimling, T.; Ablasser, A.; Hopfner, K.P.; Hornung, V. Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J. 2013, 32, 1440–1450. [Google Scholar] [CrossRef] [PubMed]
- Sidhaye, J.; Knoblich, J.A. Brain organoids: An ensemble of bioassays to investigate human neurodevelopment and disease. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-organized cerebral organoids with human-specific features predict effective drugs to combat zika virus infection. Cell Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef]
- Qiao, H.; Guo, M.; Shang, J.; Zhao, W.; Wang, Z.; Liu, N.; Li, B.; Zhou, Y.; Wu, Y.; Chen, P. Herpes simplex virus type 1 infection leads to neurodevelopmental disorder-associated neuropathological changes. PLoS Pathog. 2020, 16, e1008899. [Google Scholar] [CrossRef]
- D’Aiuto, L.; Bloom, D.C.; Naciri, J.N.; Smith, A.; Edwards, T.G.; Mcclain, L.; Callio, J.A.; Jessup, M.; Wood, J.; Chowdari, K.; et al. Modeling herpes simplex virus 1 infections in human central nervous system neuronal cells using two- and three dimensional cultures derived from induced pluripotent stem cells. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Edwards, T.G.; Bloom, D.C. Lund human mesencephalic (LUHMES) neuronal cell line supports herpes simplex virus 1 latency in vitro. J. Virol. 2019, 93, 1–14. [Google Scholar] [CrossRef]
- Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. Cloaked viruses and viral factors in cutting edge exosome-based therapies. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Conry, R.M.; Westbrook, B.; McKee, S.; Norwood, T.G. Talimogene laherparepvec: First in class oncolytic virotherapy. Hum. Vaccines Immunother. 2018, 14, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Ghonime, M.G.; Jackson, J.; Shah, A.; Roth, J.; Li, M.; Saunders, U.; Coleman, J.; Gillespie, G.Y.; Markert, J.M.; Cassady, K.A. Chimeric HCMV/HSV-1 and Δγ134.5 oncolytic herpes simplex virus elicit immune mediated antigliomal effect and antitumor memory. Transl. Oncol. 2018, 11, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical toxicology of rQNestin34.5v.2: An oncolytic herpes virus with transcriptional regulation of the ICP34.5 neurovirulence gene. Mol. Ther. Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef] [PubMed]
- Sokolowski, N.A.; Rizos, H.; Diefenbach, R.J. Oncolytic virotherapy dovepress oncolytic virotherapy using herpes simplex virus: How far have we come? Oncolytic Virotherapy 2015, 4, 207–219. [Google Scholar]
- Miao, L.; Fraefel, C.; Sia, K.C.; Newman, J.P.; Mohamed-Bashir, S.A.; Ng, W.H.; Lam, P.Y.P. The potential application of a transcriptionally regulated oncolytic herpes simplex virus for human cancer therapy. Br. J. Cancer 2014, 110, 94–106. [Google Scholar] [CrossRef][Green Version]
- Pyles, R.B.; Warnick, R.E.; Chalk, C.L.; Szanti, B.E.; Parysek, L.M. A novel multiply-mutated HSV-1 strain for the treatment of human brain tumors. Hum. Gene Ther. 1997, 8, 533–544. [Google Scholar] [CrossRef]
- Stanfield, B.A.; Stahl, J.; Chouljenko, V.N.; Subramanian, R.; Charles, A.S.; Saied, A.A.; Walker, J.D.; Kousoulas, K.G. A single intramuscular vaccination of mice with the HSV-1 VC2 virus with mutations in the glycoprotein K and the membrane protein UL20 confers full protection against lethal intravaginal challenge with virulent HSV-1 and HSV-2 strains. PLoS ONE 2014, 9, e109890. [Google Scholar] [CrossRef]
- Stanfield, B.A.; Pahar, B.; Chouljenko, V.N.; Veazey, R.; Kousoulas, K.G. Vaccination of rhesus macaques with the live-attenuated HSV-1 vaccine VC2 stimulates the proliferation of mucosal T cells and germinal center responses resulting in sustained production of highly neutralizing antibodies. Vaccine 2017, 35, 536–543. [Google Scholar] [CrossRef]





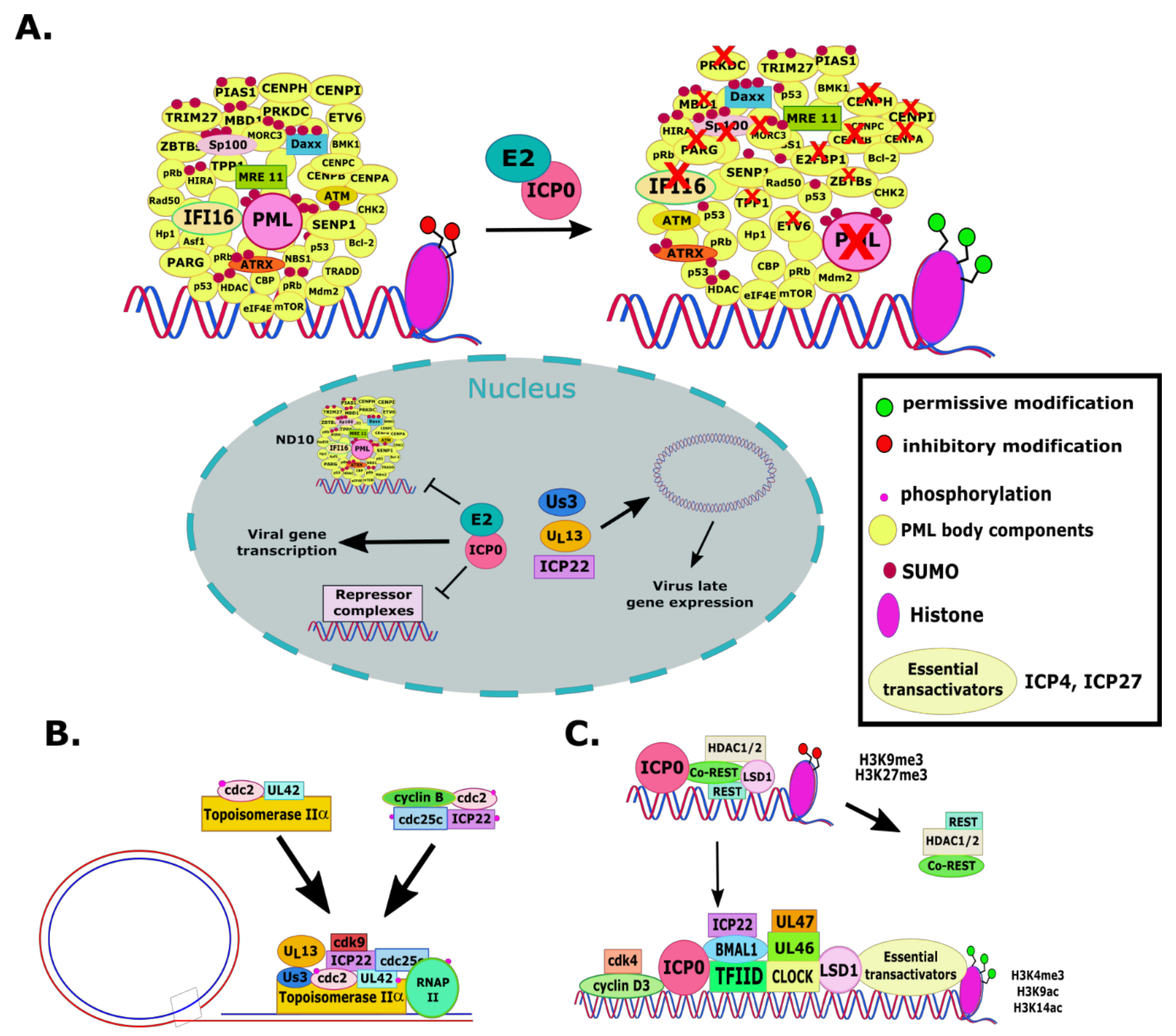{kind=link}
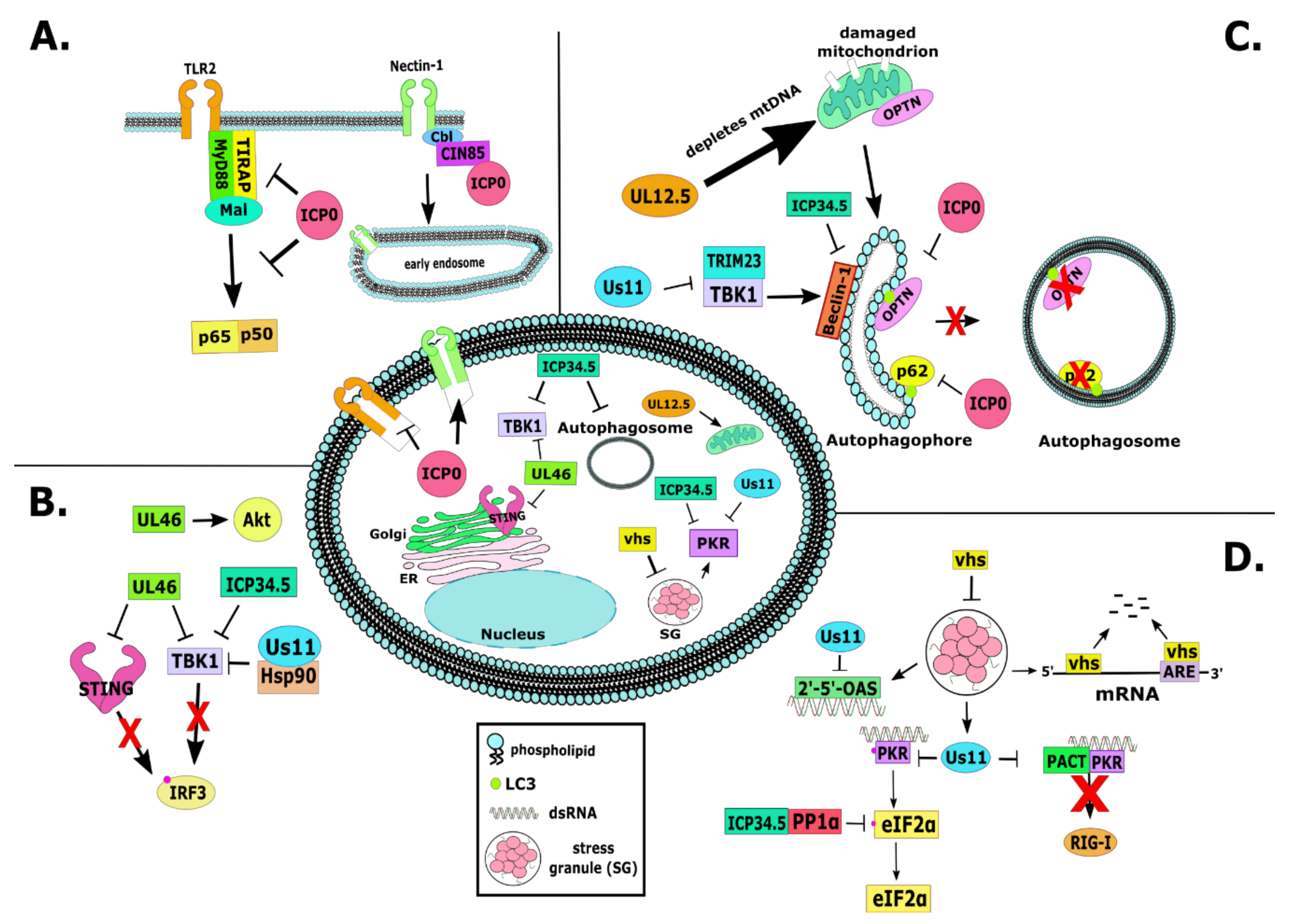{kind=link}
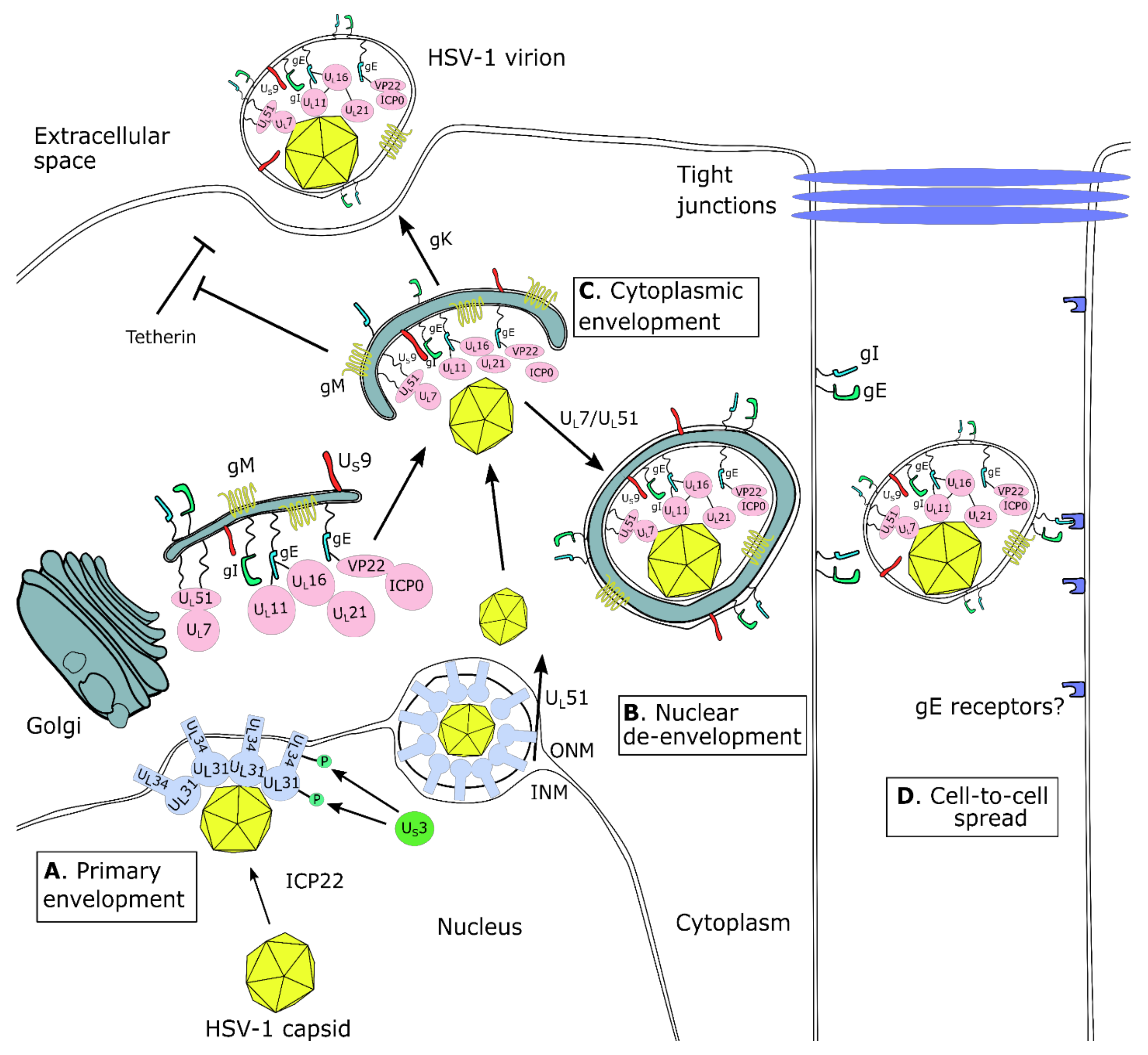{kind=link}
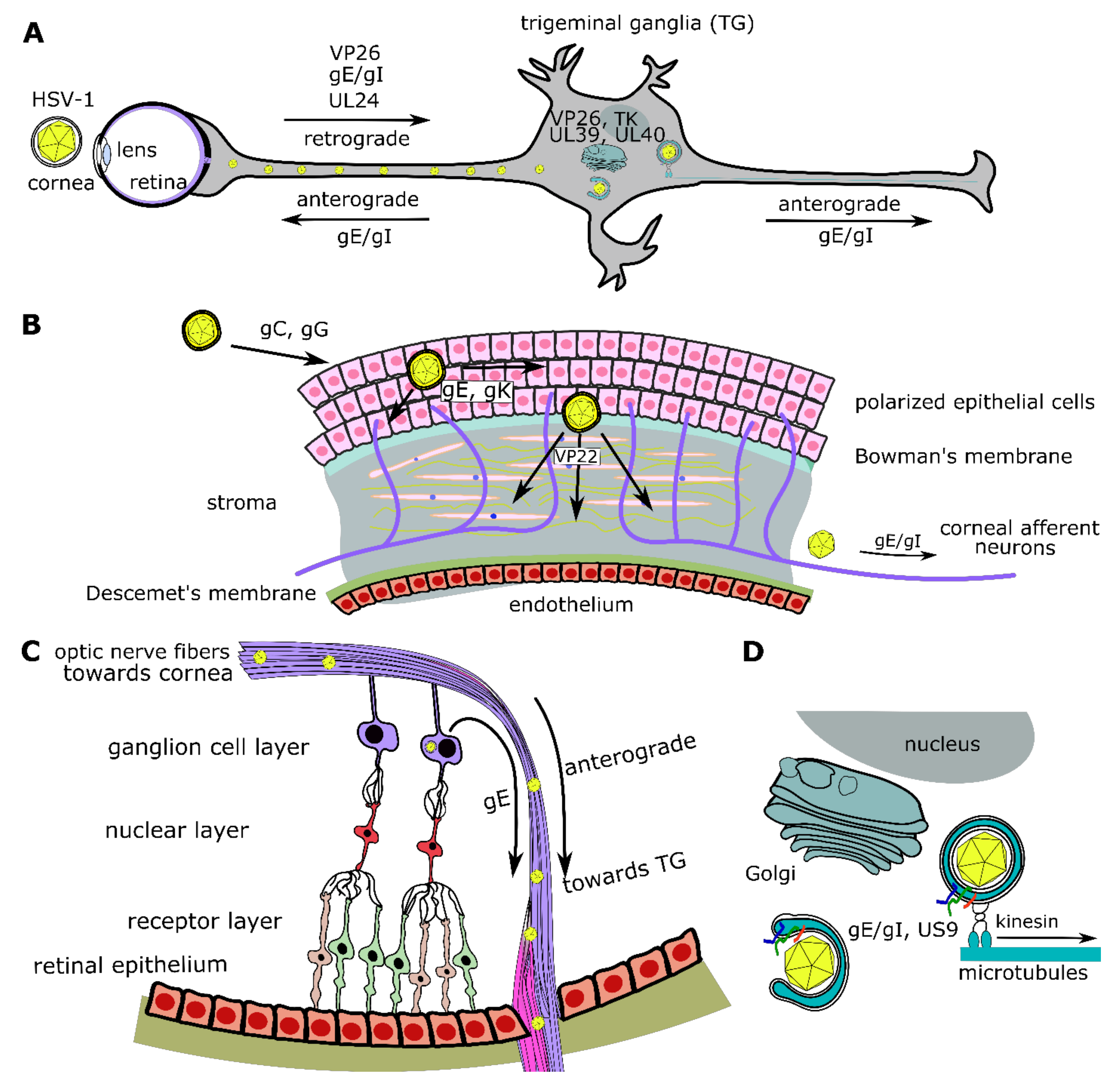{kind=link}
| Gene | Protein | Location on Virion | Function |
|---|---|---|---|
| RL1 or γ134.5 | ICP34.5 | tegument | Prevents host translational shutoff and autophagy |
| RL2 or α0 | ICP0 | tegument | Promiscuous transactivator of genes, disrupts repressor complexes, E3 ubiquitin ligase, inhibits innate immunity, modulates endocytosis, etc. |
| UL2 | uracil-DNA glycosylase | accessory | nucleic acid metabolism |
| UL3 | accessory | ||
| UL4 | accessory | ||
| UL7 | tegument | Virion assembly and egress | |
| UL10 | gM | envelope | Host and viral protein trafficking |
| UL11 | tegument | Cytoplasmic envelopment | |
| UL12 | accessory | Nucleic acid metabolism | |
| UL12.5 | accessory | Involved in depleting mtDNA | |
| UL13 | Ser/thr protein kinase | tegument | Blocking innate immune responses, supporting viral protein synthesis |
| UL16 | tegument | Cytoplasmic envelopment | |
| UL20 | envelope | Glycoprotein trafficking | |
| UL21 | tegument | Promotes capsid egress to the cytoplasm | |
| UL23 | thymidine kinase (TK) | tegument | Broad spectrum nucleoside kinase |
| UL24 | accessory | Glycoprotein trafficking, nucleolus dispersal | |
| UL31 | accessory | Component of the nuclear egress complex (NEC), promotes primary nuclear envelopment | |
| UL34 | accessory | Component of the nuclear egress complex (NEC), promotes primary nuclear envelopment | |
| UL35 | VP26 | capsid | Affects DNA packaging, mediates capsid assembly, trafficking post viral entry |
| UL39 | RR1 (ribonucleotide reductase) | accessory | Part of the ribonucleotide reductase (RR) complex, converts ribonucleotide diphosphates to corresponding deoxyribonucleotides, allowing for virus replication particularly in non-dividing cells |
| UL40 | RR2 (ribonucleotide reductase) | accessory | Part of the ribonucleotide reductase (RR) complex, converts ribonucleotide diphosphates to corresponding deoxyribonucleotides, allowing for virus replication particularly in non-dividing cells |
| UL41 | VHS | tegument | Viral RNase, degrades host transcripts and blocks antiviral responses |
| L43 | tegument | ||
| UL44 | gC | envelope | Mediates viral binding to heparan sulfate, regulates entry by a low-pH pathway |
| UL45 | envelope | Required for syncytia formation during HSV-1 gB syn infection | |
| UL46 | VP11/12 | tegument | Regulation of transcription, activates pathways for cell survival, blocks pathways for innate immunity activation |
| UL47 | VP13/14 | tegument | Regulation of transcription, modulating post-transcriptional processing of mRNAs |
| UL49 | VP22 | tegument | Facilitates viral gene expression, protein expression, and DNA replication; inhibits inflammasome |
| UL49.5 | gN | envelope | Binding partner of gM |
| UL50 | tegument | Nucleic acid metabolism | |
| UL51 | tegument | Participates in cytoplasmic envelopment; facilitates virus spread from cell-to-cell; recruits UL7 to tegument | |
| UL53 | gK | envelope | Participates in virion egress from host cell; regulates virus entry and fusogenic activity of the virion; complexes with UL20 |
| UL55 | tegument | Participates in cytoplasmic envelopment | |
| UL56 | tegument | Participates in cytoplasmic envelopment | |
| US1 | ICP22 | accessory | Regulates viral late gene expression; facilitates formation of complexes important for protein folding; participates in primary envelopment; blocks immune responses |
| US1.5 | accessory | Participates in viral gene transcription | |
| US2 | tegument | Protein trafficking | |
| US3 | Ser/thr protein kinase | tegument | Blocks apoptosis, enhances viral gene expression, facilitates capsid nuclear egress, phosphorylates numerous substrates |
| US3.5 | Ser/thr protein kinase | tegument | Phosphorylates substrates but cannot block apoptosis and does not facilitate nuclear egress |
| US4 | gG | envelope | Regulation of chemokines |
| US5 | gJ | envelope | Inhibits apoptosis and cell stress pathways |
| US7 | gI | envelope | Enhances virus spread from cell-to-cell; facilitates anterograde transport of virions after reactivation from latency; important for neurovirulence |
| US8 | gE | envelope | Enhances virus spread from cell-to-cell; facilitates anterograde transport of virions after reactivation from latency; important for neurovirulence |
| US8.5 | accessory | Localizes in the nucleoli | |
| US9 | tegument | Enhances virus spread from cell-to-cell; facilitates anterograde transport of virions after reactivation from latency; important for neurovirulence | |
| US10 | tegument | Important for neurovirulence | |
| US11 | tegument | Block PKR activation and shutoff of host translation; block IFN induction; regulation of virus genes expression; trafficking of unenveloped capsids | |
| US12 | ICP47 | accessory | Prevents MHC I antigen presentation, supports neurovirulence |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dogrammatzis, C.; Waisner, H.; Kalamvoki, M. “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses 2021, 13, 17. https://doi.org/10.3390/v13010017
Dogrammatzis C, Waisner H, Kalamvoki M. “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses. 2021; 13(1):17. https://doi.org/10.3390/v13010017
Chicago/Turabian StyleDogrammatzis, Christos, Hope Waisner, and Maria Kalamvoki. 2021. "“Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review" Viruses 13, no. 1: 17. https://doi.org/10.3390/v13010017
APA StyleDogrammatzis, C., Waisner, H., & Kalamvoki, M. (2021). “Non-Essential” Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review. Viruses, 13(1), 17. https://doi.org/10.3390/v13010017