
Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice


 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Quantification of the Virus Load
2.3. Measurement of Cytokines, Chemokines and Interferons
2.4. Immunohistochemistry
2.5. Statistical Analysis
3. Results
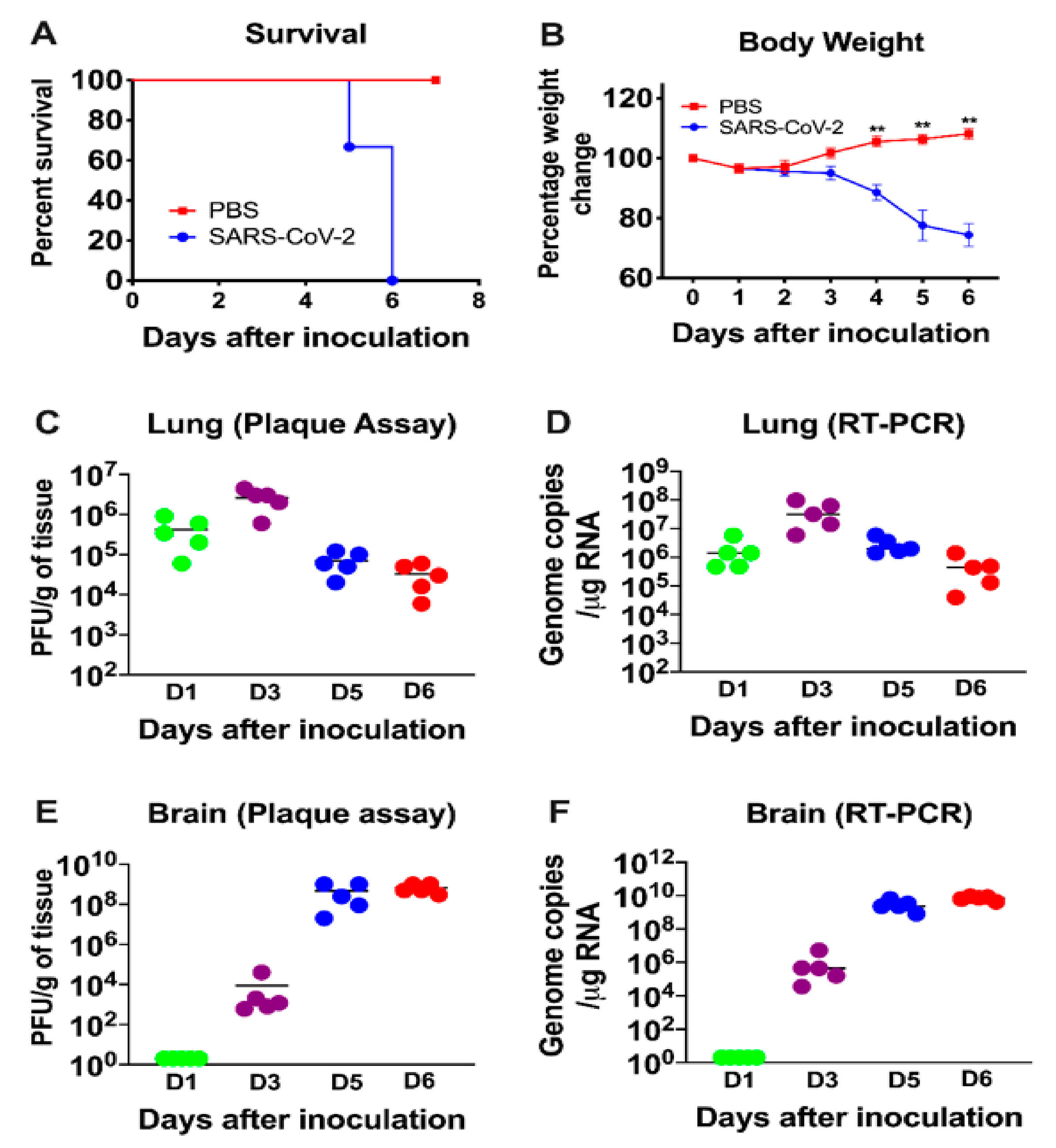
3.1. Characteristics of K18-hACE2 Mice Following SARS-CoV-2 Infection by the Intranasal Route
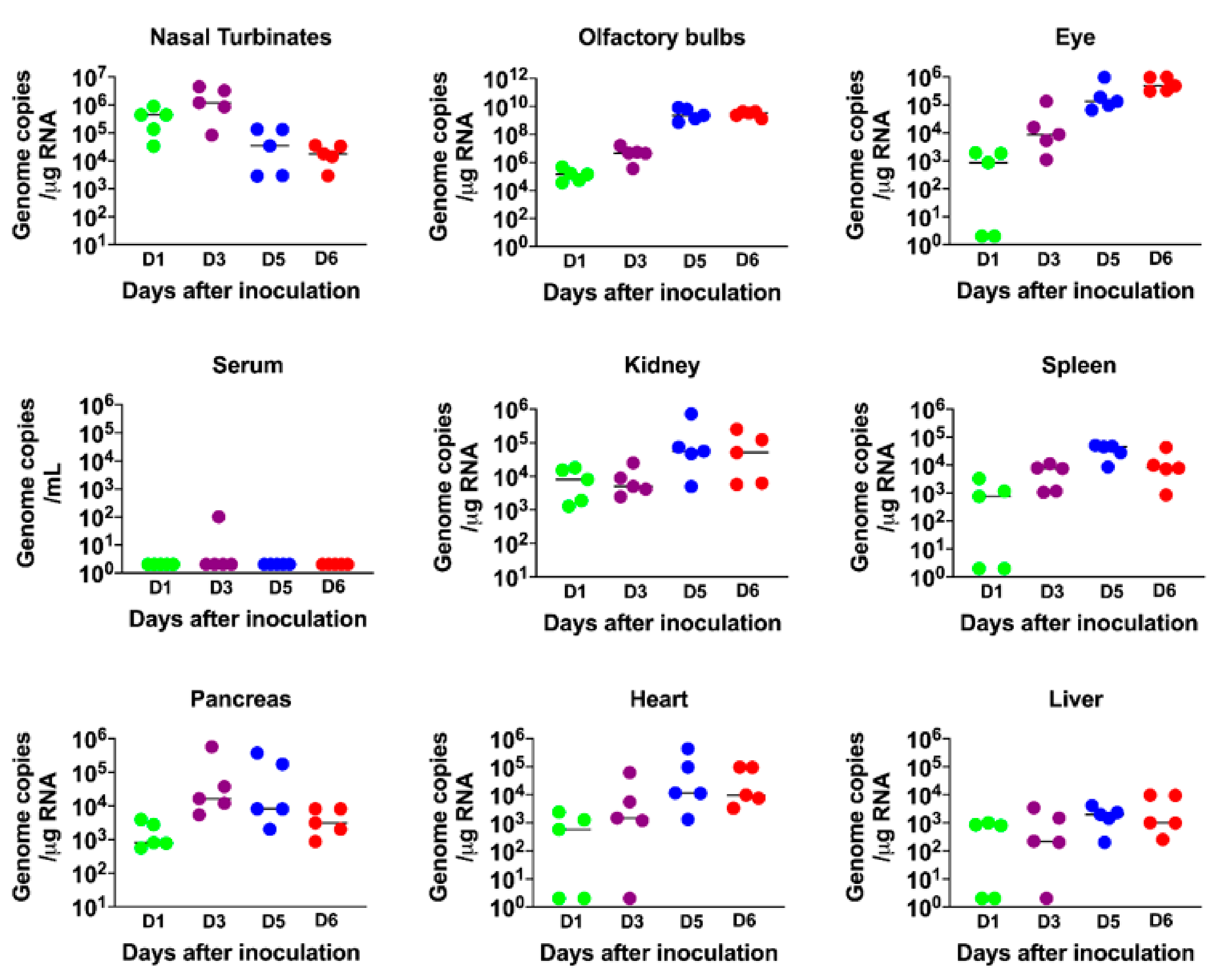
3.2. Virus Replication in the Periphery and Brains of K18-hACE2 Mice
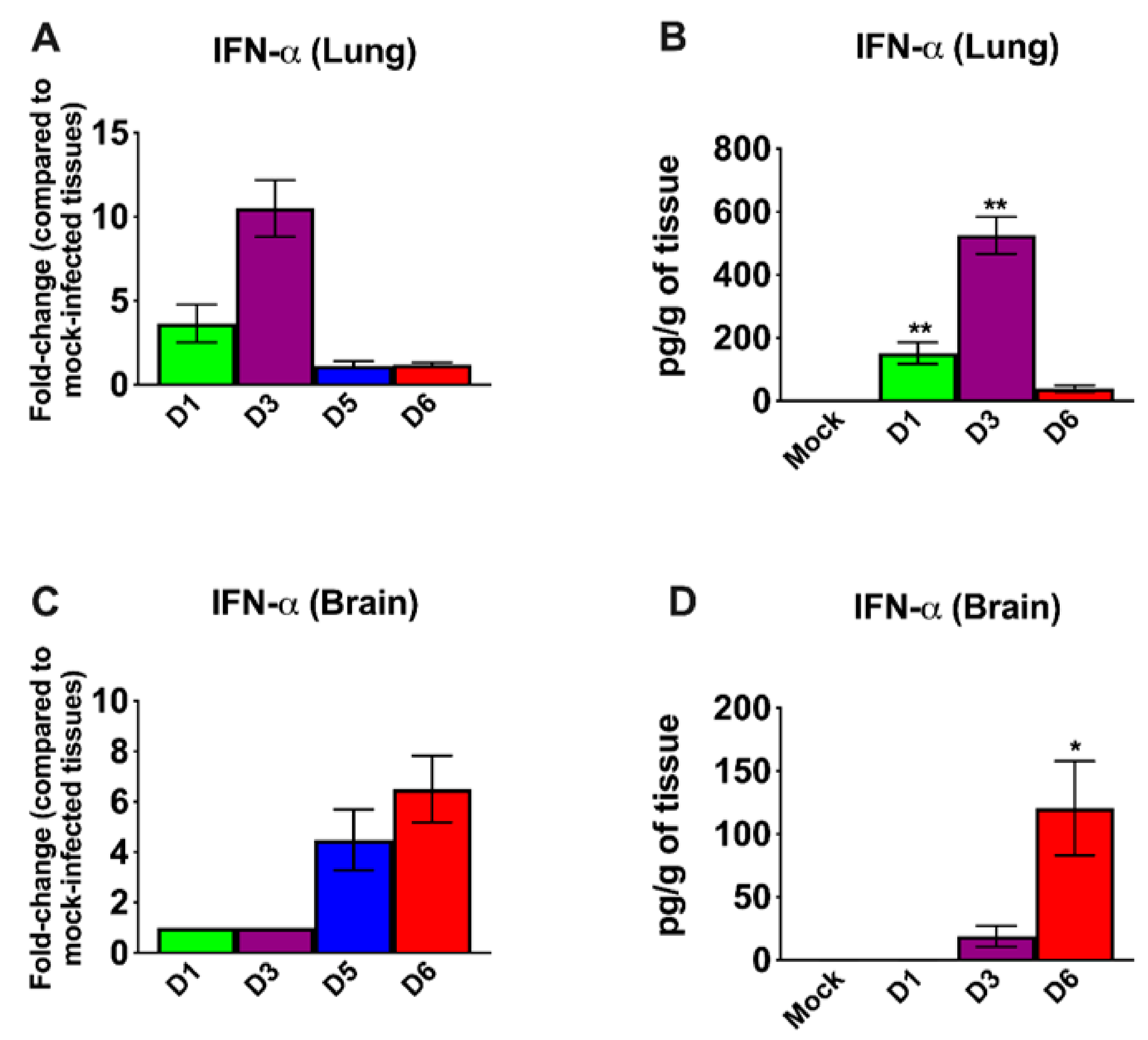
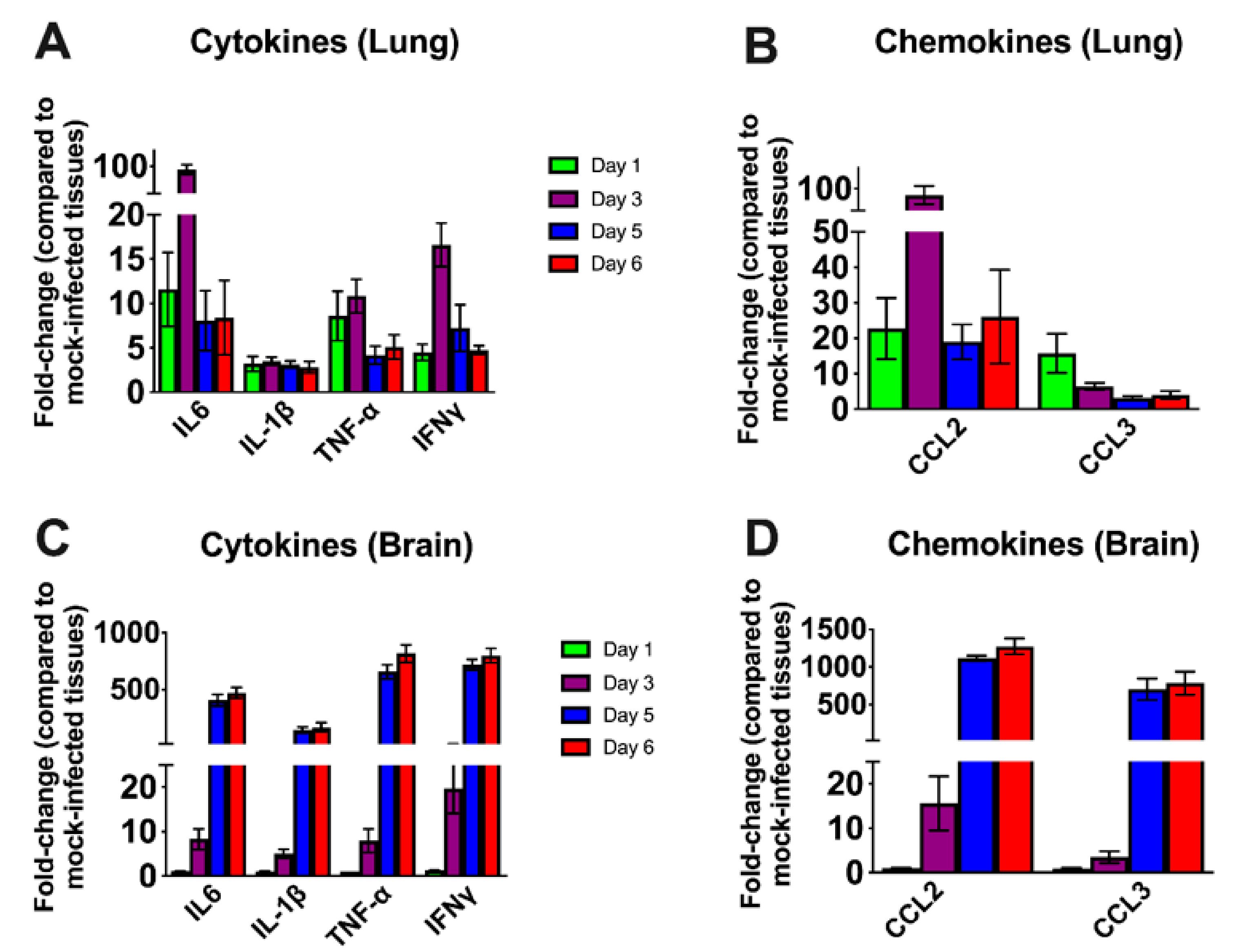
3.3. Inflammatory Changes in the Lungs and Brain of SARS-CoV-2-Infected Mice
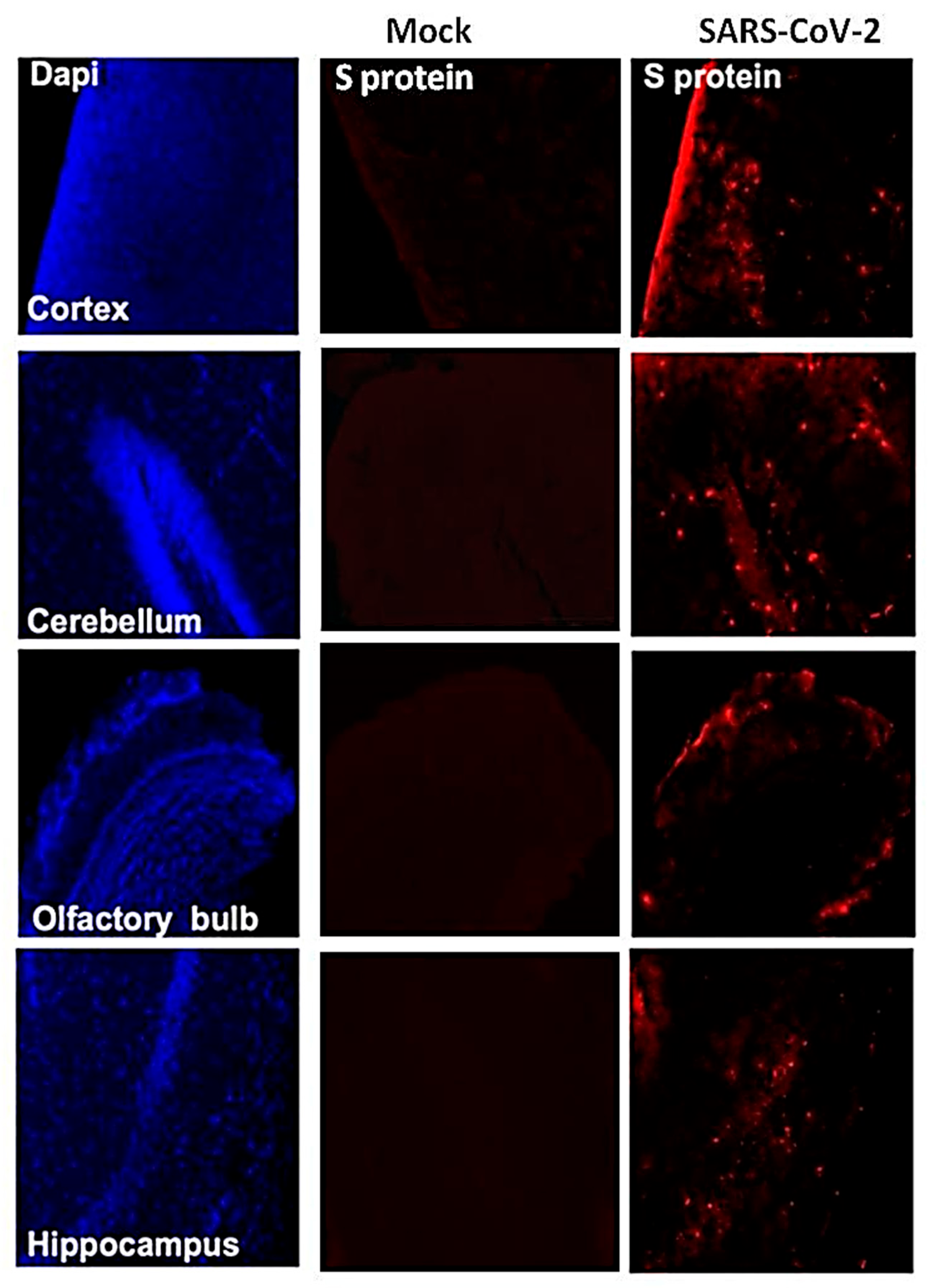
3.4. SARS-CoV-2-Induced Neuropathology in K18-hACE2 Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- Rothan, H.A.; Acharya, A.; Reid, S.P.; Kumar, M.; Byrareddy, S.N. Molecular Aspects of COVID-19 Differential Pathogenesis. Pathogens 2020, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, P.S.; Rizvi, Z.; Sharma, P.; Hindi, F.; Filatov, A. Neurological Complications of Coronavirus Disease (COVID-19): Encephalopathy, MRI Brain and Cerebrospinal Fluid Findings: Case 2. Cureus 2020, 12, e7930. [Google Scholar] [CrossRef] [PubMed]
- Filatov, A.; Sharma, P.; Hindi, F.; Espinosa, P.S. Neurological Complications of Coronavirus Disease (COVID-19): Encephalopathy. Cureus 2020, 12, e7352. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.; Latz, E.; Morgan, D.; Brown, R. Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer’s Res. Ther. 2020, 12, 69. [Google Scholar] [CrossRef]
- Mahammedi, A.; Saba, L.; Vagal, A.; Leali, M.; Rossi, A.; Gaskill, M.; Sengupta, S.; Zhang, B.; Carriero, A.; Bachir, S.; et al. Imaging in Neurological Disease of Hospitalized COVID-19 Patients: An Italian Multicenter Retrospective Observational Study. Radiology 2020, 297, E270–E273. [Google Scholar] [CrossRef] [PubMed]
- Parry, A.H.; Wani, A.H.; Yaseen, M. Neurological Dysfunction in Coronavirus Disease-19 (COVID-19). Acad. Radiol. 2020, 27, 1329–1330. [Google Scholar] [CrossRef]
- Baig, A.M.; Sanders, E.C. Potential neuroinvasive pathways of SARS-CoV-2: Deciphering the spectrum of neurological deficit seen in coronavirus disease-2019 (COVID-19). J. Med. Virol. 2020, 92, 1845–1857. [Google Scholar] [CrossRef]
- Adamczyk-Sowa, M.; Niedziela, N.; Kubicka-Baczyk, K.; Wierzbicki, K.; Jaroszewicz, J.; Sowa, P. Neurological symptoms as a clinical manifestation of COVID-19: Implications for internists. Polish Arch. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Wang, H.Y.; Li, X.L.; Yan, Z.R.; Sun, X.P.; Han, J.; Zhang, B.W. Potential neurological symptoms of COVID-19. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420917830. [Google Scholar] [CrossRef]
- Schaumburg, C.S.; Held, K.S.; Lane, T.E. Mouse hepatitis virus infection of the CNS: A model for defense, disease, and repair. Front. Biosci. 2008, 13, 4393–4406. [Google Scholar] [CrossRef] [PubMed]
- Verstrepen, K.; Baisier, L.; De Cauwer, H. Neurological manifestations of COVID-19, SARS and MERS. Acta Neurol Belg. 2020, 120, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.L.; Cassell, M.D.; Gross, K.W.; Sigmund, C.D. Adjacent expression of renin and angiotensinogen in the rostral ventrolateral medulla using a dual-reporter transgenic model. Hypertension 2004, 43, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Gowrisankar, Y.V.; Clark, M.A. Angiotensin II regulation of angiotensin-converting enzymes in spontaneously hypertensive rat primary astrocyte cultures. J. Neurochem. 2016, 138, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Pogue, A.; Hill, J.M. SARS-CoV-2 Infectivity and Neurological Targets in the Brain. Cell. Mol. Neurobiol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Solomon, I.H.; Normandin, E.; Bhattacharyya, S.; Mukerji, S.S.; Keller, K.; Ali, A.S.; Adams, G.; Hornick, J.L.; Padera, R.F., Jr.; Sabeti, P. Neuropathological Features of Covid-19. N. Engl. J. Med. 2020, 383, 989–992. [Google Scholar] [CrossRef]
- Zhang, B.Z.; Chu, H.; Han, S.; Shuai, H.; Deng, J.; Hu, Y.F.; Gong, H.R.; Lee, A.C.; Zou, Z.; Yau, T.; et al. SARS-CoV-2 infects human neural progenitor cells and brain organoids. Cell Res. 2020, 30, 928–931. [Google Scholar] [CrossRef]
- Ramani, A.; Muller, L.; Ostermann, P.N.; Gabriel, E.; Abida-Islam, P.; Muller-Schiffmann, A.; Mariappan, A.; Goureau, O.; Gruell, H.; Walker, A.; et al. SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J. 2020, 39, e106230. [Google Scholar] [CrossRef]
- von Weyhern, C.H.; Kaufmann, I.; Neff, F.; Kremer, M. Early evidence of pronounced brain involvement in fatal COVID-19 outcomes. Lancet 2020, 395, e109. [Google Scholar] [CrossRef]
- Cheng, Q.; Yang, Y.; Gao, J. Infectivity of human coronavirus in the brain. EBioMedicine 2020, 56, 102799. [Google Scholar] [CrossRef]
- Golden, J.W.; Cline, C.R.; Zeng, X.; Garrison, A.R.; Carey, B.D.; Mucker, E.M.; White, L.E.; Shamblin, J.D.; Brocato, R.L.; Liu, J.; et al. Human angiotensin-converting enzyme 2 transgenic mice infected with SARS-CoV-2 develop severe and fatal respiratory disease. JCI Insight 2020, 5, e142032. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.S.; Bailey, A.L.; Kafai, N.M.; Nair, S.; McCune, B.T.; Yu, J.; Fox, J.M.; Chen, R.E.; Earnest, J.T.; Keeler, S.P.; et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol. 2020, 21, 1327–1335. [Google Scholar] [CrossRef] [PubMed]
- Moreau, G.B.; Burgess, S.L.; Sturek, J.M.; Donlan, A.N.; Petri, W.A.; Mann, B.J. Evaluation of K18-hACE2 Mice as a Model of SARS-CoV-2 Infection. Am. J. Trop. Med. Hyg. 2020, 103, 1215–1219. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wong, L.R.; Li, K.; Verma, A.K.; Ortiz, M.; Wohlford-Lenane, C.; Leidinger, M.R.; Knudson, C.M.; Meyerholz, D.K.; McCray, P.B., Jr.; et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 2020, 1–5. [Google Scholar] [CrossRef]
- Perlman, S.; Evans, G.; Afifi, A. Effect of olfactory bulb ablation on spread of a neurotropic coronavirus into the mouse brain. J Exp. Med. 1990, 172, 1127–1132. [Google Scholar] [CrossRef]
- Netland, J.; Meyerholz, D.K.; Moore, S.; Cassell, M.; Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 2008, 82, 7264–7275. [Google Scholar] [CrossRef]
- Rothan, H.A.; Stone, S.; Natekar, J.; Kumari, P.; Arora, K.; Kumar, M. The FDA-approved gold drug auranofin inhibits novel coronavirus (SARS-COV-2) replication and attenuates inflammation in human cells. Virology 2020, 547, 7–11. [Google Scholar] [CrossRef]
- Rothan, H.A.; Arora, K.; Natekar, J.P.; Strate, P.G.; Brinton, M.A.; Kumar, M. Z-DNA-Binding Protein 1 Is Critical for Controlling Virus Replication and Survival in West Nile Virus Encephalitis. Front. Microbiol. 2019, 10, 2089. [Google Scholar] [CrossRef]
- Natekar, J.P.; Rothan, H.A.; Arora, K.; Strate, P.G.; Kumar, M. Cellular microRNA-155 Regulates Virus-Induced Inflammatory Response and Protects against Lethal West Nile Virus Infection. Viruses 2019, 12, 9. [Google Scholar] [CrossRef]
- Kumar, M.; Roe, K.; Orillo, B.; Muruve, D.A.; Nerurkar, V.R.; Gale, M., Jr.; Verma, S. Inflammasome adaptor protein Apoptosis-associated speck-like protein containing CARD (ASC) is critical for the immune response and survival in west Nile virus encephalitis. J. Virol. 2013, 87, 3655–3667. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Krause, K.K.; Azouz, F.; Nakano, E.; Nerurkar, V.R. A guinea pig model of Zika virus infection. Virol. J. 2017, 14, 75. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.; Azouz, F.; Nakano, E.; Nerurkar, V.R.; Kumar, M. Deletion of Pregnancy Zone Protein and Murinoglobulin-1 Restricts the Pathogenesis of West Nile Virus Infection in Mice. Front. Microbiol. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Roe, K.; Nerurkar, P.V.; Orillo, B.; Thompson, K.S.; Verma, S.; Nerurkar, V.R. Reduced immune cell infiltration and increased pro-inflammatory mediators in the brain of Type 2 diabetic mouse model infected with West Nile virus. J. Neuroinflamm. 2014, 11, 80. [Google Scholar] [CrossRef]
- Kumar, M.; Roe, K.; O’Connell, M.; Nerurkar, V.R. Induction of virus-specific effector immune cell response limits virus replication and severe disease in mice infected with non-lethal West Nile virus Eg101 strain. J. Neuroinflamm. 2015, 12, 178. [Google Scholar] [CrossRef]
- McCray, P.B., Jr.; Pewe, L.; Wohlford-Lenane, C.; Hickey, M.; Manzel, L.; Shi, L.; Netland, J.; Jia, H.P.; Halabi, C.; Sigmund, C.D.; et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 2007, 81, 813–821. [Google Scholar] [CrossRef]
- tenOever, B.R. The Evolution of Antiviral Defense Systems. Cell Host Microbe 2016, 19, 142–149. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Regulation of adaptive immunity by the innate immune system. Science 2010, 327, 291–295. [Google Scholar] [CrossRef]
- Roth-Cross, J.K.; Bender, S.J.; Weiss, S.R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 2008, 82, 9829–9838. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2020, 1–8. [Google Scholar] [CrossRef]
- Kanjanaumporn, J.; Aeumjaturapat, S.; Snidvongs, K.; Seresirikachorn, K.; Chusakul, S. Smell and taste dysfunction in patients with SARS-CoV-2 infection: A review of epidemiology, pathogenesis, prognosis, and treatment options. Asian Pac. J. Allergy Immunol. 2020, 38, 69–77. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef] [PubMed]
- Dube, M.; Le Coupanec, A.; Wong, A.H.M.; Rini, J.M.; Desforges, M.; Talbot, P.J. Axonal Transport Enables Neuron-to-Neuron Propagation of Human Coronavirus OC43. J. Virol. 2018, 92, e00404–e00418. [Google Scholar] [CrossRef] [PubMed]
- Swanson, P.A., 2nd; McGavern, D.B. Viral diseases of the central nervous system. Curr. Opin. Virol. 2015, 11, 44–54. [Google Scholar] [CrossRef] [PubMed]
- van Riel, D.; Verdijk, R.; Kuiken, T. The olfactory nerve: A shortcut for influenza and other viral diseases into the central nervous system. J. Pathol. 2015, 235, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Blinzinger, K.; Anzil, A.P. Neural route of infection in viral diseases of the central nervous system. Lancet 1974, 2, 1374–1375. [Google Scholar] [CrossRef]
- Durrant, D.M.; Ghosh, S.; Klein, R.S. The Olfactory Bulb: An Immunosensory Effector Organ during Neurotropic Viral Infections. ACS Chem. Neurosci. 2016, 7, 464–469. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Accession No.) | Primer Sequence (5′-3′) |
|---|---|
| IL-1β (NM_000576) | |
| Forward | AGCACCTTCTTTCCCTTCATC |
| Reverse | GGACCAGACATCACCAAGC |
| IL-6 (NM_000600) | |
| Forward | CCAGGAGCCCAGCTATGAAC |
| Reverse | CCCAGGGAGAAGGCAACTG |
| CCL3 (NM_011337) | |
| Forward | ATTCCACGCCAATTCATC |
| Reverse | ATTCAGTTCCAGGTCAGT |
| IFN-α (NM_010502) | |
| Forward | CTCTGTGCTTTCCTGATG |
| Reverse | CTGAGGTTATGAGTCTGAG |
| TNF-α (NM_013693) | |
| Forward | CCAGTCTGTATCCTTCTAA |
| Reverse | TCTTGTGTTTCTGAGTAGT |
| CCL2 (NM_011333) | |
| Forward | TCACCTGCTGCTACTCATTCACCA |
| Reverse | TACAGCTTCTTTGGGACACCTGCT |
| IFN-γ (NM_008337) | |
| Forward | TTCACTGACCAATAAGAAAC |
| Reverse | TACTACCTGACACATTCG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, P.; Rothan, H.A.; Natekar, J.P.; Stone, S.; Pathak, H.; Strate, P.G.; Arora, K.; Brinton, M.A.; Kumar, M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses 2021, 13, 132. https://doi.org/10.3390/v13010132
Kumari P, Rothan HA, Natekar JP, Stone S, Pathak H, Strate PG, Arora K, Brinton MA, Kumar M. Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses. 2021; 13(1):132. https://doi.org/10.3390/v13010132
Chicago/Turabian StyleKumari, Pratima, Hussin A. Rothan, Janhavi P. Natekar, Shannon Stone, Heather Pathak, Philip G. Strate, Komal Arora, Margo A. Brinton, and Mukesh Kumar. 2021. "Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice" Viruses 13, no. 1: 132. https://doi.org/10.3390/v13010132
APA StyleKumari, P., Rothan, H. A., Natekar, J. P., Stone, S., Pathak, H., Strate, P. G., Arora, K., Brinton, M. A., & Kumar, M. (2021). Neuroinvasion and Encephalitis Following Intranasal Inoculation of SARS-CoV-2 in K18-hACE2 Mice. Viruses, 13(1), 132. https://doi.org/10.3390/v13010132