Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Production of Virus like Particles
2.2. GII.2 Homology Model
2.3. Enzyme-Linked Immunoabsorbent Assay (EIA)
2.4. Carbohydrate Ligand Binding
2.5. Bile-Titration Assay
2.6. Carbohydrate Ligand Blockade
2.7. Quantification and Statistical Analysis
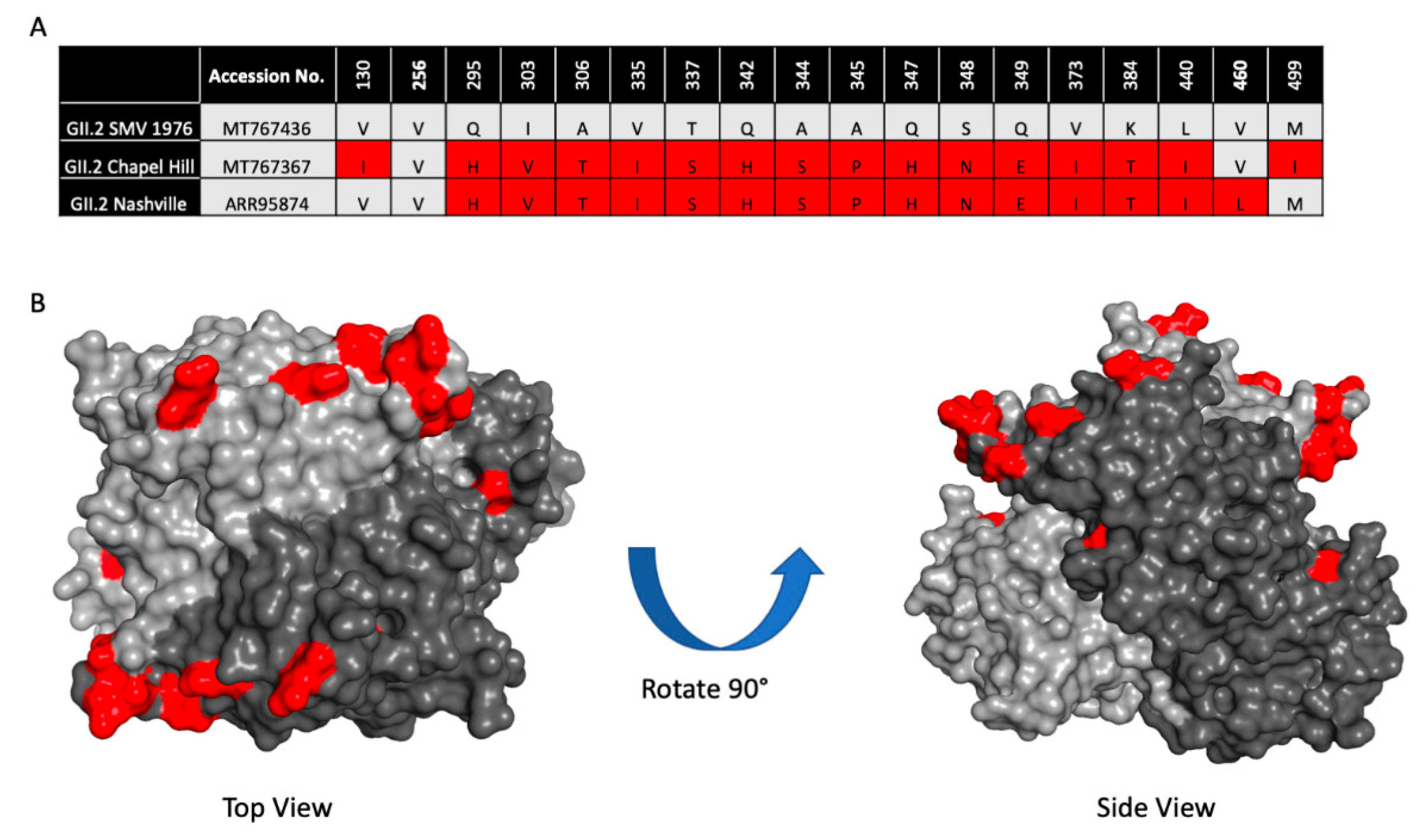
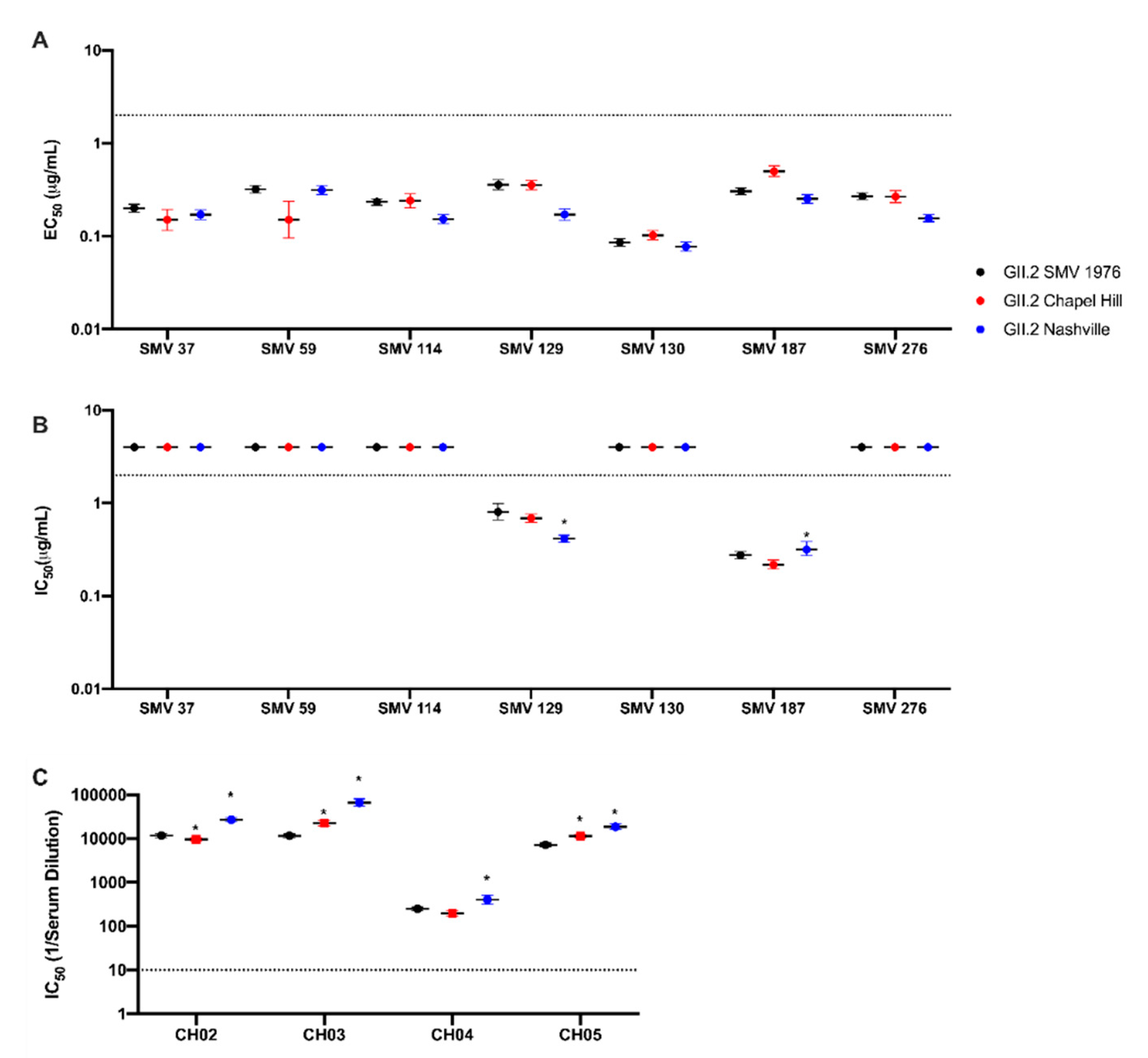
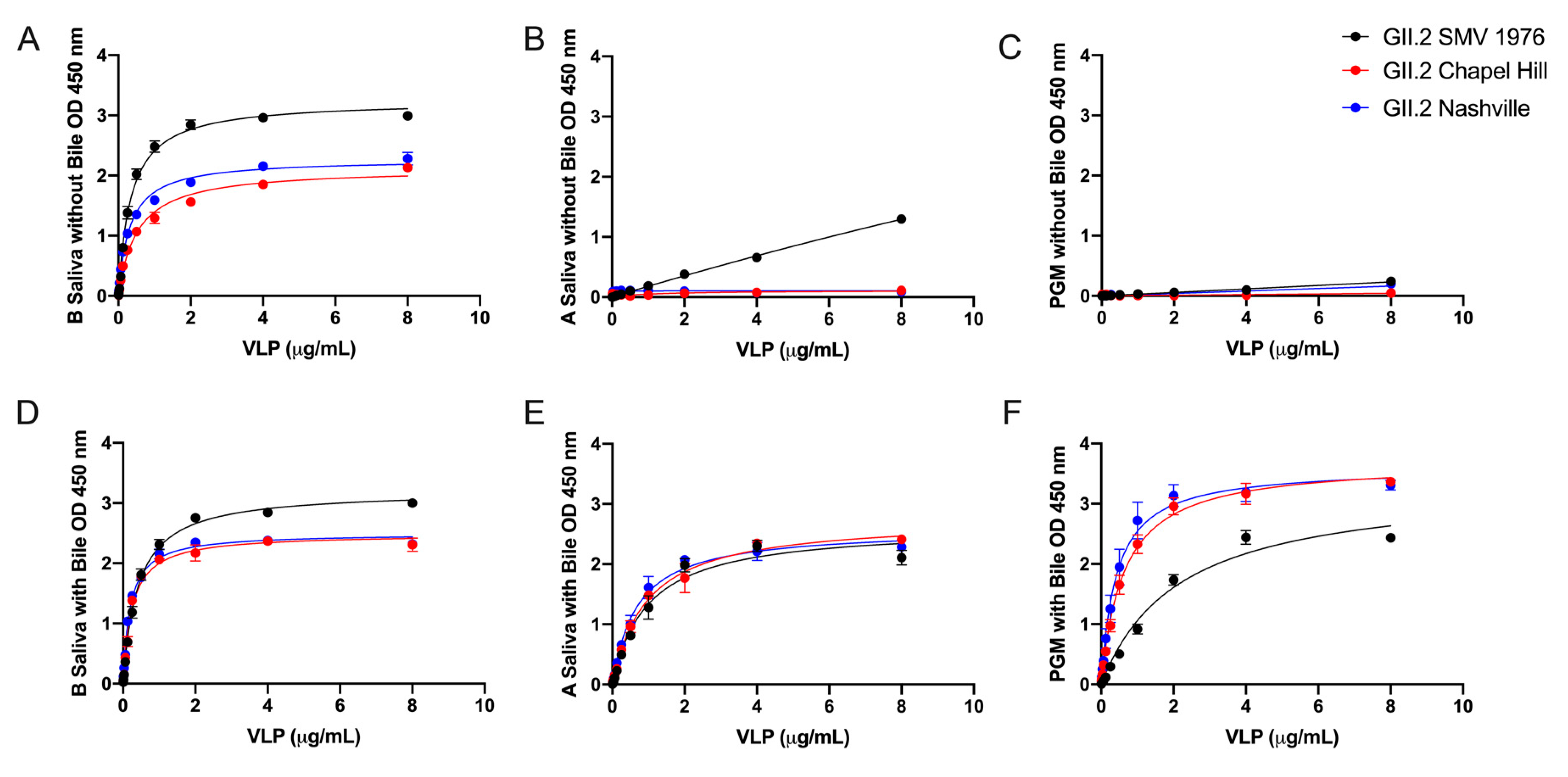
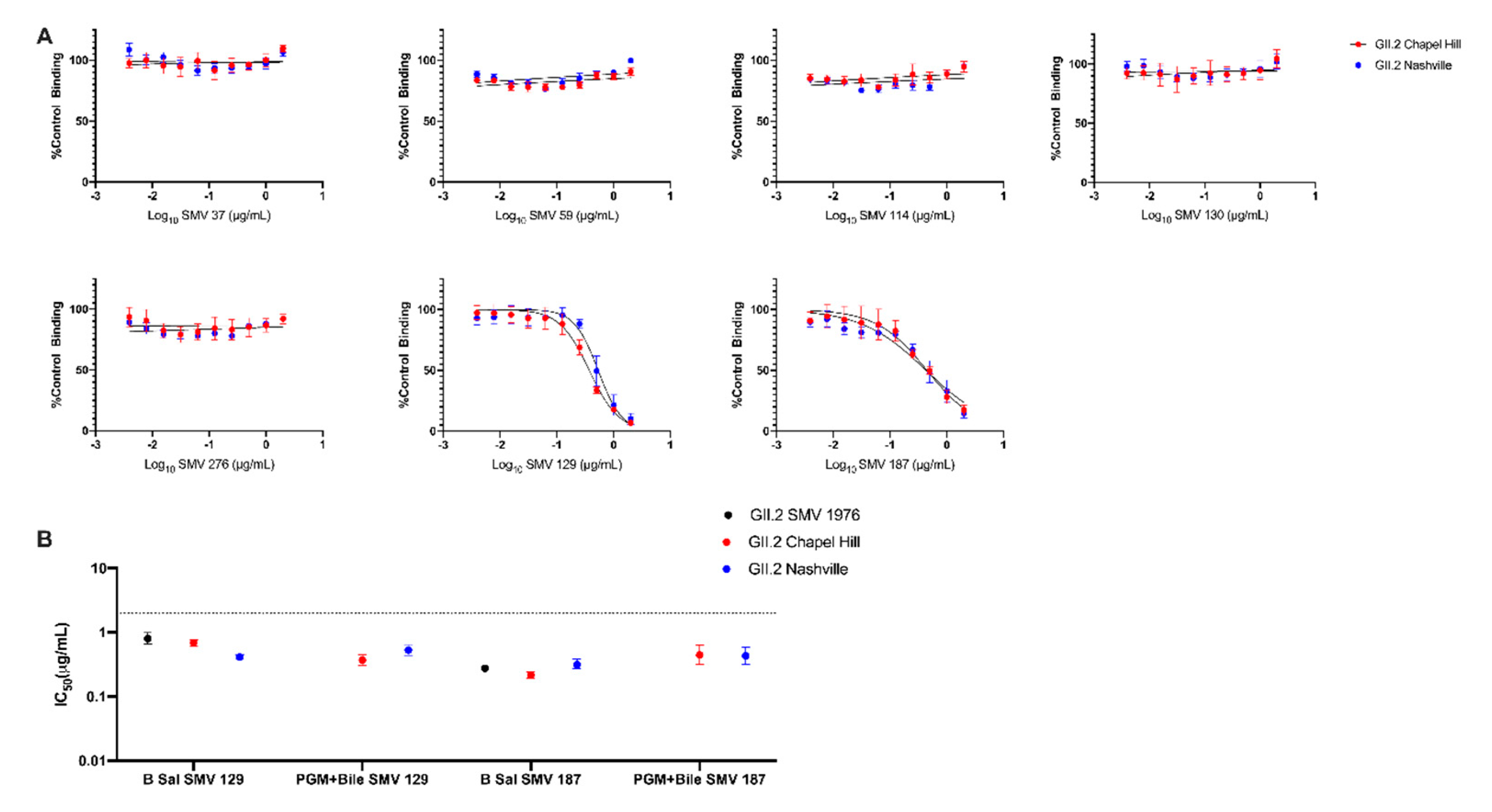
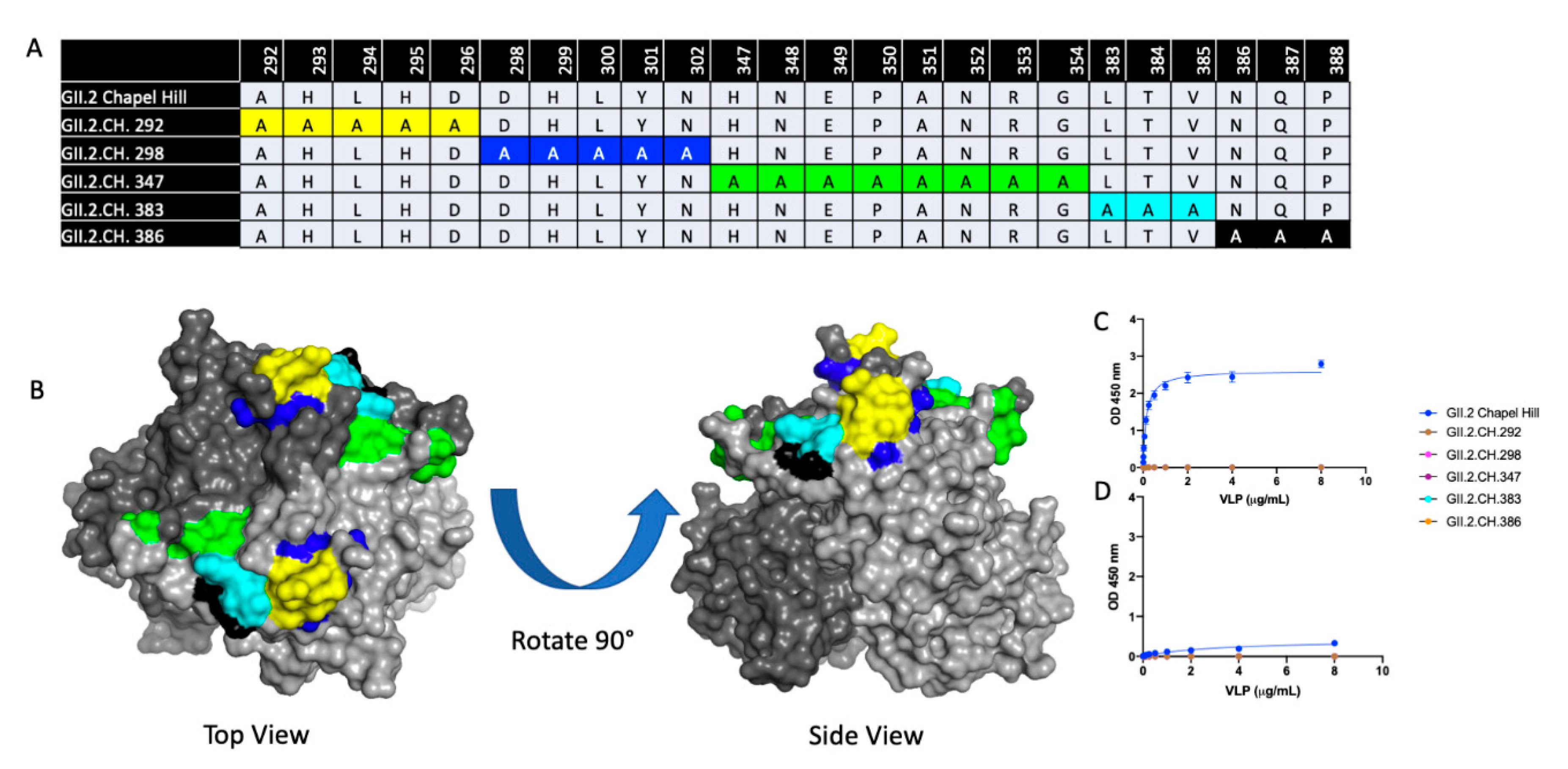
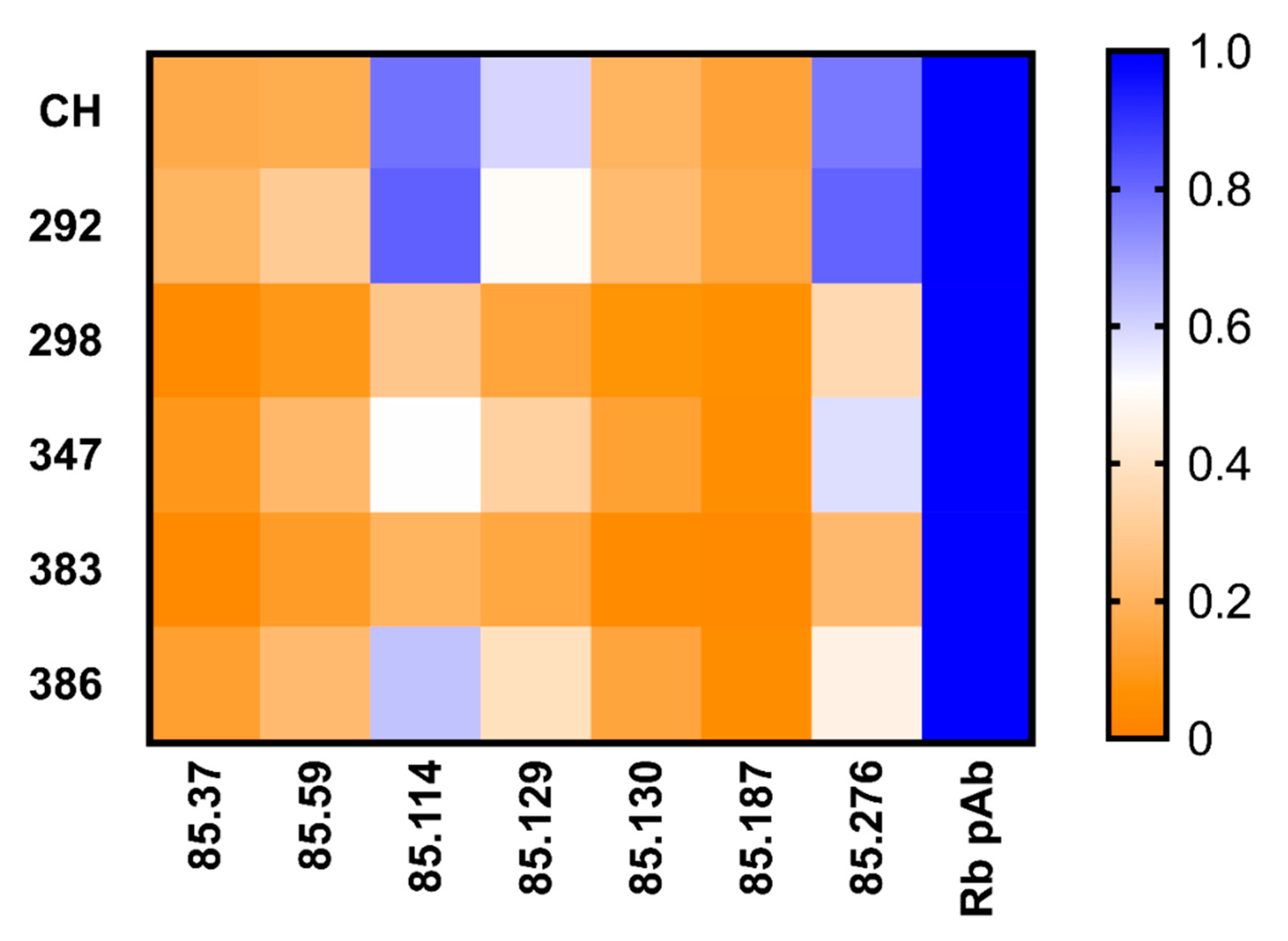
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lopman, B.; Steele, D.; Kirkwood, C.D.; Parashar, U.D. The Vast and Varied Global Burden of Norovirus: Prospects for Prevention and Control. PLoS Med. 2016, 13, e1001999. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global economic burden of norovirus gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, G.T.; Phan, K.; Teng, I.; Pu, J.; Watanabe, T. A systematic review and meta-analysis of the prevalence of norovirus in cases of gastroenteritis in developing countries. Medicine 2017, 96, e8139. [Google Scholar] [CrossRef]
- Patel, M.M.; Widdowson, M.A.; Glass, R.I.; Akazawa, K.; Vinjé, J.; Parsahar, U.D. Systematic literature review of role of noroviruses in sporadic gastroenteritis. Emerg. Infect. Dis. 2008, 14, 1224–1231. [Google Scholar] [CrossRef]
- Ahmed, S.; Hall, A.J.; Robinson, A.E.; Verhoef, L.; Premkumar, P. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 2014, 14, 725–730. [Google Scholar] [CrossRef]
- Hasing, M.E.; Lee, B.E.; Qiu, Y.; Ming, X.; Pabbaraju, K.; Wong, A.; Pang, X. Changes in norovirus genotype diversity in gastroenteritis outbreaks in Alberta, Canada: 2012–2018. BMC Infect. Dis. 2019, 19, 177. [Google Scholar] [CrossRef]
- Tran, T.N.H.; Trainor, E.; Nakagomi, T.; Cunliffe, N.; Nakagomi, O. Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: Global distribution of genogroups, genotypes and GII.4 variants. J. Clin. Virol. 2013, 56, 269–277. [Google Scholar] [CrossRef]
- Kwok, K.; Niendorf, S.; Lee, N.; Hung, T.; Chan, L.; Jacobsen, S.; Chan, M. Increased Detection of Emergent Recombinant Norovirus GII.P16-GII.2 Strains in Young Adults, Hong Kong, China, 2016–2017. Emerg. Infect. Dis. 2017, 23, 1852–1855. [Google Scholar] [CrossRef]
- Cannon, J.L.; Barclay, L.; Collins, N.R.; Wikswo, M.E.; Castro, C.J.; Magaña, L.C.; Vinjé, J. Genetic and Epidemiologic Trends of Norovirus Outbreaks in the United States from 2013 to 2016 Demonstrated Emergence of Novel GII.4 Recombinant Viruses. J. Clin. Microbiol. 2017, 55, 2208–2221. [Google Scholar] [CrossRef]
- Iritani, N.; Vennema, H.; Siebenga, J.J.; Siezen, R.J.; Renckens, B.; Seto, Y.; Koopmans, M. Genetic Analysis of the Capsid Gene of Genotype GII.2 Noroviruses. J. Virol. 2008, 82, 7336–7345. [Google Scholar] [CrossRef][Green Version]
- Jiang, X.; Wang, M.; Wang, K.; Estes, M.K. Sequence and genomic organization of Norwalk virus. Virology 1993, 195, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.N.; Graham, D.Y.; Wang, K.N.; Estes, M.K. Norwalk virus genome cloning and characterization. Science 1990, 250, 1580–1583. [Google Scholar] [CrossRef] [PubMed]
- Prasad, B.V.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Lou, Z.; Tan, M.; Chen, Y.; Liu, Y.; Zhang, Z.; Rao, Z. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 2007, 81, 5949–5957. [Google Scholar] [CrossRef]
- Chen, R.; Neill, J.D.; Estes, M.K.; Prasad, B.V. X-ray structure of a native calicivirus: Structural insights into antigenic diversity and host specificity. Proc. Natl. Acad. Sci. USA 2006, 103, 8048–8053. [Google Scholar] [CrossRef]
- Debbink, K.; Donaldson, E.F.; Lindesmith, L.C.; Baric, R.S. Genetic mapping of a highly variable norovirus GII.4 blockade epitope: Potential role in escape from human herd immunity. J. Virol. 2012, 86, 1214–1226. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Beltramello, M.; Donaldson, E.F.; Corti, D.; Swanstrom, J.; Debbink, K.; Baric, R.S. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 2012, 8, e1002705. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Debbink, K.; Swanstrom, J.; Vinje, J.; Constantini, V.; Baric, R.S.; Donaldson, E.F. Monoclonal antibody-based antigenic mapping of norovirus GII.4-2002. J. Virol. 2011, 86, 873–883. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Lobue, A.D.; Cannon, J.L.; Zheng, D.P.; Vinje, J.; Baric, R.S. Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 2008, 5, e31. [Google Scholar] [CrossRef]
- Lochridge, V.P.; Jutila, K.L.; Graff, J.W.; Hardy, M.E. Epitopes in the P2 domain of norovirus VP1 recognized by monoclonal antibodies that block cell interactions. J. Gen. Virol. 2005, 86, 2799–2806. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Baric, R.S. Norovirus GII.4 strain antigenic variation. J. Virol. 2011, 85, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Costantini, V.; Swanstrom, J.; Agnihothram, S.; Vinjé, J.; Baric, R.S.; Lindesmith, L. Human Norovirus Detection and Production, Quantification, and Storage of Virus-Like Particles. Curr. Protoc. Microbiol. 2013, 31, 15K.1.1–15K.1.45. [Google Scholar] [CrossRef]
- Agnihothram, S.; Menachery, V.D.; Yount, B.L.; Lindesmith, L.C.; Scobey, T.; Whitmore, A.; Baric, R.S. Development of a Broadly Accessible Venezuelan Equine Encephalitis Virus Replicon Particle Vaccine Platform. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.L.; Zhen, S.S.; Wang, J.X.; Zhang, C.J.; Qiu, C.; Wang, S.M.; Wang, X.Y. Burden of acute gastroenteritis caused by norovirus in China: A systematic review. J. Infect. 2017, 75, 216–224. [Google Scholar] [CrossRef]
- Vinjé, J.; Green, J.; Lewis, D.C.; Gallimore, C.I.; Brown, D.W.G.; Koopmans, M.P.G. Genetic polymorphism across regions of the three open reading frames of “Norwalk-like viruses”. Arch. Virol. 2000, 145, 223–241. [Google Scholar] [CrossRef]
- Zheng, D.; Ando, T.; Frankhauser, R.L.; Beard, R.S.; Glass, R.I.; Monroe, S.S. Norovirus classification and proposed strain nomenclature. Virology 2006, 346, 312–323. [Google Scholar] [CrossRef]
- Burke, R.M.; Shah, M.P.; Wikswo, M.E.; Barclay, L.; Kambhampati, A.; Marsh, Z.; Hall, A.J. The Norovirus Epidemiologic Triad: Predictors of Severe Outcomes in US Norovirus Outbreaks, 2009–2016. J. Infect. Dis. 2019, 219, 1364–1372. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Vennema, H.; Renckens, B.; De Bruin, E.; Van Der Veer, B.; Siezen, R.J.; Koopmans, M. Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 2007, 81, 9932–9941. [Google Scholar] [CrossRef]
- Mallory, M.L.; Lindesmith, L.C.; Graham, R.L.; Baric, R.S. GII.4 Human Norovirus: Surveying the Antigenic Landscape. Viruses 2019, 11, 177. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; McDaniel, J.R.; Changela, A.; Verardi, R.; Kerr, S.A.; Costantini, V.; Baric, R.S. Sera Antibody Repertoire Analyses Reveal Mechanisms of Broad and Pandemic Strain Neutralizing Responses after Human Norovirus Vaccination. Immunity 2019, 50, 1530–1541. [Google Scholar] [CrossRef]
- Swanstrom, J.; Lindesmith, L.C.; Donaldson, E.F.; Yount, B.; Baric, R.S. Characterization of Blockade Antibody Responses in GII.2.1976 Snow Mountain Virus-Infected Subjects. J. Virol. 2014, 88, 829–837. [Google Scholar] [CrossRef]
- Han, J.; Wu, X.; Chen, L.; Fu, Y.; Xu, D.; Zhang, P.; Ji, L. Emergence of norovirus GII.P16-GII.2 strains in patients with acute gastroenteritis in Huzhou, China, 2016–2017. BMC Infect. Dis. 2018, 18, 342. [Google Scholar] [CrossRef]
- Niendorf, S.; Jacobsen, S.; Faber, M.; EisHübinger, A.M.; Hofmann, J.; Zimmermann, O.; Bock, C.T. Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Eurosurveillance 2017, 22, 30447. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic Analyses Suggest that Factors Other than the Capsid Protein Play a Role in the Epidemic Potential of GII.2 Norovirus. MSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Iritani, N.; Kaida, A.; Abe, N.; Sekiguchi, J.; Kubo, H.; Takakura, K.; Seto, Y. Increase of GII.2 norovirus infections during the 2009–2010 season in Osaka City, Japan. J. Med. Virol. 2012, 84, 517–525. [Google Scholar] [CrossRef]
- Barclay, L.; Cannon, J.L.; Wikswo, M.E.; Phillips, A.R.; Browne, H.; Montmayeur, A.M.; Vinjé, J. Emerging Novel GII.P16 Noroviruses Associated with Multiple Capsid Genotypes. Viruses 2019, 11, 535. [Google Scholar] [CrossRef] [PubMed]
- Ruis, C.; Roy, S.; Brown, J.R.; Allen, D.J.; Goldstein, R.A.; Breuer, J. The emerging GII.P16-GII.4 Sydney 2012 norovirus lineage is circulating worldwide, arose by late-2014 and contains polymerase changes that may increase virus transmission. PLoS ONE 2017, 12, e0179572. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Farkas, T.; Zhong, W.; Tan, M.; Thorton, S.; Morrow, A.L.; Jiang, X. Norovirus and Histo-Blood Group Antigens: Demonstration of a Wide Spectrum of Strain Specificities and Classification of Two Major Binding Groups among Multiple Binding Patterns. J. Virol. 2005, 79, 6714–6722. [Google Scholar] [CrossRef]
- Harrington, P.; Lindesmith, L.; Yount, B.; Moe, C.; Baric, R. Binding of Norwalk Virus-Like Particles to ABH Histo-Blood Group Antigens Is Blocked by Antisera from Infected Human Volunteers or Experimentally Vaccinated Mice. J. Virol. 2002, 76, 12335–12343. [Google Scholar] [CrossRef]
- Lindesmith, L.; Moe, C.; Lependu, J.; Frelinger, J.A.; Treanor, J.; Baric, R.S. Cellular and Humoral Immunity Following Snow Mountain Virus Challenge. J. Virol. 2005, 79, 2900–2909. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Brewer-Jensen, P.B.; Mallory, M.L.; Jensen, K.; Yount, B.L.; Constantini, V.; Baric, R.S. Virus-Host Interactions between Nonsecretors and Human Norovirus. Cell Mol. Gastroenterol. Hepatol. 2020, 10, 245–267. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.; Orchard, R.C.; Kim, A.S.; Stegman, R.A.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 9201–9210. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Tenge, V.R.; Karandikar, U.C.; Lin, S.; Ramani, S.; Ettayebi, K.; Estes, M. Bile acids and ceramide overcome the entry restriction for GII.3 human norovirus replication in human intestinal enteroids. Proc. Natl. Acad. Sci. USA 2020, 117, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Mallory, M.L.; Debbink, K.; Donaldson, E.F.; Brewer-Jensen, P.D.; Swann, E.W.; Baric, R.S. Conformational Occlusion of Blockade Antibody Epitopes, a Novel Mechanism of GII.4 Human Norovirus Immune Evasion. MSphere 2018, 3, e00518–e00617. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Brewer-Jensen, P.D.; Mallory, M.L.; Yount, B.; Collins, M.H.; Debbink, K.; Baric, R.S. Human Norovirus Epitope D Plasticity Allows Escape from Antibody Immunity without Loss of Capacity for Binding Cellular Ligands. J. Virol. 2019, 93, e01813–e01818. [Google Scholar] [CrossRef] [PubMed]
- Lindesmith, L.C.; Beltramello, M.; Swanstom, J.; Jones, T.A.; Corti, D.; Lanzavecchia, A.; Baric, R.S. Serum Immunoglobulin A Cross-Strain Blockade of Human Noroviruses. Open Forum Infect. Dis. 2015, 2, ofv084. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Donaldson, E.F.; Beltramello, M.; Pintus, S.; Corti, D.; Swanstom, J.; Baric, R.S. Particle conformation regulates antibody access to a conserved GII.4 norovirus blockade epitope. J. Virol. 2014, 88, 8826–8842. [Google Scholar] [CrossRef]
- Lochridge, V.P.; Hardy, M.E. Snow Mountain Virus Genome Sequence and Virus-like Particle Assembly. Virus Genes 2003, 26, 71–82. [Google Scholar] [CrossRef]
- Treanor, J.; Madore, H.P. Production of a monoclonal antibody against the Snow Mountain agent of gastroenteritis by in vitro immunization of murine spleen cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3613–3617. [Google Scholar] [CrossRef]
- Alvarado, G.; Ettayebi, K.; Atmar, R.L.; Bombardi, R.G.; Kose, N.; Estes, M.K.; Crowe, J.E. Human Monoclonal Antibodies That Neutralize Pandemic GII.4 Noroviruses. Gastroenterology 2018, 155, 1898–1907. [Google Scholar] [CrossRef]
- Shirato, H. ELISA-Based Methods to Detect and Quantify Norovirus Virus-Like Particle Attachment to Histo-Blood Group Antigens. In Methods in Molecular Biology; Humana: New York, NY, USA, 2020; Volume 2132. [Google Scholar] [CrossRef]
- Kilic, T.; Koromyslova, A.; Hansman, G.S. Structural Basis for Human Norovirus Capsid Binding to Bile Acids. J. Virol. 2019, 93, e01581–e01618. [Google Scholar] [CrossRef] [PubMed]
- Shivana, V.; Kim, Y.; Chang, K.O. The crucial role of bile acids in the entry of porcine enteric calicivirus. Virology 2014, 456, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Haga, K.; Ettayebi, K.; Tenge, V.R.; Karandikar, U.C.; Lewis, M.A.; Lin, S.; Estes, M.K. Genetic Manipulation of Human Intestinal Enteroids Demonstrates the Necessity of a Functional Fucosyltransferase 2 Gene for Secretor-Dependent Human Norovirus Infection. MBio 2020, 11, e00251–e00320. [Google Scholar] [CrossRef] [PubMed]
- Todd, K.V.; Tripp, R.A. Vero Cells as a Mammalian Cell Substrate for Human Norovirus. Viruses 2020, 12, 439. [Google Scholar] [CrossRef]
- Kolawole, A.O.; Smith, H.Q.; Svoboda, S.A.; Lewis, M.S.; Sherman, M.B.; Lynch, G.C.; Pettitt, B.M.; Smith, T.J.; Wobus, C.E. Norovirus Escape from Broadly Neutralizing Antibodies Is Limited to Allostery-Like Mechanisms. MSphere 2017, 2, e00317–e00334. [Google Scholar] [CrossRef]
- Jung, J.; Grant, T.; Thomas, D.R.; Diehnelt, C.W.; Grigorieff, N.; Joshua-Tor, L. High-resolution cryo-EM structures of outbreak strain human norovirus shells reveal size variations. Proc. Natl. Acad. Sci. USA 2019, 116, 12828–12832. [Google Scholar] [CrossRef]
- Ruis, C.; Lindemsith, L.; Mallory, M.; Brewer-Jensen, P.; Bryant, J.; Costantini, V.; Monit, C.; Vinje, J.; Baric, R.; Goldstein, R.; et al. Preadaptation of pandemic GII.4 noroviruses in hidden virus reservoirs years before emergence. bioRxiv 2019, 658765. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallory, M.L.; Lindesmith, L.C.; Brewer-Jensen, P.D.; Graham, R.L.; Baric, R.S. Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains. Viruses 2020, 12, 989. https://doi.org/10.3390/v12090989
Mallory ML, Lindesmith LC, Brewer-Jensen PD, Graham RL, Baric RS. Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains. Viruses. 2020; 12(9):989. https://doi.org/10.3390/v12090989
Chicago/Turabian StyleMallory, Michael L., Lisa C. Lindesmith, Paul D. Brewer-Jensen, Rachel L. Graham, and Ralph S. Baric. 2020. "Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains" Viruses 12, no. 9: 989. https://doi.org/10.3390/v12090989
APA StyleMallory, M. L., Lindesmith, L. C., Brewer-Jensen, P. D., Graham, R. L., & Baric, R. S. (2020). Bile Facilitates Human Norovirus Interactions with Diverse Histoblood Group Antigens, Compensating for Capsid Microvariation Observed in 2016–2017 GII.2 Strains. Viruses, 12(9), 989. https://doi.org/10.3390/v12090989