Impact of Viral Lysis on the Composition of Bacterial Communities and Dissolved Organic Matter in Deep-Sea Sediments


 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Origin of Sediments
2.2. Experimental Setup and Subsampling Procedure
2.3. Enumeration of Prokaryotic Cells and Virus-Like Particles
2.4. Nucleic Acid Extraction and Sequencing
2.5. Analysis of the Illumina Data Sets
2.6. Quantification of Amino Acids, Carbohydrates and Dissolved Organic Carbon
2.7. Molecular Characterization of Dissolved Organic Matter
2.8. Statistical Analyses of DOM Data Set
3. Results
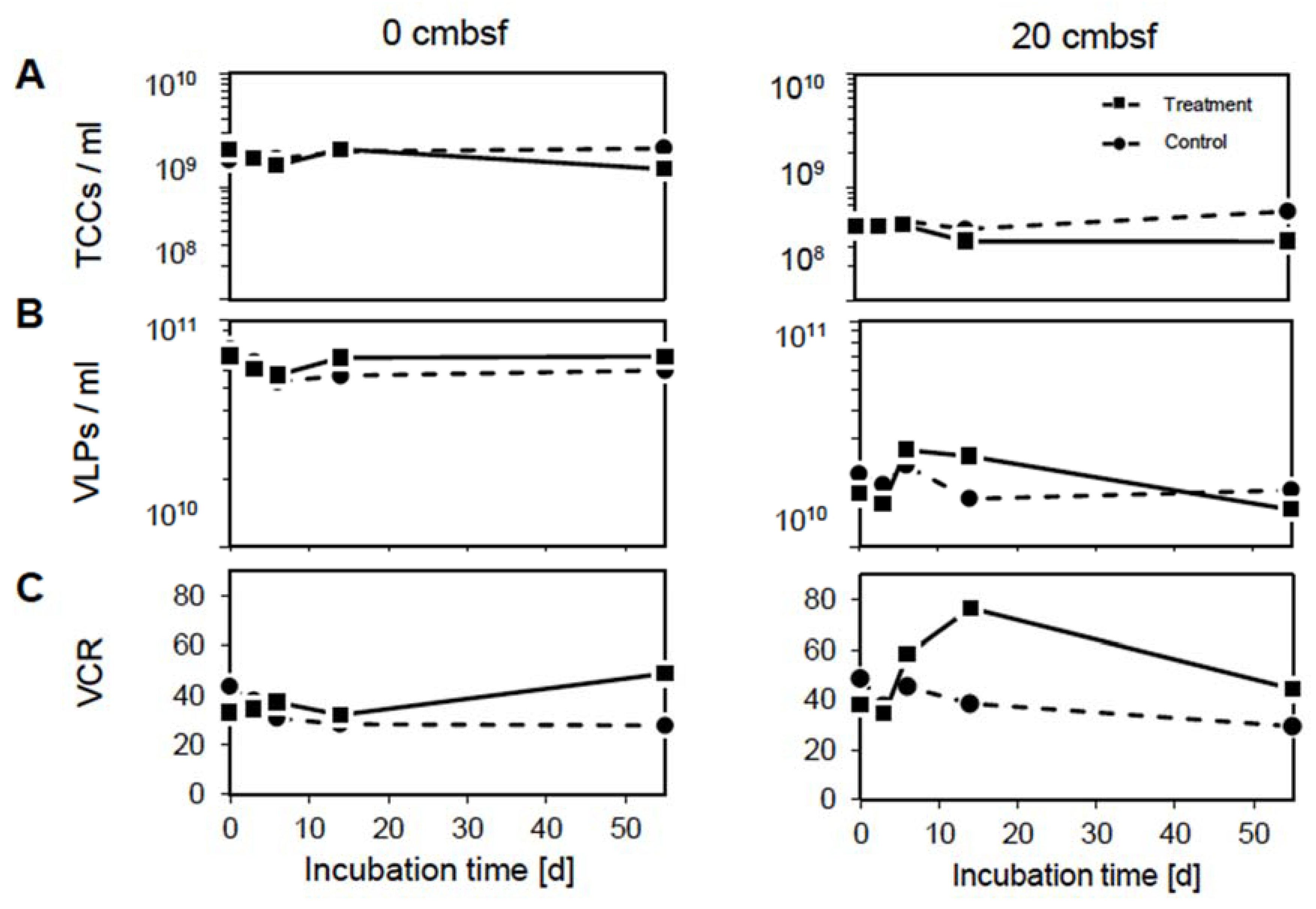
3.1. The Virus-to-Cell Ratio Indicates Prophage Induction at 20 cmbsf
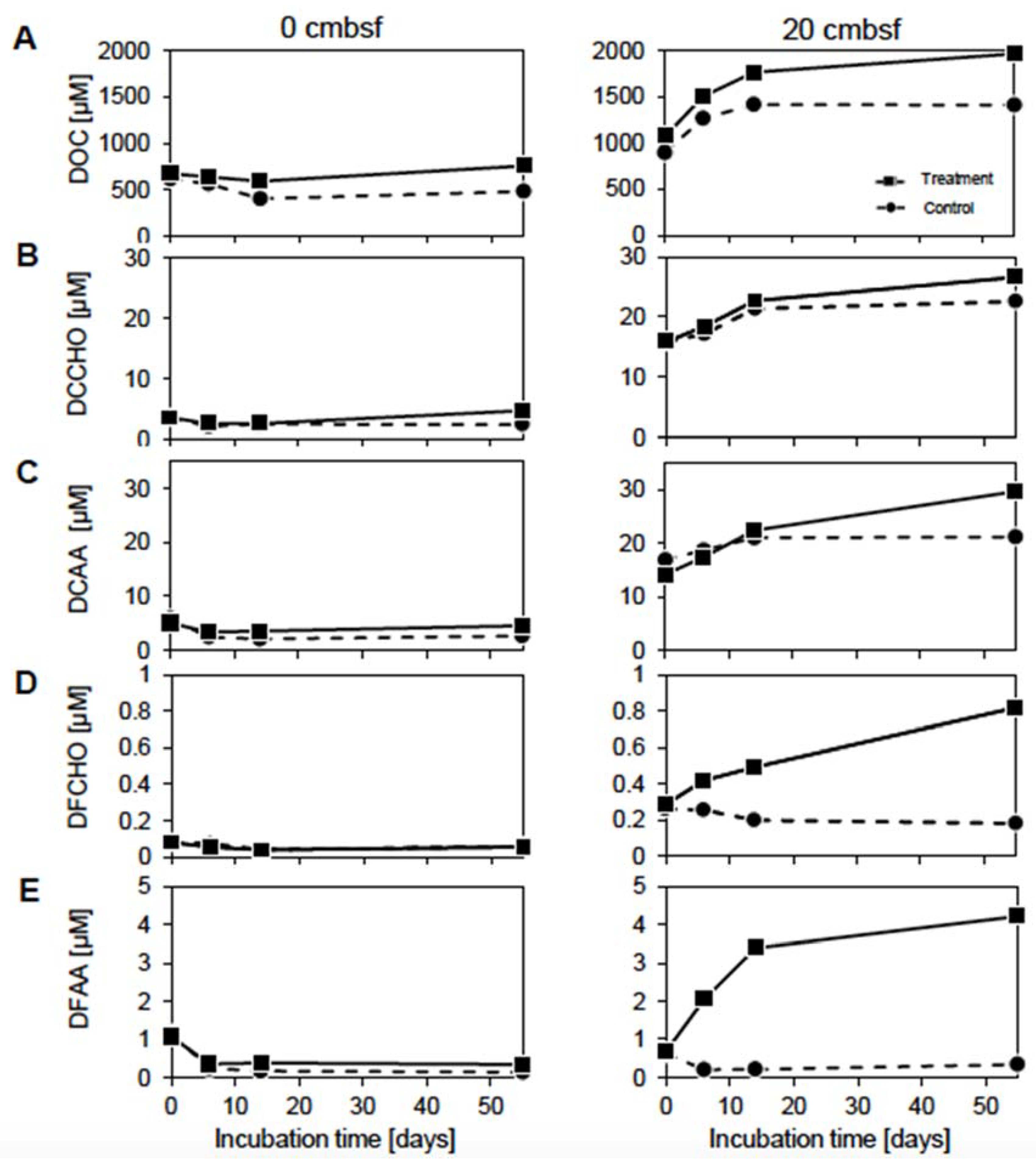
3.2. Prophage Induction Affects the DOM Pool
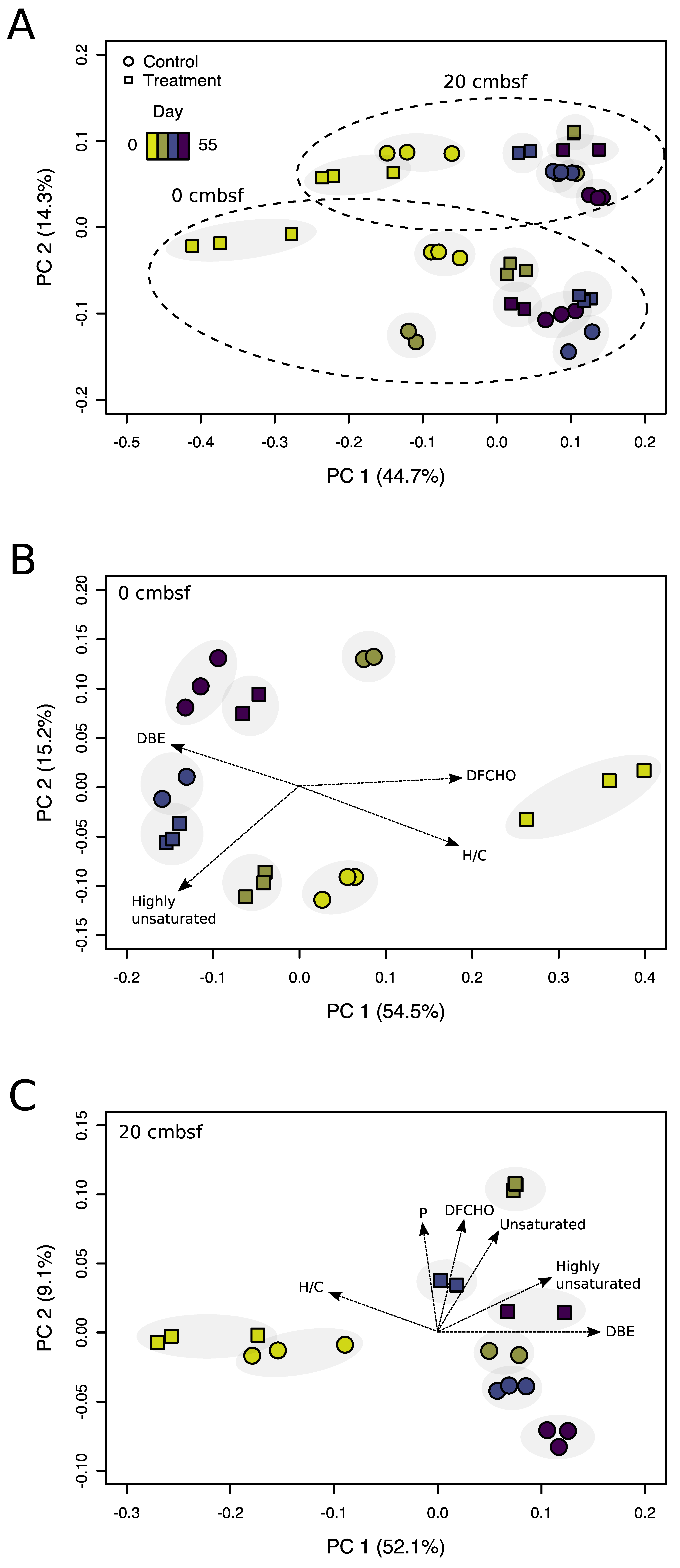
3.3. Virus-Induction in the 20 cmbsf Slurries Released Transient and Persistent DOM Components
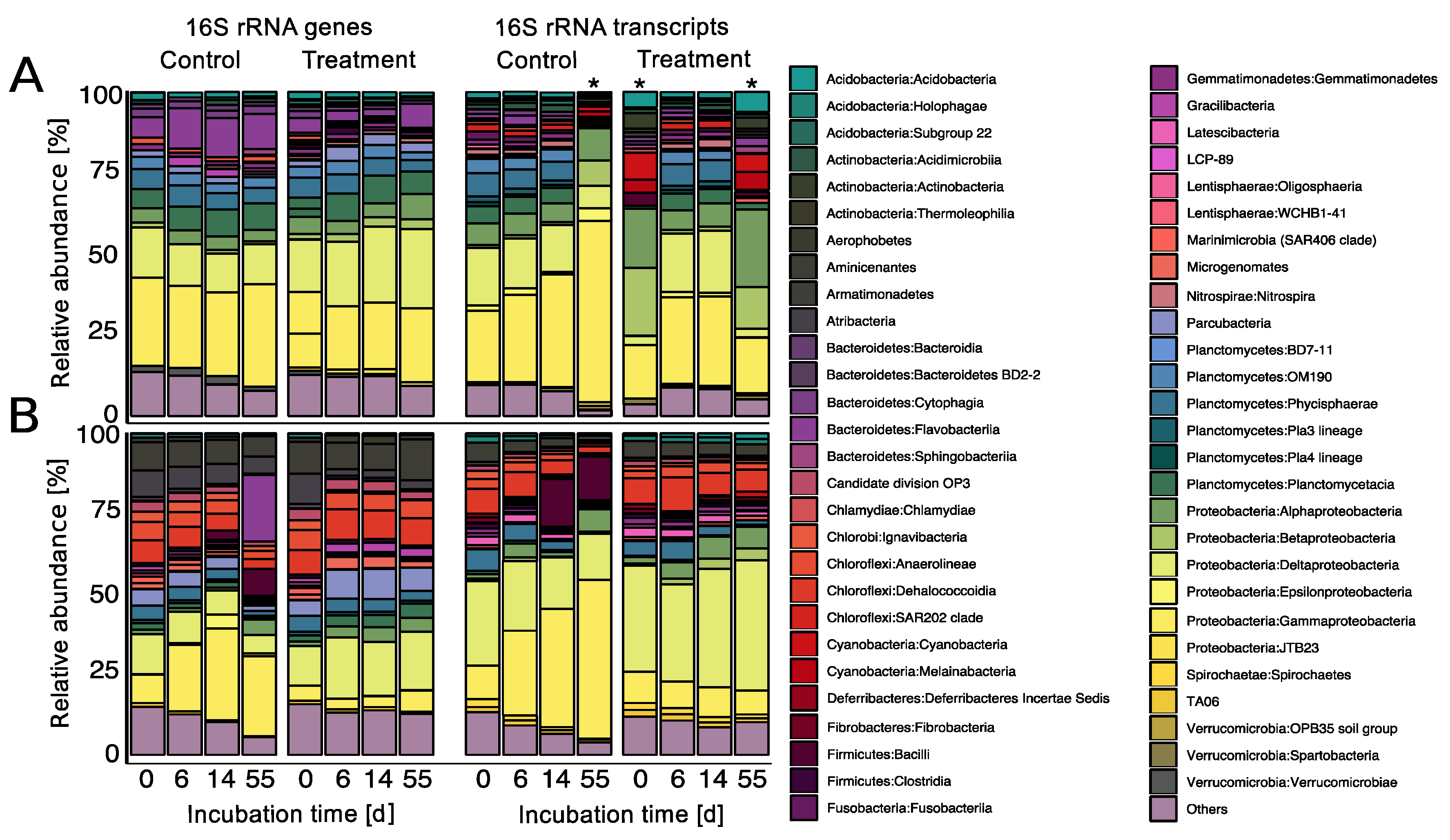
3.4. Prophage Induction Shaped the Bacterial Community Composition
4. Discussion
4.1. The Productivity of the Bering Sea Supports High Numbers of Benthic Bacteria and Viruses
4.2. Methodological Considerations
4.2.1. Experimental Setup
4.2.2. Potential Effects of the Mitomycin C Treatment
4.3. The Accumulation of Labile Organic Compounds Infers a Reduced Metabolic Potential of the Communities from 20 cmbsf
4.4. Viral Lysis and Subsequent Microbial Processes Were Imprinted in the DOM Composition
4.5. Prophage Induction Indicates a Predominance of Temperate Viruses in 20 cmbsf
4.6. Prophage Induction Stabilized Bacterial Community Composition According to the “Killing the Winner” Model
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kallmeyer, J.; Pockalny, R.; Adhikari, R.R.; Smith, D.C.; D’Hondt, S. Global distribution of microbial abundance and biomass in subseafloor sediment. Proc. Natl. Acad. Sci. USA 2012, 109, 16213–16216. [Google Scholar] [CrossRef] [PubMed]
- D’Hondt, S.; Pockalny, R.; Fulfer, V.M.; Spivack, A.J. Subseafloor life and its biogeochemical impacts. Nat. Commun. 2019, 10, 3519. [Google Scholar] [CrossRef] [PubMed]
- Orcutt, B.N.; Sylvan, J.B.; Knab, N.J.; Edwards, K.J. Microbial ecology of the dark ocean above, at, and below the seafloor. Microbiol. Mol. Biol. Rev. 2011, 75, 361–422. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Dell’Anno, A.; Corinaldesi, C.; Magagnini, M.; Noble, R.; Tamburini, C.; Weinbauer, M. Major viral impact on the functioning of benthic deep-sea ecosystems. Nature 2008, 454, 1084–1087. [Google Scholar] [CrossRef] [PubMed]
- Dell’Anno, A.; Corinaldesi, C.; Danovaro, R. Virus decomposition provides an important contribution to benthic deep-sea ecosystem functioning. Proc. Natl. Acad. Sci. USA 2015, 112, E2014–E2019. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea. BioScience 1999, 40, 781–788. [Google Scholar] [CrossRef]
- Ackermann, H.W.; DuBow, M.S. Viruses of Prokaryotes: General Properties of Bacteriophages; CRC Press Inc.: Boca Raton, FL, USA, 1987; Volume 1, pp. 49–85. [Google Scholar]
- Touchon, M.; Bernheim, A.; Rocha, E.P. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016, 10, 2744–2754. [Google Scholar] [CrossRef]
- Nanda, A.M.; Thormann, K.; Frunzke, J. Impact of spontaneous prophage induction on the fitness of bacterial populations and host-microbe interactions. J. Bacteriol. 2015, 197, 410–419. [Google Scholar] [CrossRef]
- Paul, J.H. Prophages in marine bacteria: Dangerous molecular time bombs or the key to survival in the seas? ISME J. 2008, 2, 579–589. [Google Scholar] [CrossRef]
- Zhao, Z.; Gonsior, M.; Schmitt-Kopplin, P.; Zhan, Y.; Zhang, R.; Jiao, N.; Chen, F. Microbial transformation of virus-induced dissolved organic matter from picocyanobacteria: Coupling of bacterial diversity and DOM chemodiversity. ISME J. 2019, 13, 2551–2565. [Google Scholar] [CrossRef]
- Zark, M.; Christoffers, J.; Dittmar, T. Molecular properties of deep-sea dissolved organic matter are predictable by the central limit theorem: Evidence from tandem FT-ICR-MS. Mar. Chem. 2017, 191, 9–15. [Google Scholar] [CrossRef]
- Hansell, D.A.; Carlson, C.A.; Repeta, D.J.; Schlitzer, R. Dissolved organic matter in the ocean. Oceanography 2009, 22, 202–211. [Google Scholar] [CrossRef]
- Azam, F.; Malfatti, F. Microbial structuring of marine ecosystems. Nat. Rev. Microbiol. 2007, 5, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Tanoue, E. Dissolved organic matter in oceanic waters. J. Oceanogr. 2003, 59, 129–147. [Google Scholar] [CrossRef]
- Dunne, J.P.; Sarmiento, J.L.; Gnanadesikan, A. A synthesis of global particle export from the surface ocean and cycling through the ocean interior and on the seafloor. Glob. Biogeochem. Cycles 2007, 21, GB4006. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; D’Hondt, S. A starving majority deep beneath the seafloor. Science 2006, 314, 932–934. [Google Scholar] [CrossRef]
- Middelboe, M.; Jorgensen, N.O.G. Viral Lysis of Bacteria: An important source of dissolved amino acids and cell wall compounds. J. Mar. Biol. Assoc. UK 2006, 86, 605–612. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Waterbury, J.B.; Chisholm, S.W. Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature 2003, 424, 1047–1051. [Google Scholar] [CrossRef]
- Thingstad, T.F.; Lignell, R. Theoretical models for the control of bacterial growth rate, abundance, diversity and carbon demand. Aquat. Microb. Ecol. 1997, 13, 19–27. [Google Scholar] [CrossRef]
- Winter, C.; Bouvier, T.; Weinbauer, M.G.; Thingstad, T.F. Trade-offs between competition and defense specialists among unicellular planktonic organisms: The “killing the winner” hypothesis revisited. Microbiol. Mol. Biol. Rev. 2010, 74, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, T.; del Giorgio, P.A. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environ. Microbiol. 2007, 9, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Hewson, I.; Fuhrman, J.A. Viral impacts upon marine bacterioplankton assemblage composition. J. Mar. Biol. Assoc. UK 2006, 86, 577–589. [Google Scholar] [CrossRef]
- Chen, F.; Wang, K.; Stewart, J.; Belas, R. Induction of multiple prophages from a marine bacterium: A genomic approach. Appl. Environ. Microbiol. 2006, 72, 4995–5001. [Google Scholar] [CrossRef] [PubMed]
- Sambrotto, R.N.; Mordy, C.; Zeeman, S.I.; Stabeno, P.J.; Macklin, S.A. Physical forcing and nutrient conditions associated with patterns of Chl a and phytoplankton productivity in the southeastern Bering Sea during summer. Deep Sea Res. Part II Top. Stud. Oceanogr. 2008, 55, 1745–1760. [Google Scholar] [CrossRef]
- Takahashi, K. The Bering and Okhotsk Sea: Modern and past paleooceanographic changes and gateway impact. J. Asian Earth Sci. 1998, 16, 49–58. [Google Scholar] [CrossRef]
- Wehrmann, L.M.; Risgaard-Petersen, N.; Schrum, H.N.; Walsh, E.A.; Huh, Y.; Ikehara, M.; Pierre, C.; D’Hondt, S.; Ferdelman, T.G.; Ravelo, A.C.; et al. Coupled organic and inorganic carbon cycling in the deep subseafloor sediment of the northeastern Bering Sea Slope (IODP Exp. 323). Chem. Geol. 2011, 284, 251–261. [Google Scholar] [CrossRef]
- Weinbauer, M.G.; Fuks, D.; Peduzzi, P. Distribution of viruses and dissolved DNA along a coastal trophic gradient in the Northern Adriatic Sea. Appl. Environ. Microbiol. 1993, 59, 4074–4082. [Google Scholar] [CrossRef]
- Hertkorn, N.; Frommberger, M.; Witt, M.; Koch, B.P.; Schmitt-Kopplin, P.; Perdue, E.M. Natural organic matter and the event horizont of mass spectrometry. Anal. Chem. 2008, 80, 8908–8919. [Google Scholar] [CrossRef]
- Pohlner, M.; Degenhardt, J.; von Hoyningen-Huene, A.J.E.; Wemheuer, B.; Erlmann, N.; Schnetger, B.; Badewien, T.H.; Engelen, B. The biogeographical distribution of benthic roseobacter group members along a Pacific transect is structured by mutrient availability within the sediments and primary production in different oceanic provinces. Front. Microbiol. 2017, 8, 2550. [Google Scholar] [CrossRef]
- Zech, H.; Thole, S.; Schreiber, K.; Kalhofer, D.; Voget, S.; Brinkhoff, T.; Simon, M.; Schomburg, D.; Rabus, R. Growth phase-dependent global protein and metabolite profiles of Phaeobacter gallaeciensis strain DSM 17395, a member of the marine Roseobacter-clade. Proteomics 2009, 9, 3677–3697. [Google Scholar] [CrossRef] [PubMed]
- Lunau, M.; Lemke, A.; Walther, K.; Martens-Habbena, W.; Simon, M. An improved method for counting bacteria from sediments and turbid environments by epifluorescence microscopy. Environ. Microbiol. 2005, 7, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Danovaro, R.; Middelboe, M. Manual of Aquatic Viral Ecology; Wilhelm, S.W., Weinbauer, M.G., Suttle, C.A., Eds.; American Society of Limnology and Oceanography: Waco, TX, USA, 2010; Chapter 8; pp. 74–81. [Google Scholar]
- Suttle, C.A.; Fuhrman, J.A. Manual of Aquatic Viral Ecology; Wilhelm, S.W., Weinbauer, M.G., Suttle, C.A., Eds.; American Society of Limnology and Oceanography: Waco, TX, USA, 2010; Chapter 15; pp. 145–153. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Granzow, S.; Kaiser, K.; Wemheuer, B.; Pfeiffer, B.; Daniel, R.; Vidal, S.; Wemheuer, F. The effects of cropping regimes on fungal and bacterial communities of wheat and faba bean in a greenhouse pot experiment differ between plant species and compartment. Front. Microbiol. 2017, 8, 902. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017; Available online: http://www.R-project.org (accessed on 30 July 2020).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Ordination Methods, Diversity Analysis and Other Functions for Community and Vegetation Ecologists Version 2.4-3. 2017. Available online: https://cran.r-project.org/web/packages/vegan (accessed on 5 March 2020).
- Lindroth, P.; Mopper, K. High performance liquid chromatographic determination of subpicomole amounts of amino acids by precolumn fluorescence derivatization with o-Phthaldialdehyde. Anal. Chem. 1979, 51, 1667–1674. [Google Scholar] [CrossRef]
- Lunau, M.; Lemke, A.; Dellwig, O.; Simon, M. Physical and biogeochemical controls of microaggregate dynamics in a tidally affected coastal ecosystem. Limnol. Oceanogr. 2006, 51, 847–859. [Google Scholar] [CrossRef]
- Mopper, K.; Schultz, C.A.; Chevolot, L.; Germain, C.; Revuelta, R.; Dawson, R. Determination of sugars in unconcentrated seawater and other natural waters by liquid chromatography and pulsed amperometric detection. Environ. Sci. Technol. 1992, 26, 133–138. [Google Scholar] [CrossRef]
- Dittmar, T.; Koch, B.P.; Hertkorn, N.; Kattner, G. A simple and efficient method for the solid-phase extraction of dissolved organic matter (SPE-DOM) from seawater. Limnol. Oceanogr. Meth. 2008, 6, 230–235. [Google Scholar] [CrossRef]
- Seidel, M.; Beck, M.; Riedel, T.; Waska, H.; Suryaputra, I.G.N.A.; Schnetger, B.; Niggemann, J.; Simon, M.; Dittmar, T. Biogeochemistry of dissolved organic matter in an anoxic intertidal creek bank. Geochim. Cosmochim. Acta 2014, 140, 418–434. [Google Scholar] [CrossRef]
- Rossel, P.E.; Vähätalo, A.V.; Witt, M.; Dittmar, T. Molecular composition of dissolved organic matter from a wetland plant (Juncus effusus) after photochemical and microbial decomposition (1.25 yr): Common features with deep sea dissolved organic matter. Org. Geochem. 2013, 60, 62–71. [Google Scholar] [CrossRef]
- Koch, B.P.; Dittmar, T. From mass to structure: An aromaticity index for high-resolution mass data of natural organic matter. Rapid Commun. Mass. Spectrom. 2006, 20, 926–932. [Google Scholar] [CrossRef]
- Hertkorn, M.; Benner, R.; Frommberger, M.; Schmitt-Kopplin, P.; Witt, M.; Kaiser, K.; Kettrup, A.; Hedges, J.I. Characterization of a major refractory component of marine dissolved organic matter. Geochim. Cosmochim. Acta 2006, 70, 2990–3010. [Google Scholar] [CrossRef]
- Riedel, T.; Dittmar, T. A method detection limit for the analysis of natural organic matter via Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2014, 86, 8376–8382. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Legendre, P.; Gallagher, E.D. Ecologically meaningful transformations for ordination of species data. Oecologia 2001, 129, 271–280. [Google Scholar] [CrossRef]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of Southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Kim, S.; Kramer, R.W.; Hatcher, P.G. Graphical method for analysis of ultrahigh-resolution broadband mass spectra of natural organic matter, the van Krevelen diagram. Anal. Chem. 2003, 75, 5336–5344. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.R.; Berelson, W.; Demaster, D.J.; Dobbs, F.C.; Hammond, D.; Hoover, D.J.; Pope, R.H.; Stephens, M. Latitudinal variations in benthic processes in the abyssal Equatorial Pacific: Control by biogenic particle flux. Deep Sea Res. Part II 1997, 44, 2295–2317. [Google Scholar] [CrossRef]
- Parkes, R.J.; Cragg, B.A.; Wellsbury, P. Recent studies on bacterial populations and processes in subseafloor sediments: A Review. Hydrogeol. J. 2000, 8, 11–28. [Google Scholar] [CrossRef]
- Bird, D.F.; Juniper, S.K.; Ricciardi-Rigault, M.; Martineu, P.; Prairie, Y.T.; Calvert, S.E. Subsurface viruses and bacteria in Holocene/Late Pleistocene sediments of Saanich Inlet, BC: ODP Holes 1033B and 1034B, Leg 169S. Mar. Geol. 2001, 174, 227–239. [Google Scholar] [CrossRef]
- Jiang, S.C.; Paul, J.H. Occurrence of lysogenic bacteria in marine microbial communities as determined by prophage induction. Mar. Ecol. Prog. Ser. 1996, 142, 27–38. [Google Scholar] [CrossRef]
- Tomasz, M. Mitomycin C: Small, fast and deadly (but very selective). Chem. Biol. 1995, 2, 575–579. [Google Scholar] [CrossRef]
- Novitsky, J.A. Degradation of dead microbial biomass in a marine sediment. Appl. Environ. Microbiol. 1986, 52, 504–509. [Google Scholar] [CrossRef]
- Noll, D.M.; McGregor, M.T.; Miller, P.S. Formation and repair of interstrand cross-links in DNA. Chem. Rev. 2006, 106, 277–301. [Google Scholar] [CrossRef]
- Jørgensen, B.B.; Marshall, I.P. Slow microbial life in the seabed. Ann. Rev. Mar. Sci. 2016, 8, 311–332. [Google Scholar] [CrossRef]
- Ogawa, H.; Amagai, Y.; Koike, I.; Kaiser, K.; Benner, R. Production of refractory dissolved organic matter by bacteria. Science 2001, 292, 917–920. [Google Scholar] [CrossRef]
- Hansen, J.W.; Thamdrup, B.; Jørgensen, B.B. Anoxic incubation of sediment in gas-tight plastic bags: A method for biogeochemical process studies. Mar. Ecol. Prog. Ser. 2000, 208, 273–282. [Google Scholar] [CrossRef]
- McKew, B.A.; Dumbrell, A.J.; Taylor, J.D.; McGenity, T.J.; Underwood, G.J. Differences between aerobic and anaerobic degradation of microphytobenthic biofilm-derived organic matter within intertidal sediments. FEMS Microbiol. Ecol. 2013, 84, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.Z.; Ting, L.; Ertan, H.; Johnson, J.; et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar] [CrossRef]
- Cai, R.; Zhou, W.; He, C.; Tang, K.; Guo, W.; Shi, Q.; Gonsior, M.; Jiao, N. Microbial processing of sediment-derived dissolved organic matter: Implications for its subsequent biogeochemical cycling in overlying seawater. J. Geophys. Res. Biogeosciences 2019, 124, 3479–3490. [Google Scholar] [CrossRef]
- Ma, X.; Coleman, M.L.; Waldbauer, J.R. Distinct molecular signatures in dissolved organic matter produced by viral lysis of marine cyanobacteria. Environ. Microbiol. 2018, 20, 3001–3011. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.P.; Witt, M.; Engbrodt, R.; Dittmar, T.; Kattner, G. Molecular formulae of marine and terrigenous dissolved organic matter detected by electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Geochim. Cosmochim. Acta 2005, 69, 3299–3308. [Google Scholar] [CrossRef]
- Raeke, J.; Lechtenfeld, O.J.; Wagner, M.; Herzsprung, P.; Reemtsma, T. Selectivity of solid phase extraction of freshwater dissolved organic matter and its effect on ultrahigh resolution mass spectra. Environ. Sci. Process. Impacts 2016, 18, 918–927. [Google Scholar] [CrossRef]
- Blackburn, J.W.T.; Kew, W.; Graham, M.C.; Uhrin, D. Laser desorption/ionization coupled to FT-ICR mass spectrometry for studies of natural organic matter. Anal. Chem. 2017, 89, 4382–4386. [Google Scholar] [CrossRef]
- Benner, R.; Amon, R.M. The size-reactivity continuum of major bioelements in the ocean. Ann. Rev. Mar. Sci. 2015, 7, 185–205. [Google Scholar] [CrossRef]
- Flerus, R.; Lechtenfeld, O.J.; Koch, B.P.; McCallister, S.L.; Schmitt-Kopplin, P.; Benner, R.; Kaiser, K.; Kattner, G. A molecular perspective on the ageing of marine dissolved organic matter. Biogeosciences 2012, 9, 1935–1955. [Google Scholar] [CrossRef]
- Jover, L.F.; Effler, T.C.; Buchan, A.; Wilhelm, S.W.; Weitz, J.S. The elemental composition of virus particles: Implications for marine biogeochemical cycles. Nat. Rev. Microbiol. 2014, 12, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.T.; Fuhrman, J.A. Breakdown and microbial uptake of marine viruses and other lysis products. Aquat. Microb. Ecol. 1999, 20, 1–11. [Google Scholar] [CrossRef][Green Version]
- Karl, D.M.; Björkman, K.M. Biogeochemistry of Marine Dissolved Organic Matter, 2nd ed.; Hansell, D.A., Carlson, C.A., Eds.; Academic Press: New York, NY, USA, 2015; Chapter 5; pp. 233–334. [Google Scholar] [CrossRef]
- Wikner, J.; Vallino, J.J.; Steward, G.F.; Smith, D.C.; Azam, F. Nucleic acids from the host bacterium as a major source of nucleotides for three marine bacteriophages. FEMS Microbiol. Ecol. 1993, 12, 237–248. [Google Scholar] [CrossRef]
- Zeng, Q.; Chisholm, S.W. Marine viruses exploit their host’s two-component regulatory system in response to resource limitation. Curr. Biol. 2012, 22, 124–128. [Google Scholar] [CrossRef]
- Lin, P.; Guo, L.; Chen, M.; Tong, J.; Lin, F. The distribution and chemical speciation of dissolved and particulate phosphorus in the Bering Sea and the Chukchi–Beaufort Seas. Deep Sea Res. Part II Top. Stud. Oceanogr. 2012, 81, 79–94. [Google Scholar] [CrossRef]
- Engelhardt, T.; Kallmeyer, J.; Cypionka, H.; Engelen, B. High virus-to-cell ratios indicate ongoing production of viruses in deep subsurface sediments. ISME J. 2014, 8, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, T.; Sahlberg, M.; Cypionka, H.; Engelen, B. Induction of prophages from deep-subseafloor bacteria. Environ. Microbiol. Rep. 2011, 3, 459–465. [Google Scholar] [CrossRef]
- Walsh, E.A.; Kirkpatrick, J.B.; Rutherford, S.D.; Smith, D.C.; Sogin, M.; D’Hondt, S. Bacterial diversity and community composition from seasurface to subseafloor. ISME J. 2016, 10, 979–989. [Google Scholar] [CrossRef]
- Ferguson, R.L.; Buckley, E.N.; Palumbo, A.V. Response of marine bacterioplankton to differential filtration and confinement. Appl. Environ. Microbiol. 1984, 47, 49–55. [Google Scholar] [CrossRef]
- Pernthaler, J.; Amann, R. Fate of heterotrophic microbes in pelagic habitats: Focus on populations. Microbiol. Mol. Biol. Rev. 2005, 69, 440–461. [Google Scholar] [CrossRef]
- Katsuki, K.; Takahashi, K. Diatoms as paleoenvironmental proxies for seasonal productivity, sea-ice and surface circulation in the Bering Sea during the late Quaternary. Deep Sea Res. Part II Top. Stud. Oceanogr. 2005, 52, 2110–2130. [Google Scholar] [CrossRef]
- Thingstad, T.F.; Vage, S.; Storesund, J.E.; Sandaa, R.A.; Giske, J. A theoretical analysis of how strain-specific viruses can control microbial species diversity. Proc. Natl. Acad. Sci. USA 2014, 111, 7813–7818. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Tan, S.; Xu, J.; Guo, W.; Xia, X.; Cheung, S.Y. Interactive regulations by viruses and dissolved organic matter on the bacterial community. Limnol. Oceanogr. 2017, 62, S364–S380. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Incubation | Day | No. of OTUs | Shannon Diversity Index | Pielou’s Evenness | ||
|---|---|---|---|---|---|---|
| 0 cmbsf | DNA | Control | 0 | 3019 ± 27 | 6.9 ± 2.6 | 0.93 ± 0.13 |
| 6 | 2956 ± 29 | 6.8 ± 2.6 | 0.91 ± 0.12 | |||
| 14 | 2817 ± 24 | 6.7 ± 2.4 | 0.90 ± 0.12 | |||
| 55 | 2691 ± 28 | 6.8 ± 2.6 | 0.89 ± 0.12 | |||
| Treatment | 0 | 2962 ± 25 | 7.0 ± 2.6 | 0.93 ± 0.12 | ||
| 6 | 3153 ± 22 | 7.0 ± 2.4 | 0.94 ± 0.12 | |||
| 14 | 2928 ± 23 | 6.8 ± 2.5 | 0.92 ± 0.13 | |||
| 55 | 2642 ± 29 | 6.3 ± 2.2 | 0.86 ± 0.12 | |||
| RNA | Control | 0 | 3503 ± 19 | 7.1 ± 2.1 | 0.95 ± 0.13 | |
| 6 | 3426 ± 19 | 7.0 ± 2.3 | 0.94 ± 0.13 | |||
| 14 | 2947 ± 21 | 6.4 ± 2.1 | 0.86 ± 0.11 | |||
| 55 | 296 ± 0 * | 4.4 ± 0 | 0.59 ± 0.08 | |||
| Treatment | 0 | 123 ± 2 * | 3.9 ± 0 | 0.54 ± 0.09 | ||
| 6 | 3615 ± 17 | 7.0 ± 2.2 | 0.94 ± 0.13 | |||
| 14 | 3020 ± 22 | 6.7 ± 2.2 | 0.91 ± 0.13 | |||
| 55 | 247 ± 1 * | 4.7 ± 0 | 0.62 ± 0.08 | |||
| 20 cmbsf | DNA | Control | 0 | 2702 ± 25 | 6.4 ± 1.9 | 0.86 ± 0.12 |
| 6 | 2455 ± 25 | 6.0 ± 1.8 | 0.81 ± 0.12 | |||
| 14 | 2131 ± 26 | 5.5 ± 1.3 | 0.73 ± 0.10 | |||
| 55 | 1500 ± 20 | 4.4 ± 0.8 | 0.59 ± 0.08 | |||
| Treatment | 0 | 2747 ± 22 | 6.4 ± 2.3 | 0.87 ± 0.12 | ||
| 6 | 2605 ± 22 | 6.6 ± 2.2 | 0.89 ± 0.11 | |||
| 14 | 2629 ± 25 | 6.7 ± 2.4 | 0.89 ± 0.12 | |||
| 55 | 2465 ± 18 | 6.3 ± 2.0 | 0.84 ± 0.11 | |||
| RNA | Control | 0 | 2261 ± 22 | 6.4 ± 2.2 | 0.84 ± 0.09 | |
| 6 | 1986 ± 19 | 5.7 ± 1.5 | 0.76 ± 0.1 | |||
| 14 | 1490 ± 18 | 4.7 ± 0.8 | 0.63 ± 0.09 | |||
| 55 | 792 ± 12 | 3.7 ± 0 | 0.50 ± 0.07 | |||
| Treatment | 0 | 1943 ± 17 | 6.2 ± 2 | 0.83 ± 0.11 | ||
| 6 | 1940 ± 22 | 6.2 ± 2.1 | 0.84 ± 0.12 | |||
| 14 | 973 ± 9 | 5.6 ± 1 | 0.75 ± 0.09 | |||
| 55 | 833 ± 5 | 5.4 ± 0.7 | 0.75 ± 0.11 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heinrichs, M.E.; Tebbe, D.A.; Wemheuer, B.; Niggemann, J.; Engelen, B. Impact of Viral Lysis on the Composition of Bacterial Communities and Dissolved Organic Matter in Deep-Sea Sediments. Viruses 2020, 12, 922. https://doi.org/10.3390/v12090922
Heinrichs ME, Tebbe DA, Wemheuer B, Niggemann J, Engelen B. Impact of Viral Lysis on the Composition of Bacterial Communities and Dissolved Organic Matter in Deep-Sea Sediments. Viruses. 2020; 12(9):922. https://doi.org/10.3390/v12090922
Chicago/Turabian StyleHeinrichs, Mara E., Dennis A. Tebbe, Bernd Wemheuer, Jutta Niggemann, and Bert Engelen. 2020. "Impact of Viral Lysis on the Composition of Bacterial Communities and Dissolved Organic Matter in Deep-Sea Sediments" Viruses 12, no. 9: 922. https://doi.org/10.3390/v12090922
APA StyleHeinrichs, M. E., Tebbe, D. A., Wemheuer, B., Niggemann, J., & Engelen, B. (2020). Impact of Viral Lysis on the Composition of Bacterial Communities and Dissolved Organic Matter in Deep-Sea Sediments. Viruses, 12(9), 922. https://doi.org/10.3390/v12090922