Novel Escherichia coli RNA Polymerase Binding Protein Encoded by Bacteriophage T5

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phage and Plasmids
2.2. Isolation of RNAP Core
2.3. Trypsin Digestion and Mass Spectrometry
2.4. Cloning, Expression, and Purification of gpT5.026
2.5. Far-Western Blotting Analysis
2.6. Bacterial Two-Hybrid System
2.7. Obtaining T5 Phage with Amber Mutation in Gene T5.026
2.8. One-Step Growth Experiment
2.9. Primer Extension Reactions
2.10. Fluorometric Assays
3. Results
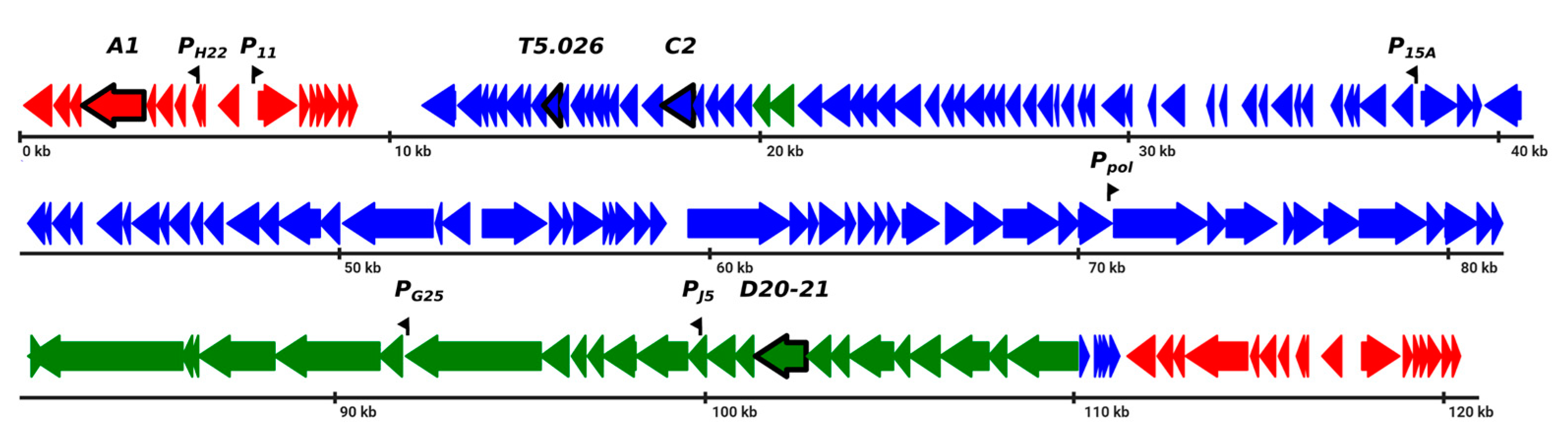
3.1. Detection and Identification of Proteins Associated with host RNAP in T5-Infected Cells
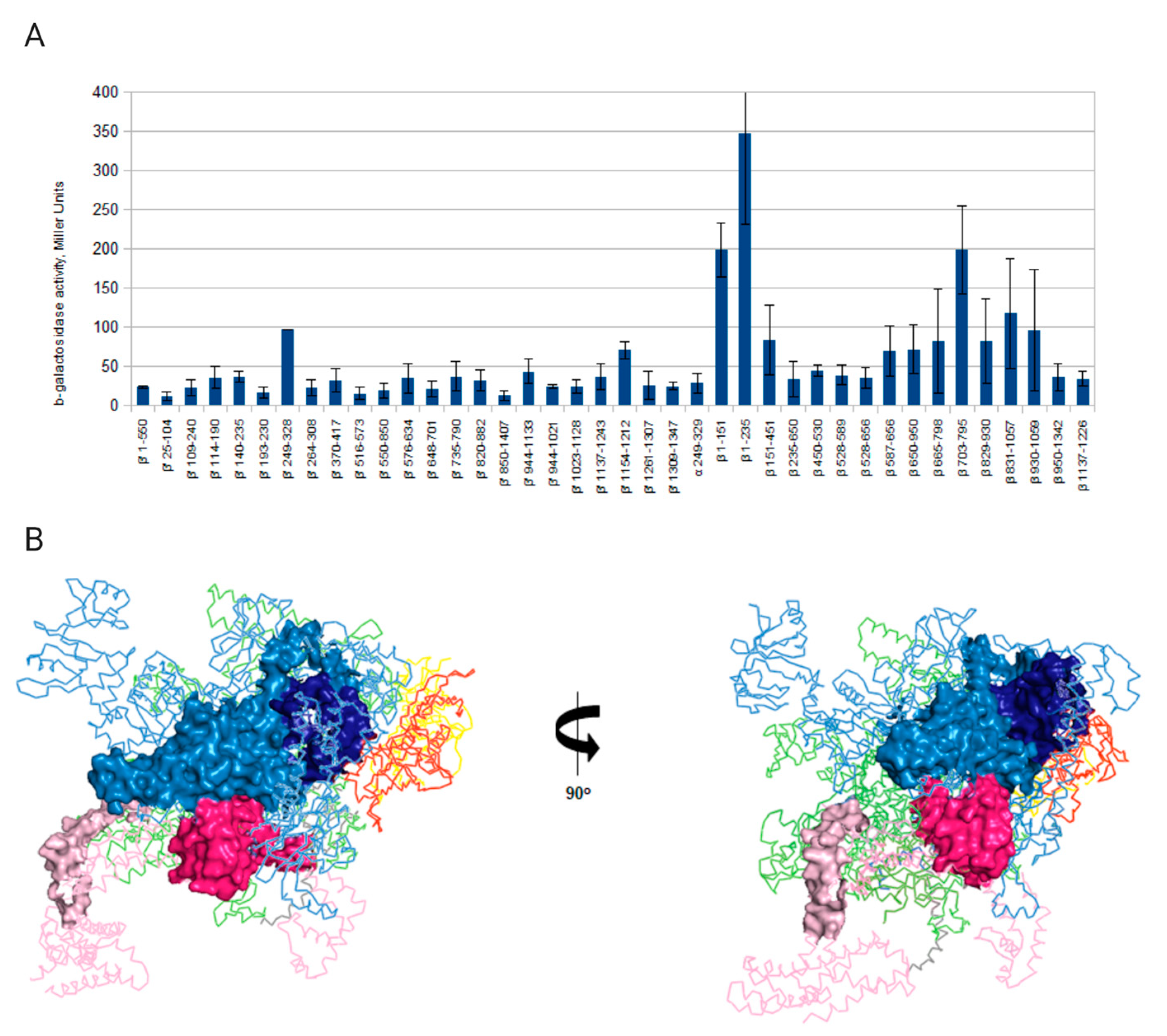
3.2. Interaction of gpT5.026 with RNAP Core Enzyme
3.3. The Role of gpT5.026 in T5 Transcription Regulation in Vivo
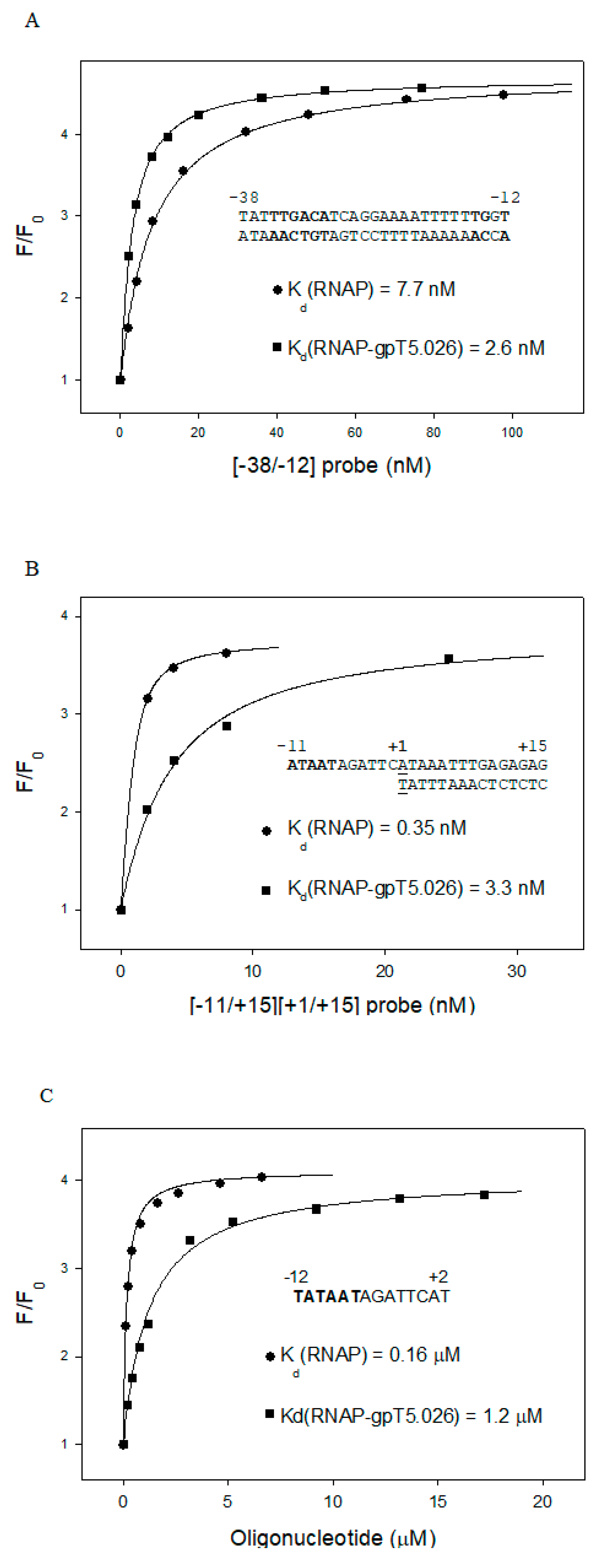
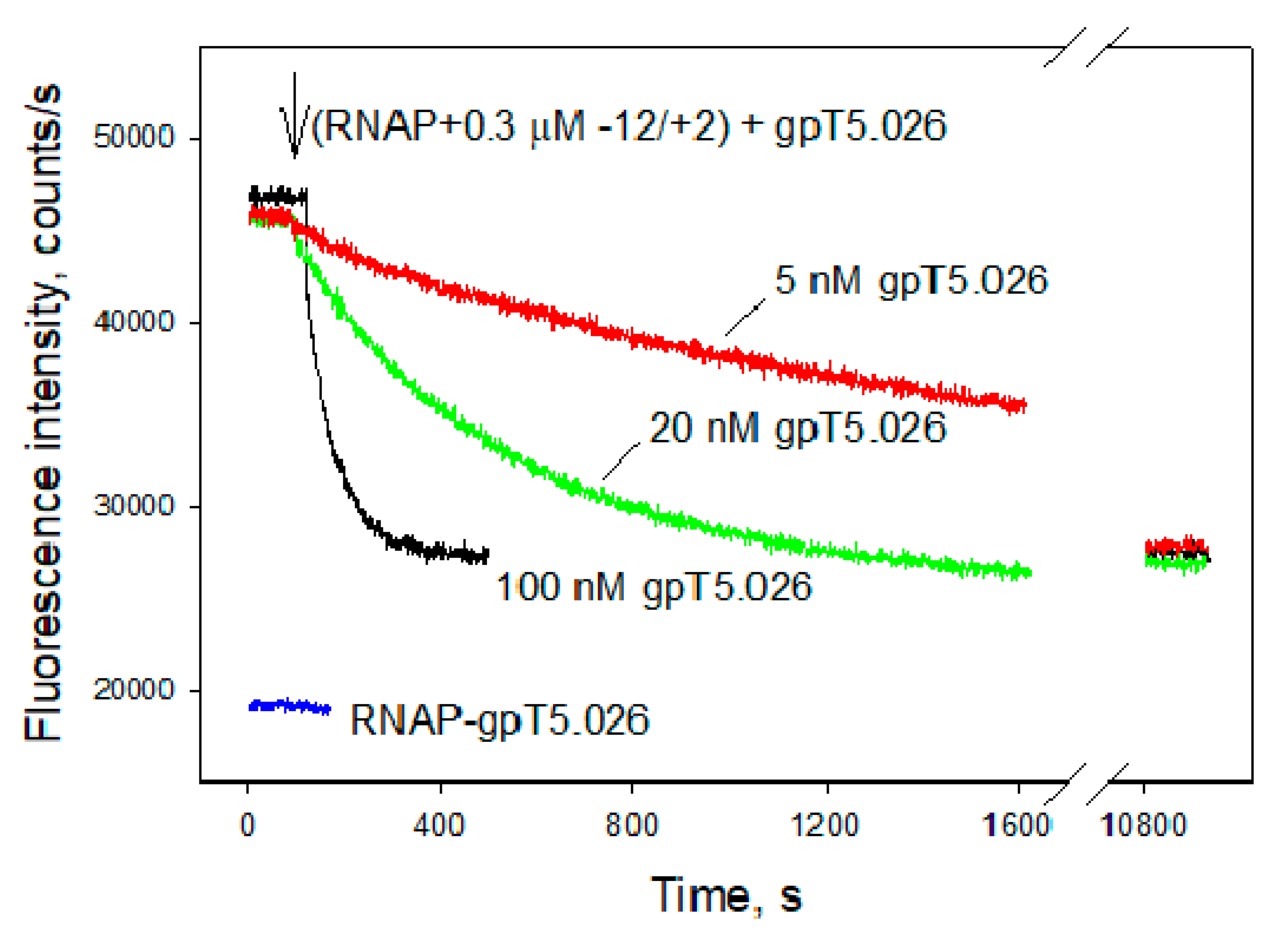
3.4. Influence of gpT5.026 on the DNA Binding Properties of the σ70 RNAP Holoenzyme, and Quantitative Analysis of gpT5.026–RNAP Interaction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lanni, T.Y. First-Step-Transfer Deoxyribonucleic Acid of Bacteriophage T5. Bacteriol. Rev. 1968, 32, 227–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, J.R. Bacteriophage T5. In The Bacteriophages; Calendar, R., Ed.; Oxford University Press: Oxford, UK, 2006; pp. 268–276. ISBN 0-19-514850-9. [Google Scholar]
- Hendrickson, H.E.; Mccorquodale, D.J.; Hendrickson, H.E.; Mccorquodale, D.J.; Anderson, G.C. Genetic and Physiological Studies of Bacteriophage T5 I. An Expanded Genetic Miap of T51. J. Virol. 1971, 7, 612–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Talaer, J.Y.; Kermici, M.; Jeanteur, P. Isolation of Escherichia coli RNA polymerase binding sites on T5 and T7 DNA: Further evidence for sigma-dependent recognition of A-T-rich DNA sequences. Proc. Natl. Acad. Sci. USA 1973, 70, 2911–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stüber, D.; Delius, H.; Bujard, H. Electron microscopic analysis of in vitro transcriptional complexes: Mapping of promoters of the coliphage T5 genome. Mol. Gen. Genet. 1978, 166, 141–149. [Google Scholar] [CrossRef]
- Gentz, R.; Langner, A.; Chang, A.C.; Cohen, S.N.; Bujard, H. Cloning and analysis of strong promoters is made possible by the downstream placement of a RNA termination signal. Proc. Natl. Acad. Sci. USA 1981, 78, 4936–4940. [Google Scholar] [CrossRef] [Green Version]
- Ksenzenko, V.N.; Kamynina, T.P.; Pustoshilova, N.M.; Kryukov, V.M.; Bayev, A.A. Cloning of bacteriophage T5 promoters. Mol. Gen. Genet. 1982, 185, 520–522. [Google Scholar] [CrossRef]
- Brunel, F.; Thi, V.H.; Pilaete, M.F.; Davison, J. Transcription regulatory elements in the late region of bacteriophage T5 DNA. Nucleic Acids Res. 1983, 11, 7649–7658. [Google Scholar] [CrossRef]
- Rommens, J.; MacKnight, D.; Pomeroy-Cloney, L.; Jay, E. Gene expression: Chemical synthesis and molecular cloning of a bacteriophage T5 (T5P25) early promoter. Nucleic Acids Res. 1983, 11, 5921–5940. [Google Scholar] [CrossRef] [Green Version]
- Beckman, L.D.; Hoffman, M.S.; McCorquodale, D.J. Pre-early proteins of bacteriophage T5: Structure and function. J. Mol. Biol. 1971, 62, 551–564. [Google Scholar] [CrossRef]
- Snyder, C.E. Bacteriophage T5 Gene A2 Protein Alters the Outer Membrane of Escherichia coli. J. Bacteriol. 1984, 160, 1191–1195. [Google Scholar] [CrossRef] [Green Version]
- Snyder, C.E.; Benzinger, R.H. Second-Step Transfer of Bacteriophage T5 DNA: Purification and Characterization of the T5 Gene A2 Protein. J. Virol. 1981, 40, 248–257. [Google Scholar] [CrossRef] [Green Version]
- Mccorquodale, D.J.; Chen, C.W.; Joseph, M.K.; Woychik, R. Modification of RNA Polymerase from Escherichia coli by Pre-Early Gene Products of Bacteriophage T5. J. Virol. 1981, 40, 958–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, D.H.; Dunn, G.B.; Mccorquodale, D.J. Identification of the Gene Controlling the Synthesis of the Major Bacteriophage T5 Membrane Protein. J. Virol. 1976, 18, 542–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Moyer, R.W. Purification and Properties of a Bacteriophage T5-Modified Form of Escherichia coli RNA Polymerase. J. Virol. 1975, 15, 1042–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C.; Dharmgrongartama, B.; Moyer, R.W. The regulation of transcription in bacteriophage T5-infected Escherichia coli. Biochemistry 1975, 14, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Chinnadurai, G.; McCorquodale, D.J. Regulation of expression of late genes of bacteriophage T5. J. Virol. 1974, 13, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY USA, 1989; ISBN 13: 978-0879693091. [Google Scholar]
- Severinov, K.; Darst, S.A. A mutant RNA polymerase that forms unusual open promoter complexes. Proc. Natl. Acad. Sci. USA 1997, 94, 13481–13486. [Google Scholar] [CrossRef] [Green Version]
- Klimuk, E.; Akulenko, N.; Makarova, K.S.; Ceyssens, P.J.; Volchenkov, I.; Lavigne, R.; Severinov, K. Host RNA polymerase inhibitors encoded by φKMV-like phages of pseudomonas. Virology 2013, 436, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.R.; Arthur, T.M.; Pietz, B.C. Mapping protein-protein interaction domains using ordered fragment ladder far-Western analysis of hexahistidine-tagged fusion proteins. Methods Enzymol. 2000, 328, 141–157. [Google Scholar] [CrossRef]
- Cámara, B.; Liu, M.; Reynolds, J.; Shadrin, A.; Liu, B.; Kwok, K.; Simpson, P.; Weinzierl, R.; Severinov, K.; Cota, E.; et al. T7 phage protein Gp2 inhibits the Escherichia coli RNA polymerase by antagonizing stable DNA strand separation near the transcription start site. Proc. Natl. Acad. Sci. USA 2010, 107, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Goede, B.; Naji, S.; von Kampen, O.; Ilg, K.; Thomm, M. Protein-protein interactions in the archaeal transcriptional machinery: Binding studies of isolated RNA polymerase subunits and transcription factors. J. Biol. Chem. 2006, 281, 30581–30592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dove, S.L.; Hochschild, A. Bacterial two-hybrid analysis of interactions between region 4 of the sigma(70) subunit of RNA polymerase and the transcriptional regulators Rsd from Escherichia coli and AlgQ from Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 6413–6421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.H. Experiments in Molecular Genetics; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY USA, 1972. [Google Scholar]
- Lanni, Y.T. Invasion by bacteriophage T5. III. Stages revealed by changes in susceptibility of early complexes to abortive infection. Virology 1961, 15, 127–135. [Google Scholar] [CrossRef]
- Decker, K.; Krauel, V.; Meesmann, A.; Heller, K.J. Lytic conversion of Escherichia coli by bacteriophage T5: Blocking of the FhuA receptor protein by a lipoprotein expressed early during infection. Mol. Microbiol. 1994, 12, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Mekler, V.; Severinov, K. RNA polymerase molecular beacon as tool for studies of RNA polymerase-promoter interactions. Methods 2015, 86, 19–26. [Google Scholar] [CrossRef] [Green Version]
- Mekler, V.; Pavlova, O.; Severinov, K. Interaction of Escherichia coli RNA polymerase σ70 subunit with promoter elements in the context of free σ70, RNA polymerase holoenzyme, and the β′-σ70 complex. J. Biol. Chem. 2011, 286, 270–279. [Google Scholar] [CrossRef] [Green Version]
- McCorquodale, D.J.; Buchanan, J.M. Patterns of protein synthesis in T5-infected Escherichia coli. J. Biol. Chem. 1968, 243, 2550–2559. [Google Scholar]
- McCorquodale, D.J.; Lanni, Y.T. Patterns of protein synthesis in Escherichia coli infected by amber mutants in the first-step-transfer DNA of T5. J. Mol. Biol. 1970, 48, 133–143. [Google Scholar] [CrossRef]
- Lanni, Y.T. Functions of two genes in the first-step-transfer DNA of bacteriophage T5. J. Mol. Biol. 1969, 44, 173–183. [Google Scholar] [CrossRef]
- Zweig, M.; Cummings, D.J. Structural proteins of bacteriophage T5. Virology 1973, 51, 443–453. [Google Scholar] [CrossRef]
- Zweig, M.; Cummings, D.J. Cleavage of head and tail proteins during bacteriophage T5 assembly: Selective host involvement in the cleavage of a tail protein. J. Mol. Biol. 1973, 80, 505–518. [Google Scholar] [CrossRef]
- Nechaev, S.; Severinov, K. Inhibition of Escherichia coli RNA Polymerase by Bacteriophage T7 Gene 2 Protein. J. Mol. Biol. 1999, 289, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Dove, S.L.; Joung, J.K.; Hochschild, A. Activation of prokaryotic transcription through arbitrary protein–protein contacts. Nature 1997, 386, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Bae, B.; Davis, E.; Brown, D.; Campbell, E.A.; Wigneshweraraj, S.; Darst, S.A. Phage T7 Gp2 inhibition of Escherichia coli RNA polymerase involves misappropriation of σ70 domain 1.1. Proc. Natl. Acad. Sci. USA 2013, 110, 19772–19777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentz, R.; Bujard, H. Promoters recognized by Escherichia coli RNA polymerase selected by function: Highly efficient promoters from bacteriophage T5. J. Bacteriol. 1985, 164, 70–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimuk, E.; Mekler, V.; Akulenko, N.; Severinov, K. Study of Transcription Regulation by gpT5.026; Institute of Molecular Genetics: Moscow, Russia, 2013. [Google Scholar]
- Marr, M.T.; Roberts, J.W. Promoter recognition as measured by binding of polymerase to nontemplate strand oligonucleotide. Science 1997, 276, 1258–1260. [Google Scholar] [CrossRef]
- Helmann, J.D.; DeHaseth, P.L. Protein-nucleic acid interactions during open complex formation investigated by systematic alteration of the protein and DNA binding partners. Biochemistry 1999, 38, 5959–5967. [Google Scholar] [CrossRef]
- Mekler, V.; Minakhin, L.; Severinov, K. A critical role of downstream RNA polymerase-promoter interactions in the formation of initiation complex. J. Biol. Chem. 2011, 286, 22600–22608. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, Y.; Chatterjee, S.; Tuske, S.; Ho, M.X.; Arnold, E.; Ebright, R.H. Structural basis of transcription initiation. Science 2012, 338, 1076–1080. [Google Scholar] [CrossRef] [Green Version]
- Haugen, S.P.; Berkmen, M.B.; Ross, W.; Gaal, T.; Ward, C.; Gourse, R.L. rRNA Promoter Regulation by Nonoptimal Binding of σ Region 1.2: An Additional Recognition Element for RNA Polymerase. Cell 2006, 125, 1069–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawley, D.K.; Mcclure, W.R. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 1983, 11, 2237–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Nucleotide Sequence (5’-3’) |
|---|---|
| A1 | CCTGAGAAGCTTTTTACAAAATACTCACCATC |
| A2 | ACTGGATAAACTAGTCGTTAAATTGGTTAT |
| B1 | GACTAGTTTATCCAGTTATTTGTCGAAACG |
| B2 | GGTTCCGGATCCGTTGTTAAAAATATTGAAAC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klimuk, E.; Mekler, V.; Lavysh, D.; Serebryakova, M.; Akulenko, N.; Severinov, K. Novel Escherichia coli RNA Polymerase Binding Protein Encoded by Bacteriophage T5. Viruses 2020, 12, 807. https://doi.org/10.3390/v12080807
Klimuk E, Mekler V, Lavysh D, Serebryakova M, Akulenko N, Severinov K. Novel Escherichia coli RNA Polymerase Binding Protein Encoded by Bacteriophage T5. Viruses. 2020; 12(8):807. https://doi.org/10.3390/v12080807
Chicago/Turabian StyleKlimuk, Evgeny, Vladimir Mekler, Darya Lavysh, Marina Serebryakova, Natalia Akulenko, and Konstantin Severinov. 2020. "Novel Escherichia coli RNA Polymerase Binding Protein Encoded by Bacteriophage T5" Viruses 12, no. 8: 807. https://doi.org/10.3390/v12080807
APA StyleKlimuk, E., Mekler, V., Lavysh, D., Serebryakova, M., Akulenko, N., & Severinov, K. (2020). Novel Escherichia coli RNA Polymerase Binding Protein Encoded by Bacteriophage T5. Viruses, 12(8), 807. https://doi.org/10.3390/v12080807