Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses


Abstract
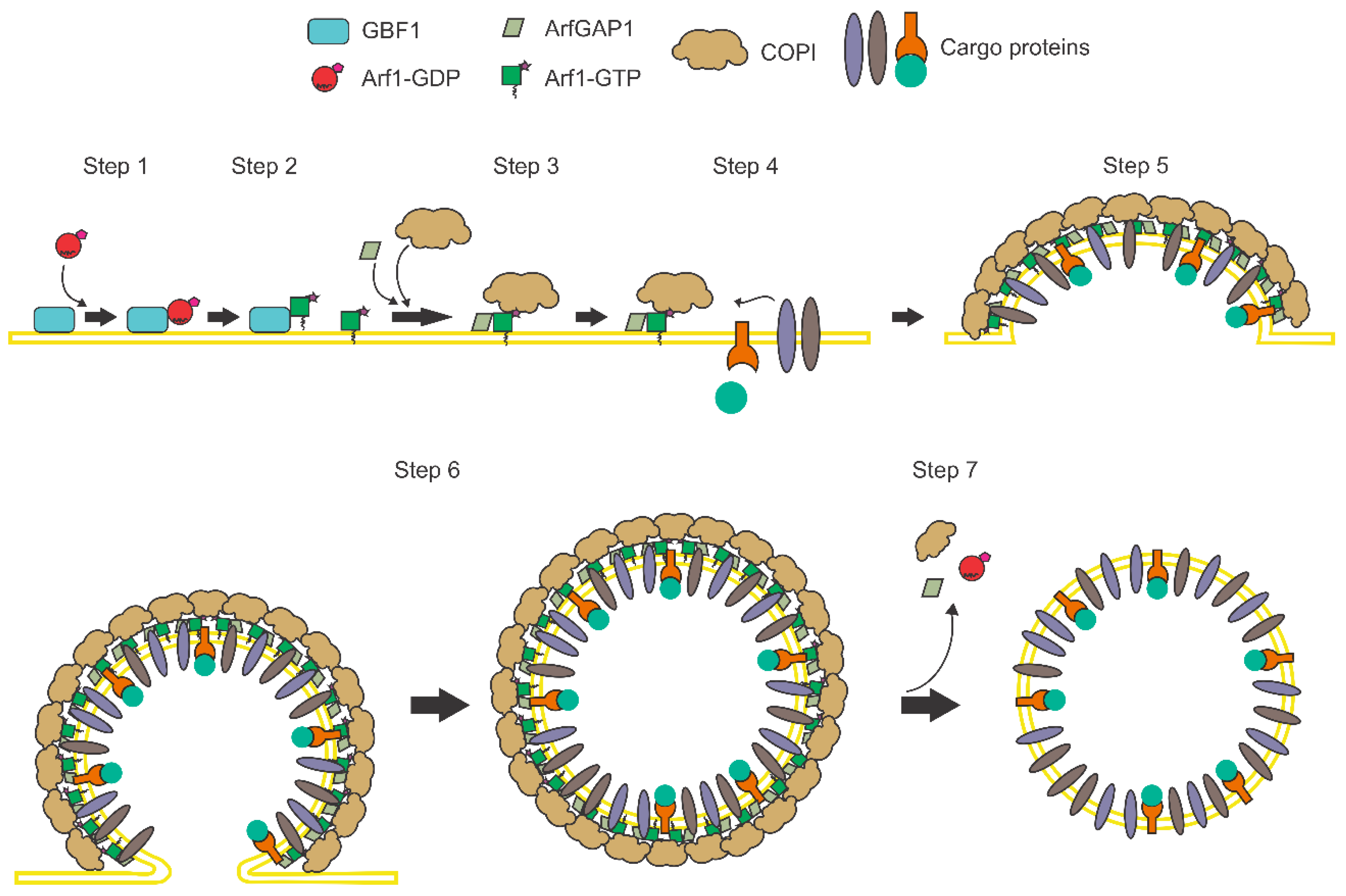
1. Introduction
2. Positive-Sense RNA Viruses
2.1. Family Flaviviridae
2.1.1. Flavivirus
2.1.2. Pestivirus
2.1.3. Hepacivirus
2.2. Family Picornaviridae
Enterovirus
2.3. Family Togaviridae
Alphavirus
2.4. Family Hepeviridae
2.5. Family Coronaviridae
3. Negative-Sense RNA Viruses
3.1. Family Rhabdoviridae
Vesiculovirus
3.2. Family Orthomyxoviridae
3.2.1. Influenza Virus
3.2.2. Other Negative-Sense RNA Viruses
4. Double-Stranded RNA Viruses
Family Reoviridae
Rotavirus
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Roberts, K.; Johnson, A.; Lewis, J.; Walter, P.; Raff, M.; Alberts, B. Intracellular Vesicular Traffic. In Molecular Biology of the Cell, 6th ed.; Alberts, B., Johnson, A., Lewis, J., Morgan, D., Raff, M., Roberts, K., Walter, P., Eds.; Garland Science: New York, NY, USA, 2019; pp. 749–812. [Google Scholar]
- Szul, T.; Grabski, R.; Lyons, S.; Morohashi, Y.; Shestopal, S.; Lowe, M.; Sztul, E. Dissecting the role of the Arf guanine nucleotide exchange factor GBF1 in Golgi biogenesis and protein trafficking. J. Cell Sci. 2007, 120, 3929–3940. [Google Scholar] [CrossRef] [PubMed]
- Manolea, F.; Claude, A.; Chun, J.; Rosas, J.; Melanc, P. Distinct functions for Arf guanine nucleotide exchange factors at the Golgi complex: GBF1 and BIGs are required for assembly and maintenance of the Golgi Stack and Trans- Golgi network, Respectively. Mol. Biol. Cell. 2008, 19, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Spang, A. Retrograde traffic from the Golgi to the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Boal, F.; Stephens, D.J. Specific functions of BIG1 and BIG2 in endomembrane organization. PLoS ONE 2010, 5, e9898. [Google Scholar] [CrossRef]
- Ward, T.H.; Polishchuk, R.S.; Caplan, S.; Hirschberg, K.; Lippincott-Schwartz, J. Maintenance of Golgi structure and function depends on the integrity of ER export. J. Cell Biol. 2001, 155, 557–570. [Google Scholar] [CrossRef]
- Joshi, A.S.; Zhang, H.; Prinz, W.A. Organelle biogenesis in the endoplasmic reticulum. Nat. Cell Biol. 2017, 19, 876–882. [Google Scholar] [CrossRef]
- Springer, S.; Spang, A.; Schekman, R. Primer on vesicle budding. Cell 1999, 97, 145–148. [Google Scholar] [CrossRef]
- Schekman, R.; Orci, L. Coat proteins and vesicle budding. Science 1996, 271, 1526–1533. [Google Scholar] [CrossRef]
- Rothman, J.E.; Wieland, F.T. Protein sorting by transport vesicles. Science 1996, 272, 227–234. [Google Scholar] [CrossRef]
- Kirchhausen, T. Three ways to make a vesicle. Nat. Rev. Mol. Cell Biol. 2000, 1, 187–198. [Google Scholar] [CrossRef]
- Wieland, F.; Hartert, C. Mechanisms of vesicle formation: Insights from the COP system. Curr. Opin. Cell Biol. 1999, 11, 440–446. [Google Scholar] [CrossRef]
- Barlowe, C.; Orci, L.; Yeung, T.; Hosobuchi, M.; Hamamoto, S.; Salama, N.; Rexach, M.F.; Ravazzola, M.; Amherdt, M.; Schekman, R. COPII: A membrane coat formed by set proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895–907. [Google Scholar] [CrossRef]
- Lee, M.C.S.; Miller, E.A.; Goldberg, J.; Orci, L.; Schekman, R. Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 2004, 20, 87–123. [Google Scholar] [CrossRef] [PubMed]
- Popoff, V.; Adolf, F.; Brugger, B.; Wieland, F. COPI budding within the Golgi stack. Cold Spring Harb. Perspect. Biol. 2011, 3, a005231. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.; Pepperkok, R.; Locker, J.K.; Kreis, T.E. Immunocytochemical localization of beta-COP to the ER-Golgi boundary and the TGN. J. Cell Sci. 1995, 108, 2839–2856. [Google Scholar]
- Duden, R.; Griffiths, G.; Frank, R.; Argos, P.; Kreis, T.E. Beta-COP, a 110 kd protein associated with non-clathrin-coated vesicles and the Golgi complex, shows homology to beta-adaptin. Cell 1991, 64, 649–665. [Google Scholar] [CrossRef]
- Puertollano, R. Clathrin-mediated transport: Assembly required. Workshop on molecular mechanisms of vesicle selectivity. EMBO Rep. 2004, 5, 942–946. [Google Scholar] [CrossRef]
- Gu, F.; Aniento, F.; Parton, R.G.; Gruenberg, J. Functional dissection of COP-I Subunits in the biogenesis of multivesicular endosomes. J Cell Biol. 1997, 139, 1183–1195. [Google Scholar] [CrossRef]
- Daro, E.; Sheff, D.; Gomez, M.; Kreis, T.; Mellman, I. Inhibition of endosome function in CHO Cells bearing a temperature-sensitive defect in the Coatomer (COPI) component-COP. J Cell Biol. 1997, 139, 1747–1759. [Google Scholar] [CrossRef]
- Botelho, R.J.; Hackam, D.J.; Schreiber, A.D.; Grinstein, S. Role of COPI in phagosome maturation. J. Biol. Chem. 2000, 275, 15717–15727. [Google Scholar] [CrossRef]
- Razi, M.; Chan, E.Y.W.; Tooze, S.A. Early endosomes and endosomal coatomer are required for autophagy. J. Cell Biol. 2009, 185, 305–321. [Google Scholar] [CrossRef] [PubMed]
- Lay, D.; Gorgas, K.; Just, W.W. Peroxisome biogenesis: Where Arf and coatomer might be involved. Biochim. Biophys. Acta–Mol. Cell Res. 2006, 1763, 1678–1687. [Google Scholar] [CrossRef] [PubMed]
- Beller, M.; Sztalryd, C.; Southall, N.; Bell, M.; Jäckle, H.; Auld, D.S.; Oliver, B. COPI complex is a regulator of lipid homeostasis. PLoS Biol. 2008, 6, 2530–2549. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Walther, T.C.; Rao, M.; Stuurman, N.; Goshima, G.; Terayama, K.; Wong, J.S.; Vale, R.D.; Walter, P.; Farese, R.V. Functional genomic screen reveals genes involved in lipid-droplet formation and utilization. Nature 2008, 453, 657–661. [Google Scholar] [CrossRef]
- Wilfling, F.; Thiam, A.R.; Olarte, M.-J.; Wang, J.; Beck, R.; Gould, T.J.; Allgeyer, E.S.; Pincet, F.; Bewersdorf, J.; Farese, R.V.; et al. Arf1/COPI machinery acts directly on lipid droplets and enables their connection to the ER for protein targeting. Elife 2014, 3, e01607. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Jackson, C.L. Arf family G proteins and their regulators: Roles in membrane transport, development and disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362–375. [Google Scholar] [CrossRef]
- Kahn, R.A.; Cherfils, J.; Elias, M.; Lovering, R.C.; Munro, S.; Schurmann, A. Nomenclature for the human Arf family of GTP-binding proteins: Arf, ARL, and SAR proteins. J. Cell Biol. 2006, 172, 645–650. [Google Scholar] [CrossRef]
- Chavrier, P.; Ménétrey, J. Toward a structural understanding of Arf family: Effector specificity. Structure 2010, 18, 1552–1558. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Li, Y.; Zhang, C.-J.; Kahn, R.A. Isoform-selective effects of the depletion of ADP- ribosylation factors 1–5 on membrane traffic. Mol. Biol. Cell 2005, 16, 4495–4508. [Google Scholar] [CrossRef]
- Antonny, B.; Beraud-Dufour, S.; Chardin, P.; Chabre, M. N-terminal hydrophobic residues of the G-protein ADP-ribosylation factor-1 insert into membrane phospholipids upon GDP to GTP exchange. Biochemistry 1997, 36, 4675–4684. [Google Scholar] [CrossRef]
- Hsu, V.W.; Yang, J.-S. Mechanisms of COPI vesicle formation. FEBS Lett. 2009, 583, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Beck, R.; Ravet, M.; Wieland, F.T.; Cassel, D.; Cassel, D. The COPI system: Molecular mechanisms and function. FEBS Lett. 2009, 583, 2701–2709. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, B.; Verbavatz, J.M.; Jackson, C.L. GBF1 and Arf1 function in vesicular trafficking, lipid homoeostasis and organelle dynamics. Biol. Cell 2017, 109, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, S.; Golinelli-Cohen, M.-P.; Contremoulins, V.; Jackson, C.L. Targeting of the Arf-GEF GBF1 to lipid droplets and Golgi membranes. J. Cell Sci. 2013, 126, 4794–4805. [Google Scholar] [CrossRef] [PubMed]
- Mossessova, E.; Corpina, R.A.; Goldberg, J. Crystal structure of Arf1*Sec7 complexed with brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol. Cell 2003, 12, 1403–1411. [Google Scholar] [CrossRef]
- Goldberg, J. Structural basis for activation of Arf GTPase: Mechanisms of guanine nucleotide exchange and GTP-myristoyl switching. Cell 1998, 95, 237–248. [Google Scholar] [CrossRef]
- Claude, A.; Zhao, B.-P.; Kuziemsky, C.E.; Dahan, S.; Berger, S.J.; Yan, J.-P.; Armold, A.D.; Sullivan, E.M.; Melançon, P. GBF1: A Novel Golgi-associated BFA-resistant Guanine Nucleotide Exchange Factor That displays specificity for ADP-ribosylation factor 5. J. Cell Biol. 1999, 146, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.; Pfeifer, A.C.; Lippincott-schwartz, J.; Jackson, C.L. Dynamics of GBF1, a brefeldin A-sensitive Arf1 Exchange Factor at the Golgi. Mol. Biol. Cell 2005, 16, 1213–1222. [Google Scholar] [CrossRef]
- Szul, T.; Garcia-Mata, R.; Brandon, E.; Shestopal, S.; Alvarez, C.; Sztul, E. Dissection of membrane dynamics of the Arf-guanine nucleotide exchange factor GBF1. Traffic 2005, 6, 374–385. [Google Scholar] [CrossRef]
- Anders, N.; Jürgens, G. Large Arf guanine nucleotide exchange factors in membrane trafficking. Cell. Mol. Life Sci. 2008, 65, 3433–3445. [Google Scholar] [CrossRef]
- Ellong, E.N.; Soni, K.G.; Bui, Q.T.; Sougrat, R.; Golinelli-Cohen, M.P.; Jackson, C.L. Interaction between the triglyceride lipase ATGL and the Arf1 activator GBF1. PLoS ONE 2011, 6, e21889. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.M.; Bhatt, J.M.; Lee, E.; Styers, M.L.; Ivanova, A.A.; Kahn, R.A.; Sztul, E. The Arf guanine nucleotide exchange factor GBF1 is targeted to Golgi membranes through a PIP-binding domain. J. Cell Sci. 2018, 131, jcs210245. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, X.; Yao, L.; Yan, L.; Zhang, L.; Qiu, J.; Liu, X.; Jia, S.; Meng, A. Impairment of cargo transportation caused by GBF1 mutation disrupts vascular integrity and causes hemorrhage in zebrafish embryos. J. Biol. Chem. 2017, 292, 2315–2327. [Google Scholar] [CrossRef] [PubMed]
- Pokrovskaya, I.D.; Szwedo, J.W.; Goodwin, A.; Lupashina, T.V.; Nagarajan, U.M.; Lupashin, V.V. Chlamydia trachomatis hijacks intra-Golgi COG complex-dependent vesicle trafficking pathway. Cell. Microbiol. 2012, 14, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Misselwitz, B.; Dilling, S.; Vonaesch, P.; Sacher, R.; Snijder, B.; Schlumberger, M.; Rout, S.; Stark, M.; von Mering, C.; Pelkmans, L.; et al. RNAi screen of Salmonella invasion shows role of COPI in membrane targeting of cholesterol and Cdc42. Mol. Syst. Biol. 2014, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- Sáenz, J.B.; Sun, W.J.; Chang, J.W.; Li, J.; Bursulaya, B.; Gray, N.S.; Haslam, D.B. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nat. Chem. Biol. 2009, 5, 157–165. [Google Scholar] [CrossRef]
- Iglesias, N.G.; Mondotte, J.A.; Byk, L.A.; De Maio, F.A.; Samsa, M.M.; Alvarez, C.; Gamarnik, A.V. Dengue virus uses a non-canonical function of the host GBF1-Arf-COPI system for capsid protein accumulation on Lipid Droplets. Traffic 2015, 16, 962–977. [Google Scholar] [CrossRef] [PubMed]
- Hafirassou, M.L.; Meertens, L.; Umaña-Diaz, C.; Labeau, A.; Dejarnac, O.; Bonnet-Madin, L.; Kümmerer, B.M.; Delaugerre, C.; Roingeard, P.; Vidalain, P.; et al. A Global Interactome Map of the dengue Virus NS1 Identifies Virus Restriction and Dependency Host Factors. Cell Rep. 2017, 21, 3900–3913. [Google Scholar] [CrossRef]
- Vonderstein, K.; Nilsson, E.; Hubel, P.; Skalman, L.N.; Upadhyay, A.; Pasto, J.; Pichlmair, A.; Lundmark, R.; Överby, A.K. Viperin targets flavivirus virulence by inducing assembly of noninfectious capsid particles. J. Virol. 2018, 92, e01751-17. [Google Scholar] [CrossRef]
- Ferlin, J.; Farhat, R.; Belouzard, S.; Cocquerel, L.; Bertin, A.; Hober, D.; Dubuisson, J.; Rouillé, Y. Investigation of the role of GBF1 in the replication of positive-sense single-stranded RNA viruses. J. Gen. Virol. 2018, 99, 1086–1096. [Google Scholar] [CrossRef]
- Mackenzie, J.M.; Jones, M.K.; Westaway, E.G. Markers for trans-Golgi membranes and the intermediate compartment localize to induced membranes with distinct replication functions in flavivirus-infected cells. J. Virol. 1999, 73, 9555–9567. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Zheng, M.; Bao, C.; Zhang, Y. CSFV proliferation is associated with GBF1 and Rab2. J. Biosci. 2017, 42, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Goueslain, L.; Alsaleh, K.; Horellou, P.; Roingeard, P.; Descamps, V.; Duverlie, G.; Ciczora, Y.; Wychowski, C.; Dubuisson, J.; Rouille, Y. Identification of GBF1 as a cellular factor required for hepatitis C virus RNA replication. J. Virol. 2010, 84, 773–787. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A Functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hong, Z.; Lin, W.; Shao, R.X.; Goto, K.; Hsu, V.W.; Chung, R.T. Arf1 and GBF1 generate a PI4P-enriched environment supportive of hepatitis C virus replication. PLoS ONE 2012, 7, e32135. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Ehrenfeld, E. Involvement of cellular membrane traffic proteins in poliovirus replication. Cell Cycle 2007, 6, 36–38. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Feng, Q.; Nikovics, K.; Jackson, C.L.; Ehrenfeld, E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 2008, 4, e1000216. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Kovtunovych, G.; Jackson, C.L.; Ehrenfeld, E. Poliovirus replication requires the N-terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cell. Microbiol. 2010, 12, 1463–1479. [Google Scholar] [CrossRef]
- Van der Linden, L.; van der Schaar, H.M.; Lanke, K.H.W.; Neyts, J.; van Kuppeveld, F.J.M. Differential Effects of the putative GBF1 inhibitors Golgicide A and AG1478 on enterovirus replication. J. Virol. 2010, 84, 7535–7542. [Google Scholar] [CrossRef]
- Lanke, K.H.W.; van der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; van Kuppeveld, F.J.M. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for Coxsackievirus B3 RNA replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef]
- Hsu, N.Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Lin, L.; Chen, Y.; Wu, S.; Si, X.; Wu, H.; Zhai, X.; Wang, Y.; Tong, L.; Pan, B.; et al. Curcumin inhibits the replication of enterovirus 71 in vitro. Acta Pharm. Sin. B 2014, 4, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Du, J.; Jin, Q. Class I ADP-ribosylation factors are involved in enterovirus 71 replication. PLoS ONE 2014, 9, e99768. [Google Scholar] [CrossRef] [PubMed]
- Dorobantu, C.M.; Ford-Siltz, L.A.; Sittig, S.P.; Lanke, K.H.W.; Belov, G.A.; van Kuppeveld, F.J.M.; van der Schaar, H.M. GBF1- and ACBD3-independent recruitment of PI4KIIIβ to replication sites by rhinovirus 3A proteins. J. Virol. 2015, 89, 1913–1918. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, L. Key components of COPI and COPII machineries are required for chikungunya virus replication. Biochem. Biophys. Res. Commun. 2017, 493, 1190–1196. [Google Scholar] [CrossRef]
- Molina, S.; Sanz, M.A.; Madan, V.; Ventoso, I.; Castelló, A.; Carrasco, L. Differential inhibition of cellular and Sindbis virus translation by brefeldin A. Virology 2007, 363, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Farhat, R.; Ankavay, M.; Lebsir, N.; Gouttenoire, J.; Jackson, C.L.; Wychowski, C.; Moradpour, D.; Dubuisson, J.; Rouillé, Y.; Cocquerel, L. Identification of GBF1 as a cellular factor required for hepatitis E virus RNA replication. Cell. Microbiol. 2018, 20, e12804. [Google Scholar] [CrossRef]
- Verheije, M.H.; Raaben, M.; Mari, M.; Te Lintelo, E.G.; Reggiori, F.; van Kuppeveld, F.J.M.; Rottier, P.J.M.; de Haan, C.A.M. Mouse hepatitis coronavirus RNA replication depends on GBF1-mediated Arf1 activation. PLoS Pathog. 2008, 4, e1000088. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Swett-Tapia, C.; van den Worm, S.H.E.; te Velthuis, A.J.W.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J.; Kikkert, M. Integrity of the early secretory pathway promotes, but is not required for, severe acute respiratory syndrome coronavirus RNA synthesis and virus-induced remodeling of endoplasmic reticulum membranes. J. Virol. 2010, 84, 833–846. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Wannee, K.F.; Scholte, F.E.M.; Goeman, J.J.; ten Dijke, P.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. A kinome-wide small interfering RNA screen identifies proviral and antiviral host factors in severe acute respiratory syndrome coronavirus replication, including double-stranded RNA-activated protein kinase and early secretory pathway proteins. J. Virol. 2015, 89, 8318–8333. [Google Scholar] [CrossRef]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 19036–19041. [Google Scholar] [CrossRef] [PubMed]
- Sun, E.; He, J.; Zhuang, X. Dissecting the role of COPI complexes in influenza virus infection. J. Virol. 2013, 87, 2673–2685. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kawakami, E.; Shoemaker, J.E.; Lopes, T.J.S.; Matsuoka, Y.; Tomita, Y.; Kozuka-Hata, H.; Gorai, T.; Kuwahara, T.; Takeda, E.; et al. Influenza virus-host interactome screen as a platform for antiviral drug development. Cell Host Microbe 2014, 16, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Panganiban, A.T.; Honer Zu Bentrup, K.; Voss, T.G. Influenza infection modulates vesicular trafficking and induces Golgi complex disruption. Virus Disease 2016, 27, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L.; Arnoldi, F.; Schraner, E.M.; Eichwald, C.; Silva-Ayala, D.; Lee, E.; Sztul, E.; Burrone, Ó.R.; López, S.; Arias, C.F. The guanine nucleotide exchange factor GBF1 participates in rotavirus replication. J. Virol. 2019, 93, e01062-19. [Google Scholar] [CrossRef] [PubMed]
- Apte-Sengupta, S.; Sirohi, D.; Kuhn, R.J. Coupling of replication and assembly in flaviviruses. Curr. Opin. Virol. 2014, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Samsa, M.M.; Mondotte, J.A.; Iglesias, N.G.; Assunção-Miranda, I.; Barbosa-Lima, G.; Da Poian, A.T.; Bozza, P.T.; Gamarnik, A.V. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 2009, 5, e1000632. [Google Scholar] [CrossRef] [PubMed]
- Carpp, L.N.; Rogers, R.S.; Moritz, R.L.; Aitchison, J.D. Quantitative proteomic analysis of host-virus interactions reveals a role for Golgi brefeldin A resistance Factor 1 (GBF1) in dengue Infection. Mol. Cell. Proteomics 2014, 13, 2836–2854. [Google Scholar] [CrossRef] [PubMed]
- Kudelko, M.; Brault, J.B.; Kwok, K.; Li, M.Y.; Pardigon, N.; Peiris, J.S.M.; Bruzzone, R.; Despre, P.; Nal, B.; Wang, P.G. Class II ADP-ribosylation factors are required for efficient secretion of dengue viruses. J. Biol. Chem. 2012, 287, 767–777. [Google Scholar] [CrossRef]
- Farias, K.J.S.; Machado, P.R.L.; de Almeida Júnior, R.F.; Lopes da Fonseca, B.A. Brefeldin A and cytochalasin B reduce dengue virus replication in cell cultures but do not protect mice against viral challenge. Access Microbiol. 2019, 1, e000041. [Google Scholar] [CrossRef]
- Helbig, K.J.; Carr, J.M.; Calvert, J.K.; Wati, S.; Clarke, J.N.; Eyre, N.S.; Narayana, S.K.; Fiches, G.N.; McCartney, E.M.; Beard, M.R. Viperin is induced following dengue virus type-2 (DENV-2) infection and has anti-viral actions requiring the C-terminal end of viperin. PLoS Negl. Trop. Dis. 2013, 7, e2178. [Google Scholar] [CrossRef] [PubMed]
- Szretter, K.J.; Brien, J.D.; Thackray, L.B.; Virgin, H.W.; Cresswell, P.; Diamond, M.S. The Interferon-inducible gene viperin restricts West Nile virus pathogenesis. J. Virol. 2011, 85, 11557–11566. [Google Scholar] [CrossRef] [PubMed]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Chang, K.-M. Hepatitis C virus: Virology and life cycle. Clin. Mol. Hepatol. 2013, 19, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Dustin, L.B.; Bartolini, B.; Capobianchi, M.R.; Pistello, M. Hepatitis C virus: Life cycle in cells, infection and host response, and analysis of molecular markers influencing the outcome of infection and response to therapy. Clin. Microbiol. Infect. 2016, 22, 826–832. [Google Scholar] [CrossRef]
- Matto, M.; Sklan, E.H.; David, N.; Melamed-Book, N.; Casanova, J.E.; Glenn, J.S.; Aroeti, B. Role for ADP ribosylation factor 1 in the regulation of hepatitis C virus replication. J. Virol. 2010, 85, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Farhat, R.; Séron, K.; Ferlin, J.; Fénéant, L.; Belouzard, S.; Goueslain, L.; Jackson, C.L.; Dubuisson, J.; Rouillé, Y. Identification of class II ADP-ribosylation factors as cellular factors required for hepatitis C virus replication. Cell. Microbiol. 2016, 18, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yin, P.; Zhou, L.; Li, H.; Zhang, L. Arf1 activation dissociates ADRP from lipid droplets to promote HCV assembly. Biochem. Biophys. Res. Commun. 2016, 475, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Fukasawa, M. Cellular lipid droplets and hepatitis C virus life cycle. Biol. Pharm. Bull. 2010, 33, 355–359. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gastaminza, P.; Cheng, G.; Wieland, S.; Zhong, J.; Liao, W.; Chisari, F.V. Cellular determinants of hepatitis C virus assembly, maturation, degradation, and secretion. J. Virol. 2007, 82, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Farhat, R.; Goueslain, L.; Wychowski, C.; Belouzard, S.; Fénéant, L.; Jackson, C.L.; Dubuisson, J.; Rouillé, Y. Hepatitis C virus replication and Golgi function in brefeldin A-resistant hepatoma-derived cells. PLoS ONE 2013, 8, e74491. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, X.; Yang, G.; Hong, Z.; Zhou, L.; Yin, P.; Xiao, Y.; Chen, L.; Chung, R.T.; Zhang, L. Hepatitis C virus NS5A hijacks ArfGAP1 to maintain a phosphatidylinositol 4-phosphate-enriched microenvironment. J. Virol. 2014, 88, 5956–5966. [Google Scholar] [CrossRef] [PubMed]
- Balla, A.; Balla, T. Phosphatidylinositol 4-kinases: Old enzymes with emerging functions. Trends Cell Biol. 2006, 16, 351–361. [Google Scholar] [CrossRef]
- Lebsir, N.; Goueslain, L.; Farhat, R.; Callens, N.; Dubuisson, J.; Jackson, C.L.; Rouillé, Y. Functional and physical interaction between the Arf Activator GBF1 and hepatitis C virus NS3 Protein. J. Virol. 2019, 93, e01459-18. [Google Scholar] [CrossRef]
- Maynell, L.a.; Kirkegaard, K.; Klymkowsky, M.W. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 1992, 66, 1985–1994. [Google Scholar] [CrossRef]
- Cuconati, A.; Molla, A.; Wimmer, E. Brefeldin A inhibits cell-free, de novo synthesis of poliovirus. J. Virol. 1998, 72, 6456–6464. [Google Scholar] [CrossRef]
- Wang, J.; Wu, Z.; Jin, Q. COPI is required for enterovirus 71 replication. PLoS ONE 2012, 7, e38035. [Google Scholar] [CrossRef]
- Gazina, E.V.; Mackenzie, J.M.; Gorrell, R.J.; Anderson, D.A. Differential Requirements for COPI coats in formation of replication complexes among three genera of Picornaviridae. J. Virol. 2002, 76, 11113–11122. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.K.; Pacheco, J.M.; Henry, T.M.; Mason, P.W. Subcellular distribution of the foot-and-mouth disease virus 3A protein in cells infected with viruses encoding wild-type and bovine-attenuated forms of 3A. Virology 2001, 287, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 2006, 81, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Wessels, E.; Duijsings, D.; Niu, T.K.; Neumann, S.; Oorschot, V.M.; de Lange, F.; Lanke, K.H.W.; Klumperman, J.; Henke, A.; Jackson, C.L.; et al. A viral protein that blocks Arf1-mediated COP-I assembly by inhibiting the guanine nucleotide exchange factor GBF1. Dev. Cell 2006, 11, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Wessels, E.; Notebaart, R.A.; Melchers, W.J.G.; Van Kuppeveld, F.J.M.; Irol, J.V. A Proline-rich region in the coxackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum to Golgi transport. J. Virol. 2005, 79, 5163–5173. [Google Scholar] [CrossRef]
- Wessels, E.; Duijsings, D.; Lanke, K.H.W.; van Dooren, S.H.J.; Jackson, C.L.; Melchers, W.J.G.; van Kuppeveld, F.J.M. Effects of picornavirus 3A proteins on protein transport and GBF1-dependent COP-I recruitment. J. Virol. 2006, 80, 11852–11860. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wessels, E.; Duijsings, D.; Lanke, K.H.W.; Melchers, W.J.G.; Jackson, C.L.; van Kuppeveld, F.J.M. Molecular determinants of the interaction between coxsackievirus protein 3A and guanine nucleotide exchange factor GBF1. J. Virol. 2007, 81, 5238–5245. [Google Scholar] [CrossRef][Green Version]
- Belov, G.a.; Fogg, M.H.; Ehrenfeld, E. Poliovirus proteins induce membrane association of GTPase ADP-ribosylation factor. J. Virol. 2005, 79, 7207–7216. [Google Scholar] [CrossRef]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [CrossRef]
- Belov, G.A.; Sztul, E. Rewiring of cellular membrane homeostasis by picornaviruses. J. Virol. 2014, 88, 9478–9489. [Google Scholar] [CrossRef]
- Viktorova, E.G.; Nchoutmboube, J.A.; Ford-Siltz, L.A.; Iverson, E.; Belov, G.A. Phospholipid synthesis fueled by lipid droplets drives the structural development of poliovirus replication organelles. PLoS Pathog. 2018, 14, e1007280. [Google Scholar] [CrossRef] [PubMed]
- Soni, K.G.; Mardones, G.A.; Sougrat, R.; Smirnova, E.; Jackson, C.L.; Bonifacino, J.S. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 2009, 122, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Irurzun, A.; Perez, L.; Carrasco, L. Involvement of membrane traffic in the replication of poliovirus genomes: Effects of brefeldin A. Virology 1992, 191, 166–175. [Google Scholar] [CrossRef]
- Sasaki, J.; Ishikawa, K.; Arita, M.; Taniguchi, K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012, 31, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.J. Togaviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 629–650. [Google Scholar]
- Jose, J.; Taylor, A.B.; Kuhn, R.J. Spatial and temporal analysis of alphavirus replication and assembly in mammalian and mosquito cells. MBio 2017, 8, e02294-16. [Google Scholar] [CrossRef]
- Madan, V.; Sanz, M.A.; Carrasco, L. Requirement of the vesicular system for membrane permeabilization by Sindbis virus. Virology 2005, 332, 307–315. [Google Scholar] [CrossRef]
- Barletta, A.B.F.; Alves, L.R.; Nascimento Silva, M.C.L.; Sim, S.; Dimopoulos, G.; Liechocki, S.; Maya-Monteiro, C.M.; Sorgine, M.H.F. Emerging role of lipid droplets in Aedes aegypti immune response against bacteria and dengue virus. Sci. Rep. 2016, 6, 19928. [Google Scholar] [CrossRef]
- Emerson, S.U.; Purcell, R.H. Hepatitis E virus. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2242–2258. [Google Scholar]
- Masters, P.S.; Perlman, S. Coronaviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 825–858. [Google Scholar]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Dascher, C.; Balch, W.E. Dominant inhibitory mutants of Arf1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J. Biol. Chem. 1994, 269, 1437–1448. [Google Scholar]
- Lyles, D.S.; Kuzmin, I.V.; Rupprecht, C.E. Rhabdoviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 885–922. [Google Scholar]
- Irurzun, A.; Pérez, L.; Carrasco, L. Brefeldin A blocks protein glycosylation and RNA replication of vesicular stomatitis virus. FEBS Lett. 1993, 336, 496–500. [Google Scholar] [CrossRef]
- Cureton, D.K.; Burdeinick-Kerr, R.; Whelan, S.P.J. Genetic inactivation of COPI coatomer separately inhibits vesicular stomatitis virus entry and gene expression. J. Virol. 2012, 86, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Chadha, M.S.; Arankalle, V.A.; Jadi, R.S.; Joshi, M.V.; Thakare, J.P.; Mahadev, P.V.M.; Mishra, A.C. An outbreak of Chandipura virus encephalitis in the eastern districts of Gujarat state, India. Am. J. Trop. Med. Hyg. 2005, 73, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Gurav, Y.K.; Tandale, B.V.; Jadi, R.S.; Gunjikar, R.S.; Tikute, S.S.; Jamgaonkar, A.V.; Khadse, R.K.; Jalgaonkar, S.V.; Arankalle, V.A.; Mishra, A.C. Chandipura virus encephalitis outbreak among children in Nagpur division, Maharashtra, 2007. Indian J. Med. Res. 2010, 132, 395–399. [Google Scholar] [PubMed]
- Rajasekharan, S.; Rana, J.; Gulati, S.; Sharma, S.K.; Gupta, V.; Gupta, S. Predicting the host protein interactors of Chandipura virus using a structural similarity-based approach. Pathog. Dis. 2013, 69, 29–35. [Google Scholar] [PubMed]
- Heinrich, B.S.; Cureton, D.K.; Rahmeh, A.A.; Whelan, S.P.J. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog. 2010, 6, e1000958. [Google Scholar] [CrossRef]
- Andrew Whitney, J.; Gomez, M.; Sheff, D.; Kreis, T.E.; Mellman, I. Cytoplasmic coat proteins involved in endosome function. Cell 1995, 83, 703–713. [Google Scholar] [CrossRef]
- Dou, D.; Revol, R.; Östbye, H.; Wang, H.; Daniels, R. Influenza A virus cell entry, replication, virion assembly and movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Shaw, M.L.; Palese, P. Orthomyxoviridae. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1151–1185. [Google Scholar]
- Amorim, M.J.; Bruce, E.A.; Read, E.K.C.; Foeglein, Á.; Mahen, R.; Stuart, A.D.; Digard, P. A Rab11- and microtubule-dependent mechanism for cytoplasmic transport of influenza A Virus viral RNA. J. Virol. 2011, 85, 4143–4156. [Google Scholar] [CrossRef]
- Momose, F.; Sekimoto, T.; Ohkura, T.; Jo, S.; Kawaguchi, A.; Nagata, K.; Morikawa, Y. Apical transport of influenza A virus ribonucleoprotein requires Rab11-positive recycling endosome. PLoS ONE 2011, 6, e21123. [Google Scholar] [CrossRef]
- De Castro Martin, I.F.; Fournier, G.; Sachse, M.; Pizarro-Cerda, J.; Risco, C.; Naffakh, N. Influenza virus genome reaches the plasma membrane via a modified endoplasmic reticulum and Rab11-dependent vesicles. Nat. Commun. 2017, 8, 1396. [Google Scholar] [CrossRef]
- Jo, S.; Kawaguchi, A.; Takizawa, N.; Morikawa, Y.; Momose, F.; Nagata, K. Involvement of vesicular trafficking system in membrane targeting of the progeny influenza virus genome. Microbes Infect. 2010, 12, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.K.; Greenberg, H.B. Rotaviruses. In Fields Virology; Knipe, M.D., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1347–1401. [Google Scholar]
- Maass, D.; Atkinson, P.H. Rotavirus Proteins VP7, NS28, and VP4 form oligomeric structures. J. Virol. 1990, 64, 2632–2641. [Google Scholar] [CrossRef] [PubMed]
- Green, V.A.; Pelkmans, L. A systems survey of progressive host-cell reorganization during rotavirus infection. Cell Host Microbe 2016, 20, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Silva-Ayala, D.; Lopez, T.; Gutierrez, M.; Perrimon, N.; Lopez, S.; Arias, C.F. Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc. Natl. Acad. Sci. USA 2013, 110, 10270–10275. [Google Scholar] [CrossRef] [PubMed]
- Mirazimi, A.; Von Bonsdorff, C.H.; Svensson, L. Effect of brefeldin A on rotavirus assembly and oligosaccharide processing. Virology 1996, 217, 554–563. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cuadras, M.A.; Bordier, B.B.; Zambrano, J.L.; Ludert, J.E.; Greenberg, H.B. Dissecting rotavirus particle-raft interaction with small interfering RNAs: Insights into rotavirus transit through the secretory pathway. J. Virol. 2006, 80, 3935–3946. [Google Scholar] [CrossRef]
- Petrie, B.L.; Estes, M.K.; Graham, D.Y. Effects of tunicamycin on rotavirus morphogenesis and infectivity. J. Virol. 1983, 46, 270–274. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Type of Genome | Family | Virus | Viral Replication Step that Requires the GBF1 Activity | Reference |
|---|---|---|---|---|
| Non-segmented (+)ssRNA | Flaviviridae | DENV | RNA replication Virus assembly | [48,49] |
| TBEV | Virus assembly | [50] | ||
| YFV | Virus secretion | [51] | ||
| KUN | Formation of viral replication complexes | [52] | ||
| CSFV | RNA replication | [53] | ||
| HCV | RNA replication Protein expression Virus secretion | [54,55,56] | ||
| Picornaviridae | Poliovirus | RNA replication | [57,58,59] | |
| CVA21 | RNA replication | [60] | ||
| CVB3 | RNA replication | [60,61,62] | ||
| CVB4 | RNA replication | [51] | ||
| EV71 | RNA replication Protein expression | [60,63,64] | ||
| HRV2 | RNA replication | [65] | ||
| HRV14 | RNA replication | [65] | ||
| Togaviridae | CHIKV | RNA replication Early protein expression | [66] | |
| SINV | RNA replication Protein expression | [51,67] | ||
| Hepeviridae | HEV | RNA replication | [68] | |
| Coronaviridae | MHV | RNA replication | [69] | |
| SARS-CoV | RNA replication RNA transcription | [70,71] | ||
| HCoV-229E | RNA replication | [51] | ||
| Non-segmented (-)ssRNA | Rhabdoviridae | VSV | RNA replication RNA transcription Protein expression | [72] |
| Paramyxoviridae | HPIV3 | Protein expression | [72] | |
| Segmented (-)ssRNA | Orthomyxoviridae | IAV | Virus assembly | [73,74,75] |
| Arenaviridae | LCMV | Protein expression | [72] | |
| Segmented dsRNA | Reoviridae | Rotavirus | Virus assembly | [76] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, J.L.; Arias, C.F. Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses. Viruses 2020, 12, 682. https://doi.org/10.3390/v12060682
Martínez JL, Arias CF. Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses. Viruses. 2020; 12(6):682. https://doi.org/10.3390/v12060682
Chicago/Turabian StyleMartínez, José L., and Carlos F. Arias. 2020. "Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses" Viruses 12, no. 6: 682. https://doi.org/10.3390/v12060682
APA StyleMartínez, J. L., & Arias, C. F. (2020). Role of the Guanine Nucleotide Exchange Factor GBF1 in the Replication of RNA Viruses. Viruses, 12(6), 682. https://doi.org/10.3390/v12060682