Porcine Reproductive and Respiratory Syndrome Virus Antagonizes PCSK9’s Antiviral Effect via Nsp11 Endoribonuclease Activity


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viruses
2.2. Plasmid Construction
2.3. Reagents and Antibodies
2.4. Multi-Step Growth Curve of Virus
2.5. TCID50 Assay for PRRSV
2.6. Indirect Immunofluorescence Assay
2.7. RNA Extraction and RT-qPCR Assay
2.8. Co-Immunoprecipitation (Co-IP)
2.9. Confocal Microscopy
2.10. Western Blotting
2.11. Reporter Assay
2.12. Bioinformatics Prediction
2.13. Statistical Analyses
3. Results
3.1. PCSK9 Inhibits the Replication of Both Type-1 and Type-2 PRRSV Species
3.2. The C-Terminal Domain of PCSK9 Has Antiviral Activity
3.3. PCSK9 Degrades PRRSV Receptor CD163 Through Lysosome Pathway
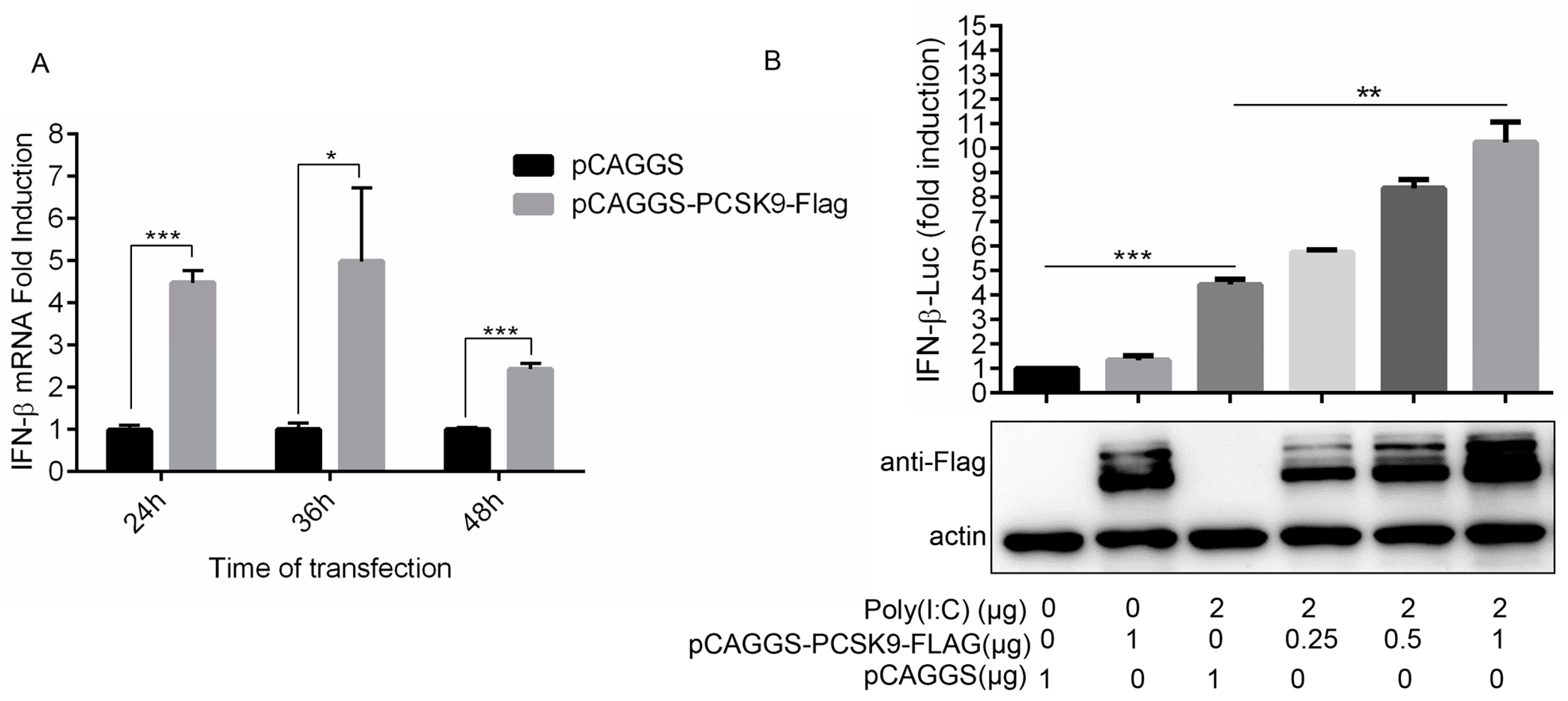
3.4. PCSK9 Promotes Interferon Production in a Dose-Dependent Manner
3.5. PRRSV Down-Regulates PCSK9 Expression Both in MARC-145 Cells and in PAM Cells
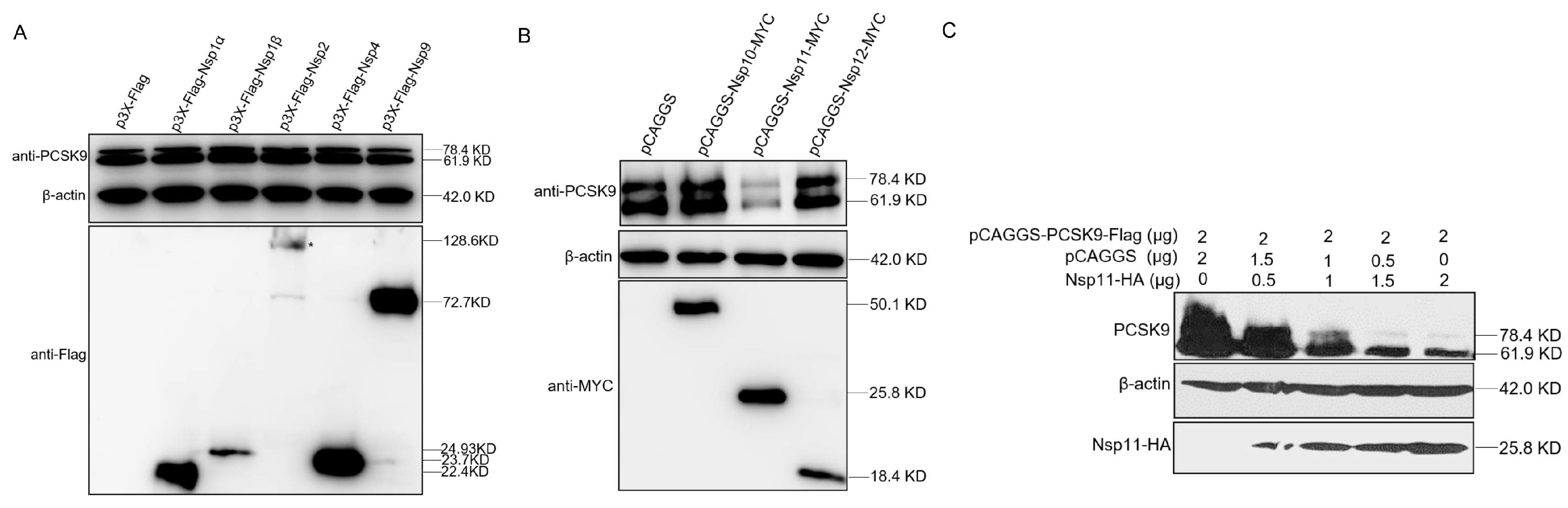
3.6. PRRSV nsp11 Negatively Regulates PCSK9 Expression
3.7. PRRSV nsp11 Inhibits PCSK9 via Its Endoribonuclease Activity
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keffaber, K.K. Reproductive failure of unknown etiology. Am. Assoc. Swine Pract. Newsl. 1989, 1, 1–9. [Google Scholar]
- Lunney, J.K.; Fang, Y.; Ladinig, A.; Chen, N.; Li, Y.; Rowland, B.; Renukaradhya, G.J. Porcine reproductive and respiratory syndrome virus (PRRSV): Pathogenesis and interaction with the immune system. Annu. Rev. Anim. Biosci. 2016, 4, 129–154. [Google Scholar] [CrossRef]
- Allende, R.; Lewis, T.L.; Lu, Z.; Rock, D.L.; Kutish, G.F.; Ali, A.; Doster, A.R.; Osorio, F.A. North American and European porcine reproductive and respiratory syndrome viruses differ in non-structural protein coding regions. J. Gen. Virol. 1999, 80 Pt 2, 307–315. [Google Scholar] [CrossRef]
- Nelsen, C.J.; Murtaugh, M.P.; Faaberg, K.S. Porcine reproductive and respiratory syndrome virus comparison: Divergent evolution on two continents. J. Virol. 1999, 73, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Wensvoort, G.; Terpstra, C.; Pol, J.M.; ter Laak, E.A.; Bloemraad, M.; De Kluyver, E.P.; Kragten, C.; Van Buiten, L.; Den Besten, A.; Wagenaar, F.; et al. Mystery swine disease in The Netherlands: The isolation of Lelystad virus. Vet. Q. 1991, 13, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Snijder, E.J.; Kikkert, M.; Fang, Y. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 2013, 94, 2141–2163. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef] [PubMed]
- Rossow, K.D. Porcine reproductive and respiratory syndrome. Vet. Pathol. 1998, 35, 1–20. [Google Scholar] [CrossRef]
- Fang, Y.; Treffers, E.E.; Li, Y.; Tas, A.; Sun, Z.; Van der Meer, Y.; De Ru, A.H.; Van Veelen, P.A.; Atkins, J.F.; Snijder, E.J.; et al. Efficient-2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc. Natl. Acad. Sci. USA 2012, 109, E2920–E2928. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E.; Zevenhoven-Dobbe, J.C.; Wills, N.M.; Go, Y.Y.; Balasuriya, U.B.; Atkins, J.F.; Snijder, E.J.; Posthuma, C.C. Discovery of a small arterivirus gene that overlaps the GP5 coding sequence and is important for virus production. J. Gen. Virol. 2011, 92, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.R.; Griggs, T.F.; Gnanandarajah, J.; Murtaugh, M.P. Novel structural protein in porcine reproductive and respiratory syndrome virus encoded by an alternative ORF5 present in all arteriviruses. J. Gen. Virol. 2011, 92, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Snijder, E.J. The PRRSV replicase: Exploring the multifunctionality of an intriguing set of nonstructural proteins. Virus Res. 2010, 154, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tas, A.; Snijder, E.J.; Fang, Y. Identification of porcine reproductive and respiratory syndrome virus ORF1a-encoded non-structural proteins in virus-infected cells. J. Gen. Virol. 2012, 93, 829–839. [Google Scholar] [CrossRef] [PubMed]
- Rascon-Castelo, E.; Burgara-Estrella, A.; Mateu, E.; Hernandez, J. Immunological features of the non-structural proteins of porcine reproductive and respiratory syndrome virus. Viruses 2015, 7, 873–886. [Google Scholar] [CrossRef]
- Nedialkova, D.D.; Ulferts, R.; van den Born, E.; Lauber, C.; Gorbalenya, A.E.; Ziebuhr, J.; Snijder, E.J. Biochemical characterization of arterivirus nonstructural protein 11 reveals the nidovirus-wide conservation of a replicative endoribonuclease. J. Virol. 2009, 83, 5671–5682. [Google Scholar] [CrossRef]
- Posthuma, C.C.; Nedialkova, D.D.; Zevenhoven-Dobbe, J.C.; Blokhuis, J.H.; Gorbalenya, A.E.; Snijder, E.J. Site-directed mutagenesis of the nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006, 80, 1653–1661. [Google Scholar] [CrossRef]
- Shi, Y.; Li, Y.; Lei, Y.; Ye, G.; Shen, Z.; Sun, L.; Luo, R.; Wang, D.; Fu, Z.F.; Xiao, S.; et al. A Dimerization-dependent mechanism drives the endoribonuclease function of porcine reproductive and respiratory syndrome virus nsp11. J. Virol. 2016, 90, 4579–4592. [Google Scholar] [CrossRef]
- Shi, X.; Wang, L.; Li, X.; Zhang, G.; Guo, J.; Zhao, D.; Chai, S.; Deng, R. Endoribonuclease activities of porcine reproductive and respiratory syndrome virus nsp11 was essential for nsp11 to inhibit IFN-beta induction. Mol. Immunol. 2011, 48, 1568–1572. [Google Scholar] [CrossRef]
- Sun, Y.; Ke, H.; Han, M.; Chen, N.; Fang, W.; Yoo, D. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus suppresses both MAVS and RIG-I expression as one of the mechanisms to antagonize type I interferon production. PLoS ONE 2016, 11, e0168314. [Google Scholar] [CrossRef]
- Sun, Y.; Li, D.; Giri, S.; Prasanth, S.G.; Yoo, D. Differential host cell gene expression and regulation of cell cycle progression by nonstructural protein 11 of porcine reproductive and respiratory syndrome virus. Biomed. Res. Int. 2014, 2014, 430508. [Google Scholar] [CrossRef]
- Yang, L.; He, J.; Wang, R.; Zhang, X.; Lin, S.; Ma, Z.; Zhang, Y. Nonstructural protein 11 of porcine reproductive and respiratory syndrome virus induces STAT2 degradation to inhibit interferon signaling. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Chen, J.; Yu, C.; Zhu, X.; Xu, S.; Fang, L.; Xiao, S. Porcine reproductive and respiratory syndrome virus nsp11 antagonizes type I interferon signaling by targeting IRF9. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fan, J.; Fang, L.; Luo, R.; Ouyang, H.; Ouyang, C.; Zhang, H.; Chen, H.; Li, K.; Xiao, S. The nonstructural protein 11 of porcine reproductive and respiratory syndrome virus inhibits NF-kappaB signaling by means of its deubiquitinating activity. Mol. Immunol. 2015, 68, 357–366. [Google Scholar] [CrossRef]
- Zhang, Q.; Yoo, D. PRRS virus receptors and their role for pathogenesis. Vet. Microbiol. 2015, 177, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiao, S.; Xiao, Y.; Wang, X.; Zhang, C.; Zhao, Q.; Nan, Y.; Huang, B.; Liu, H.; Liu, N.; et al. MYH9 is an essential factor for porcine reproductive and respiratory syndrome virus infection. Sci. Rep. 2016, 6, 25120. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Zhu, Z.; Guo, Y.; Wang, X.; Yu, P.; Xiao, S.; Chen, Y.; Cao, Y.; Liu, X. Heparanase upregulation contributes to porcine reproductive and respiratory syndrome virus release. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Ke, W.; Fang, L.; Jing, H.; Tao, R.; Wang, T.; Li, Y.; Long, S.; Wang, D.; Xiao, S. Cholesterol 25-hydroxylase inhibits porcine reproductive and respiratory syndrome virus replication through enzyme activity-dependent and -independent mechanisms. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Song, Z.; Zhang, Q.; Liu, X.; Bai, J.; Zhao, Y.; Wang, X.; Jiang, P. Cholesterol 25-hydroxylase is an interferon-inducible factor that protects against porcine reproductive and respiratory syndrome virus infection. Vet. Microbiol. 2017, 210, 153–161. [Google Scholar] [CrossRef]
- Ke, H.; Han, M.; Kim, J.; Gustin, K.E.; Yoo, D. Porcine reproductive and respiratory syndrome virus nonstructural protein 1 beta interacts with nucleoporin 62 to promote viral replication and immune evasion. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Li, J.; Guo, D.; Huang, L.; Yin, M.; Liu, Q.; Wang, Y.; Yang, C.; Liu, Y.; Zhang, L.; Tian, Z.; et al. The interaction between host Annexin A2 and viral Nsp9 is beneficial for replication of porcine reproductive and respiratory syndrome virus. Virus Res. 2014, 189, 106–113. [Google Scholar] [CrossRef]
- Zhao, Y.; Song, Z.; Bai, J.; Liu, X.; Nauwynck, H.; Jiang, P. ZAP, a CCCH-type zinc finger protein, inhibits porcine reproductive and respiratory syndrome virus replication and interacts with viral Nsp9. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Zhou, Y.; Fang, L.; Ding, Z.; Wang, D.; Ke, W.; Chen, H.; Xiao, S. DExD/H-box helicase 36 signaling via myeloid differentiation primary response gene 88 contributes to NF-kappaB activation to type 2 porcine reproductive and respiratory syndrome virus infection. Front. Immunol. 2017, 8, 1365. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Li, L.W.; Zhang, Y.J.; Jiang, Y.F.; Gao, F.; Li, G.X.; Yu, L.X.; Zhao, W.Y.; Shan, T.L.; Zhou, Y.J.; et al. MOV10 inhibits replication of porcine reproductive and respiratory syndrome virus by retaining viral nucleocapsid protein in the cytoplasm of Marc-145cells. Biochem. Biophys. Res. Commun. 2018, 504, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, K.; Gao, F.; Jiang, Y.; Shan, T.; Tong, W.; Zheng, H.; Yu, L.; Li, G.; Ma, Z.; et al. Restriction of porcine reproductive and respiratory syndrome virus replication by galectin-1. Vet. Microbiol. 2019, 235, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, Y.; Jiang, Y.; Gao, F.; Shan, T.; Zhao, K.; Zhang, Y.; Li, L.; Tong, G. Galectin-3 inhibits replication of porcine reproductive and respiratory syndrome virus by interacting with viral Nsp12 in vitro. Virus Res. 2018, 253, 87–91. [Google Scholar] [CrossRef]
- Seidah, N.G.; Chretien, M. Proprotein and prohormone convertases: A family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999, 848, 45–62. [Google Scholar] [CrossRef]
- Sanchez-Hernandez, R.M.; Di Taranto, M.D.; Benito-Vicente, A.; Uribe, K.B.; Lamiquiz-Moneo, I.; Larrea-Sebal, A.; Jebari, S.; Galicia-Garcia, U.; Novoa, F.J.; Boronat, M.; et al. The Arg499His gain-of-function mutation in the C-terminal domain of PCSK9. Atherosclerosis 2019, 289, 162–172. [Google Scholar] [CrossRef]
- Cohen, J.; Pertsemlidis, A.; Kotowski, I.K.; Graham, R.; Garcia, C.K.; Hobbs, H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 2005, 37, 161–165. [Google Scholar] [CrossRef]
- Maxwell, K.N.; Fisher, E.A.; Breslow, J.L. Overexpression of PCSK9 accelerates the degradation of the LDLR in a post-endoplasmic reticulum compartment. Proc. Natl. Acad. Sci. USA 2005, 102, 2069–2074. [Google Scholar] [CrossRef]
- Cohen, J.C.; Boerwinkle, E.; Mosley, T.H., Jr.; Hobbs, H.H. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N. Engl. J. Med. 2006, 354, 1264–1272. [Google Scholar] [CrossRef]
- Zhang, D.W.; Garuti, R.; Tang, W.J.; Cohen, J.C.; Hobbs, H.H. Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 13045–13050. [Google Scholar] [CrossRef]
- Benjannet, S.; Rhainds, D.; Essalmani, R.; Mayne, J.; Wickham, L.; Jin, W.; Asselin, M.C.; Hamelin, J.; Varret, M.; Allard, D.; et al. NARC-1/PCSK9 and its natural mutants: Zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem 2004, 279, 48865–48875. [Google Scholar] [CrossRef]
- Labonte, P.; Begley, S.; Guevin, C.; Asselin, M.C.; Nassoury, N.; Mayer, G.; Prat, A.; Seidah, N.G. PCSK9 impedes hepatitis C virus infection in vitro and modulates liver CD81 expression. Hepatology 2009, 50, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Leucker, T.M.; Weiss, R.G.; Schar, M.; Bonanno, G.; Mathews, L.; Jones, S.R.; Brown, T.T.; Moore, R.; Afework, Y.; Gerstenblith, G.; et al. Coronary endothelial dysfunction is associated with elevated serum PCSK9 levels in people with HIV independent of low-density lipoprotein cholesterol. J. Am. Heart Assoc. 2018, 7, e009996. [Google Scholar] [CrossRef] [PubMed]
- Tong, G.Z.; Zhou, Y.J.; Hao, X.F.; Tian, Z.J.; An, T.Q.; Qiu, H.J. Highly pathogenic porcine reproductive and respiratory syndrome, China. Emerg. Infect. Dis. 2007, 13, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.J.; An, T.Q.; Zhou, Y.J.; Peng, J.M.; Hu, S.P.; Wei, T.C.; Jiang, Y.F.; Xiao, Y.; Tong, G.Z. An attenuated live vaccine based on highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) protects piglets against HP-PRRS. Vet. Microbiol. 2009, 138, 34–40. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, F.; Qu, Z.; Jiang, Y.; Zhou, Y.; Yu, L.; Li, L.; Zhao, K.; Tong, G. Characterization of differentially expressed membrane proteins of pulmonary alveolar macrophages infected with porcine reproductive and respiratory syndrome virus. Chin. J. Anim. Infect. Dis. 2019, 27, 1–8. [Google Scholar]
- Park, S.W.; Moon, Y.A.; Horton, J.D. Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 2004, 279, 50630–50638. [Google Scholar] [CrossRef]
- Nassoury, N.; Blasiole, D.A.; Tebon Oler, A.; Benjannet, S.; Hamelin, J.; Poupon, V.; McPherson, P.S.; Attie, A.D.; Prat, A.; Seidah, N.G. The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 2007, 8, 718–732. [Google Scholar] [CrossRef]
- Zhang, D.W.; Lagace, T.A.; Garuti, R.; Zhao, Z.; McDonald, M.; Horton, J.D.; Cohen, J.C.; Hobbs, H.H. Binding of proprotein convertase subtilisin/kexin type 9 to epidermal growth factor-like repeat A of low density lipoprotein receptor decreases receptor recycling and increases degradation. J. Biol. Chem. 2007, 282, 18602–18612. [Google Scholar] [CrossRef]
- Demers, A.; Samami, S.; Lauzier, B.; Des Rosiers, C.; Ngo Sock, E.T.; Ong, H.; Mayer, G. PCSK9 induces CD36 degradation and affects long-chain fatty acid uptake and triglyceride metabolism in adipocytes and in mouse liver. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, Q. Proprotein convertase subtilisin/kexin type 9 inhibits interferon beta expression through interacting with ATF-2. FEBS Lett. 2018, 592, 2323–2333. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Yoo, D. The viral innate immune antagonism and an alternative vaccine design for PRRS virus. Vet. Microbiol. 2017, 209, 75–89. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Porcine reproductive and respiratory syndrome virus nsp1beta and nsp11 antagonize the antiviral activity of cholesterol-25-hydroxylase via lysosomal degradation. Vet. Microbiol. 2018, 223, 134–143. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer a | Sequence (5′-3′) b | Usage |
|---|---|---|
| Mature-PCSK9-F | TGCTCTAGAATGAGCATCCCGTGGAACCTGGAG | Amplification of mature PCSK9 (151–690aa) |
| Mature-PCSK9-R | CCGGAATTCTCACTTATCGTCGTCATCCTTGTAATCCTGGGACTCCTGGGAGGCCTC | |
| PCSK9-151-400-Flag-F | TCATTTTGGCAAAGATGAGCATCCCGTGGAACCTGG | Amplification of PCSK9 catalysis domain (151–400aa) |
| PCSK9-151-400-Flag-R | CCAGATCTGAATTTTACTTATCGTCGTCATCCTTGTAATCCGGCTCAGCCGTCAGCATC | |
| PCSK9-448-690-Flag-F | TCATTTTGGCAAAGATGGGTGGGCAGCTGTTCTGCA | Amplification of PCSK9 C-terminal domain (448–690aa) |
| PCSK9-448-690-Flag-R | TACCAGATCTGAATTTCACTTATCGTCGTCATCCTTGTAATCCTGGGACTCCTGGGAG | |
| PCSK9-Q150A-F | CGTCTTTGCGGCGAGCATCCCGTGGAACCTGGAGCGG | Amplification of mutation PCSK9(Q150A) |
| PCSK9-Q150A-R | CGGGATGCTCGCCGCAAAGACGAAGGAGTCCTCCTCG | |
| PCSK9-D182A-F | GTATCTCTTAGCCACCAGCATCCAAAGTGGCCACC | Amplification of mutation PCSK9 (D182A) |
| PCSK9-D182A-R | GGATGCTGGTGGCTAAGAGATACACCTCCACCAGG | |
| PCSK9-H222A-F | GTGTGACAGCGCCGGCACCCACCTGGCCGGGGT | Amplification of mutation PCSK9 (H222A) |
| PCSK9-H222A-R | GTGGGTGCCGGCGCTGTCACACTTGTTCGCCTG | |
| PCSK9-N313A-F | CCGCTGCTGGCGCCTTCCGGGACGACGCCTGCCTC | Amplification of mutation PCSK9 (N313A) |
| PCSK9-N313A-R | CGTCCCGGAAGGCGCCAGCAGCGGCCACCAGCACT | |
| PCSK9-S382A-F | CAGAGCGGGACGGCACAGGCTGCCGCCCATGTGGC | Amplification of mutation PCSK9 (S382A) |
| PCSK9-S382A-R | CGGCAGCCTGTGCCGTCCCGCTCTGCGACGTGAAGC | |
| PCSK9-N529A-F | CTGCCCCGGGCCGCCTGCAGCATCCACATGGCTCCA | Amplification of mutation PCSK9 (N529A) |
| PCSK9-N529A-R | TGGATGCTGCAGGCGGCCCGGGGCAGCAGGCAGCAC | |
| pGL3-PCSK9-F | CGGGGTACCTTGGCTGGTTGGTGAGGTGAG | Amplification of PCSK9 promoter |
| pGL3-PCSK9-R | CCGCTCGAGGCAGCAGTAGCAGCAGCGGCGGC | |
| Nsp11-C112A-F | GAGGTAGATGCTCGAGAGTATCTTGATGATCGGGAGC | Amplification of Nsp11 (C112A, H112K173A) |
| Nsp11-C112A-R | TACTCTCGAGCATCTACCTCAATTCGGCCGGTGCTGAAG | |
| NSP11-H129A-F | GTCCCTCCCAGCTGCCTTCATCGGCGATGTCAAAG | Amplification of Nsp11 (H129A, H129H144A, H129K173A) |
| NSP11-H129A-R | GATGAAGGCAGCTGGGAGGGACTCAGCAACCTCTC | |
| Nsp11-H144A-F | GTTGGGGGATGTGCTCACGTTACCTCCAAATACCTTC | Amplification of Nsp11 (H144A, H129H144A) |
| Nsp11-H144A-R | GAGGTAACGTGAGCACATCCCCCAACGGTGGTACCT | |
| Nsp11-K173A-F | GAAAGCCGCGGCAGCAGTTTGCACATTGACGGATGTGTAC | Amplification of Nsp11 (K173A, H112K173A, H129K173A) |
| Nsp11-K173A-R | GCAAACTGCTGCCGCGGCTTTCCCGGGGCTCGAAACCCCG | |
| EGFP-Flag-F | TCATTTTGGCAAAGATGGTGAGCAAGGGCGAGG | Amplification of EGFP-Flag |
| EGFP-Flag-R | TACCAGATCTGAATTTTACTTATCGTCGTCATCCTTGTAATCCTTGTACAGCTCGTCC | |
| q-Pig-PCSK9-F | CCACGTCCTCACAGGTTGC | qPCR for detection of pig PCSK9 |
| q-Pig-PCSK9-R | CGTGGACACTGGCCTTCTC | |
| q-Monkey-PCSK9-F | ACCCGTGTCCACTGCCATCAG | qPCR for detection of monkey PCSK9 |
| q-Monkey-PCSK9-R | ACCTCGTGGCCTCAGCACAG | |
| human-PCSK9-F | GAAGATGAGTGGCGACCT | qPCR for detection of human PCSK9 |
| human-PCSK9-R | CCGGTGGTCACTCTGTATGCT | |
| Pig-GAPDH-F | ATGGTGAAGGTCGGAGTGAAC | qPCR for detection of human GAPDH |
| Pig-GAPDH-R | CGTGGGTGGAATCATACTGG | |
| Monkey-GAPDH-F | CCTTCCGTGTCCCTACTGCCAAC | qPCR for detection of monkey GAPDH |
| Monkey-GAPDH-R | GACGCCTGCTTCACCACCTTCT | |
| PRRSV-N-F | AAAACCAGTCCAGAGGCAAG | qPCR for detection of PRRSV N |
| PRRSV-N-R | CGGATCAGACGCACAGTATG | |
| Monkey-IFN-β-F | GCAATTGAATGGAAGGCTTGA | qPCR for detection of monkey IFN-β |
| Monkey-IFN-β-R | CAGCGTCCTCCTTCTGGAACT | |
| pIFN-β-F | GGCGGTACCCTTGGCTTATGGTGGTTTTTTTTG | Amplification of PCSK9 promoter |
| pIFN-β-R | TTTCTCGAGGCTCCACTACTCAAGTGCTGAAG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Gao, F.; Li, L.; Zhao, K.; Jiang, S.; Jiang, Y.; Yu, L.; Zhou, Y.; Liu, C.; Tong, G. Porcine Reproductive and Respiratory Syndrome Virus Antagonizes PCSK9’s Antiviral Effect via Nsp11 Endoribonuclease Activity. Viruses 2020, 12, 655. https://doi.org/10.3390/v12060655
Zhang Y, Gao F, Li L, Zhao K, Jiang S, Jiang Y, Yu L, Zhou Y, Liu C, Tong G. Porcine Reproductive and Respiratory Syndrome Virus Antagonizes PCSK9’s Antiviral Effect via Nsp11 Endoribonuclease Activity. Viruses. 2020; 12(6):655. https://doi.org/10.3390/v12060655
Chicago/Turabian StyleZhang, Yujiao, Fei Gao, Liwei Li, Kuan Zhao, Shan Jiang, Yifeng Jiang, Lingxue Yu, Yanjun Zhou, Changlong Liu, and Guangzhi Tong. 2020. "Porcine Reproductive and Respiratory Syndrome Virus Antagonizes PCSK9’s Antiviral Effect via Nsp11 Endoribonuclease Activity" Viruses 12, no. 6: 655. https://doi.org/10.3390/v12060655
APA StyleZhang, Y., Gao, F., Li, L., Zhao, K., Jiang, S., Jiang, Y., Yu, L., Zhou, Y., Liu, C., & Tong, G. (2020). Porcine Reproductive and Respiratory Syndrome Virus Antagonizes PCSK9’s Antiviral Effect via Nsp11 Endoribonuclease Activity. Viruses, 12(6), 655. https://doi.org/10.3390/v12060655