The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities

 , ,
, ,  and
and 
Abstract
1. Introduction
2. Material and Methods
2.1. Genome Sequences
2.2. miR Prediction
2.3. Mutational Analysis of Potential miRNA Sites
2.4. Pathway Analysis
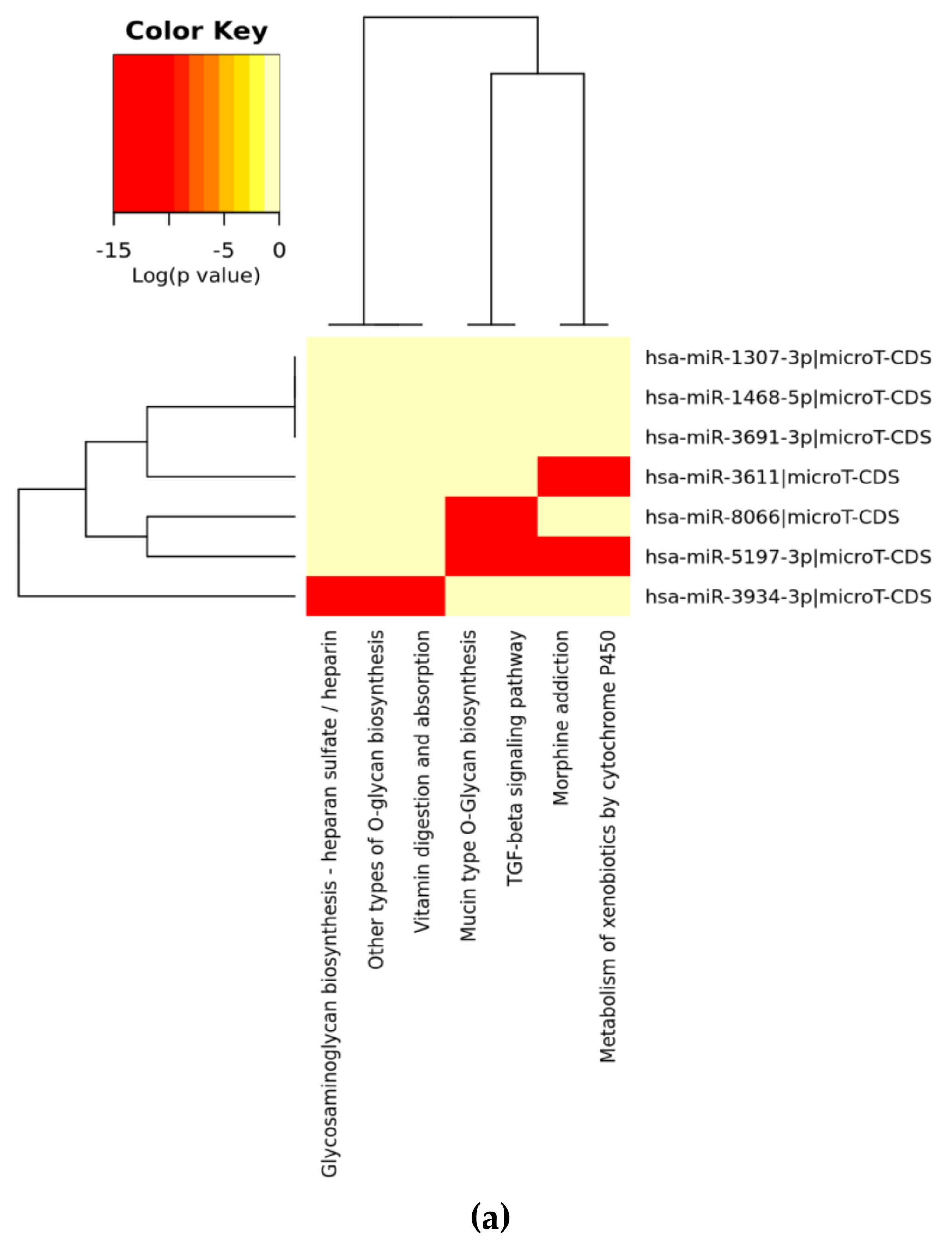
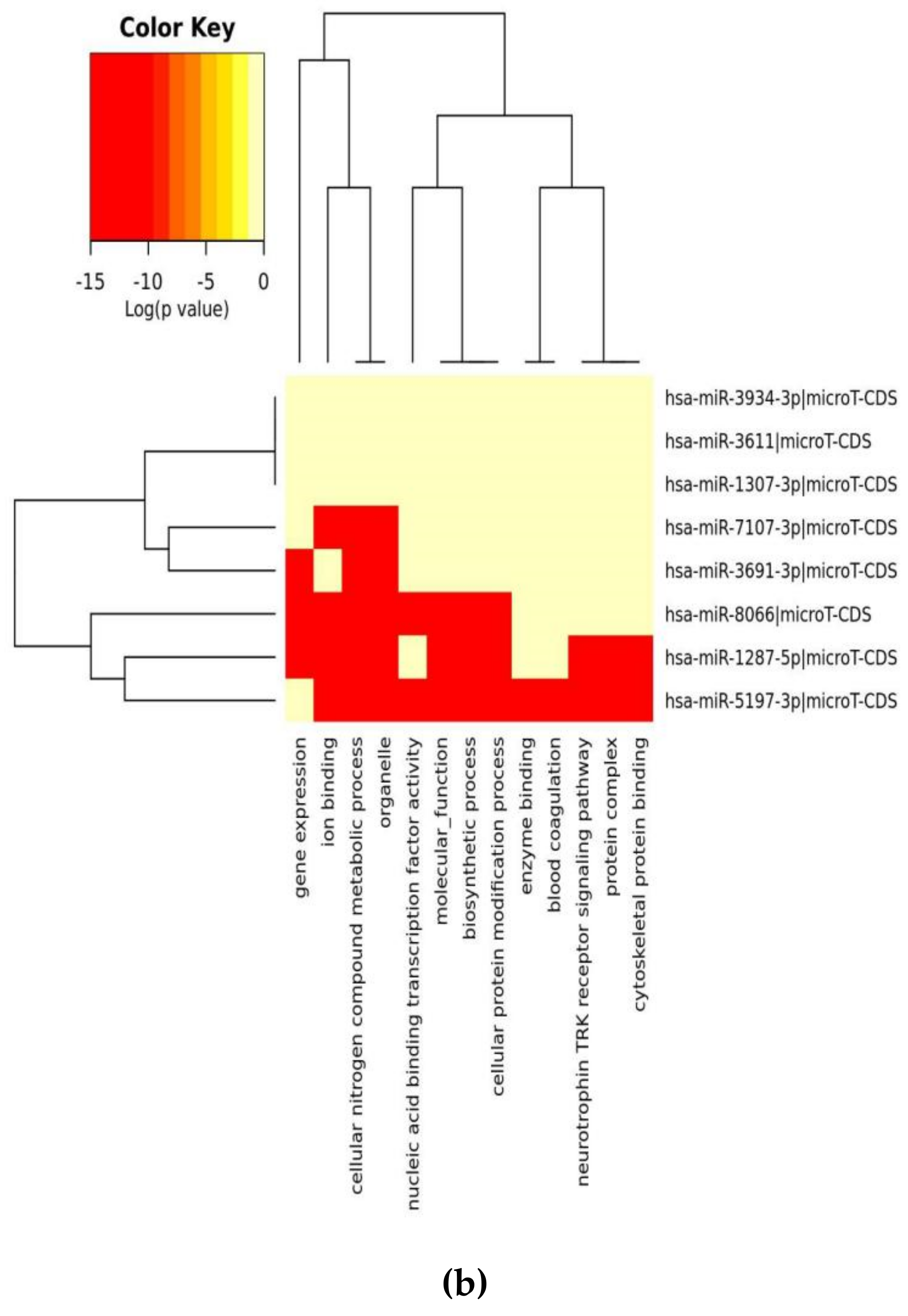
3. Results
3.1. Analysis of SARS-CoV-2 Viral Genome for miR Sequences with High Human Similarity and Functional Characterisation
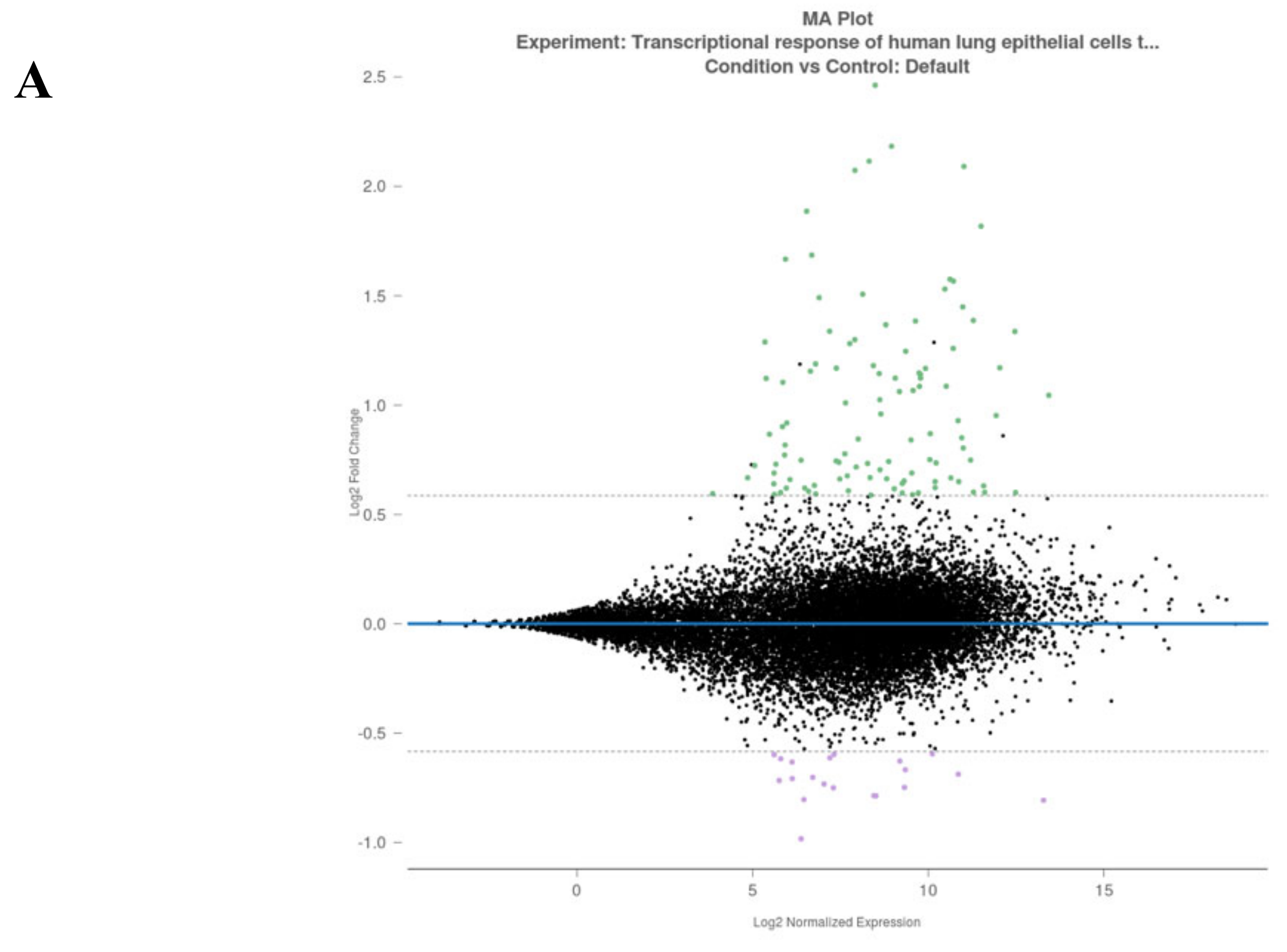
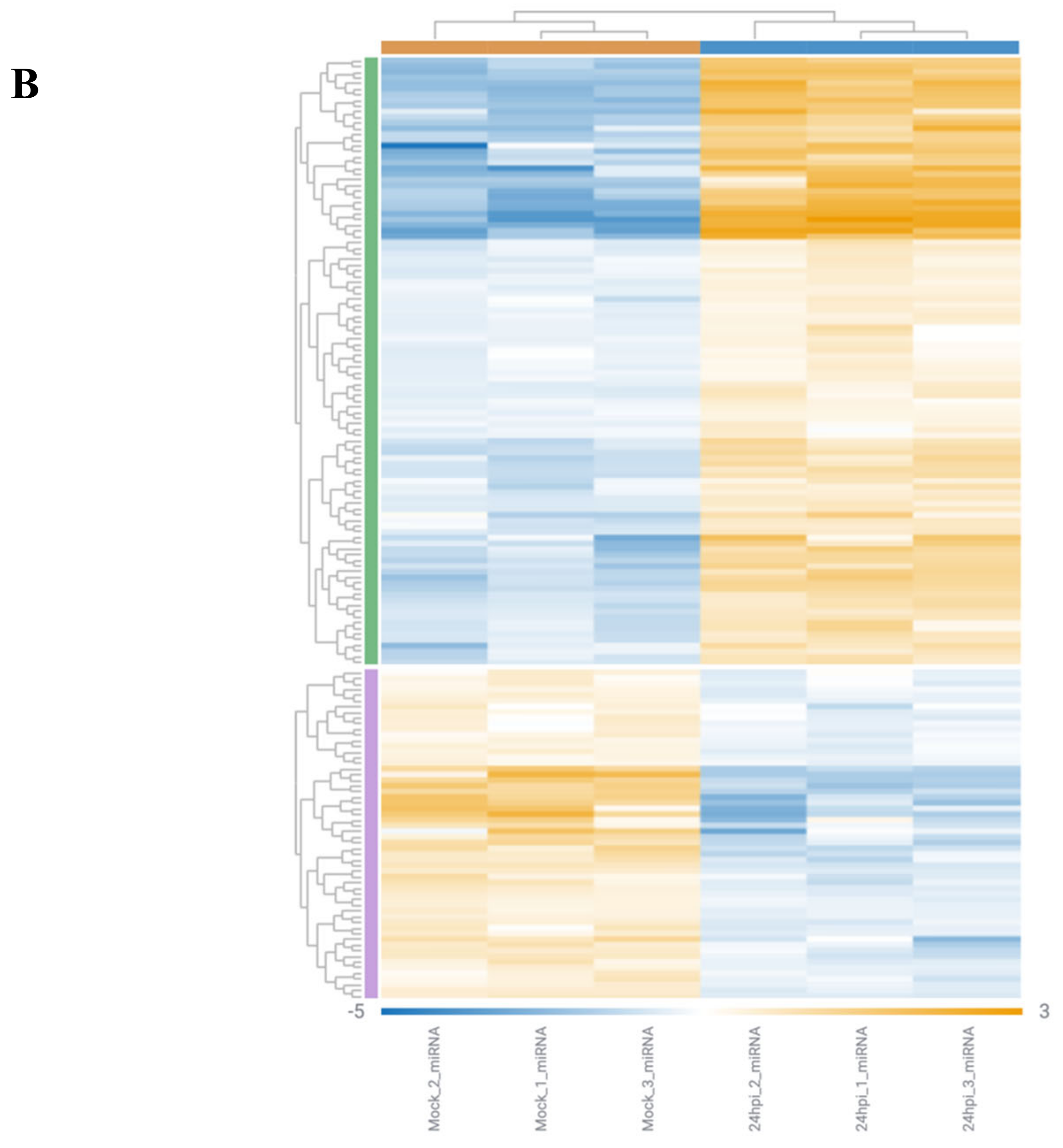
3.2. Analysis of Gene Alterations in NHEB Bronchial Epithelial and A549 Cells Due to SARS-CoV-2 Infection
4. Discussion
4.1. Biological Significance of Top Ranked miRs in Humans
4.1.1. miR-8066
4.1.2. miR-5197
4.1.3. miR-3611
4.1.4. miR-3934-3p
4.1.5. miR-1307-3p
4.1.6. miR-3691-3p
4.1.7. miR1468-5p
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Tsang, K.W.; Ooi, G.C.; Ho, P.L. Diagnosis and pharmacotherapy of severe acute respiratory syndrome: What have we learnt? Eur. Respir. J. 2004, 24, 1025–1032. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004, 5, 522–531. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Whisnant, A.W.; Kehl, T.; Bao, Q.; Materniak, M.; Kuzmak, J.; Löchelt, M.; Cullen, B.R. Identification of novel, highly expressed retroviral microRNAs in cells infected by bovine foamy virus. J. Virol. 2014, 88, 4679–4686. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S. Virus-encoded microRNAs. Virology 2011, 411, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Jongejan, A.; van Kampen, A.H.; Berkhout, B.; Das, A.T. Tat-dependent production of an HIV-1 TAR-encoded miRNA-like small RNA. Nucleic Acids Res. 2016, 44, 4340–4353. [Google Scholar] [CrossRef] [PubMed]
- Bruscella, P.; Bottini, S.; Baudesson, C.; Pawlotsky, J.M.; Feray, C.; Trabucchi, M. Viruses and miRNAs: More Friends than Foes. Front. Microbiol. 2017, 8, 824. [Google Scholar] [CrossRef] [PubMed]
- Nightingale, K.; Dry, I.; Hopkins, J.; Dalziel, R. Regulation of Ov2 by virus encoded microRNAs. Vet. Res. Commun. 2019, 43, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. MicroRNAs as mediators of viral evasion of the immune system. Nat. Immunol. 2013, 14, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Cullen, B.R. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J. Virol. 2004, 78, 12868–12876. [Google Scholar] [CrossRef] [PubMed]
- Bennasser, Y.; Yeung, M.L.; Jeang, K.-T. HIV-1 TAR RNA Subverts RNA Interference in Transfected Cells through Sequestration of TAR RNA-binding Protein, TRBP. J. Biol. Chem. 2006, 281, 27674–27678. [Google Scholar] [CrossRef] [PubMed]
- Harwig, A.; Das, A.T.; Berkhout, B. Retroviral microRNAs. Curr. Opin. Virol. 2014, 7, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Luna, J.M.; Scheel, T.K.H.; Rice, C.M.; Darnell, R.B. Hepatitis C Virus RNA Functionally Sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef]
- Piedade, D.; Azevedo-Pereira, J.M. The Role of microRNAs in the Pathogenesis of Herpesvirus Infection. Viruses 2016, 8, 156. [Google Scholar] [CrossRef]
- Sanghvi, V.R.; Steel, L.F. RNA silencing as a cellular defence against HIV-1 infection: Progress and issues. FASEB J. 2012, 26, 3937–3945. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, Y.; Bisaro, D.M.; Parris, D.S. Herpes simplex virus type 1 suppresses RNA-induced gene silencing in mammalian cells. J. Virol. 2009, 83, 6652–6663. [Google Scholar] [CrossRef]
- Hussain, M.; Asgari, S. MicroRNA-like viral small RNA from Dengue virus 2 autoregulates its replication in mosquito cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2746–2751. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, I.M.; Cheng, G.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Van Rooij, E.; Kauppinen, S. Development of microRNA therapeutics is coming of age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data-from vision to reality. EuroSurveillance 2017, 22. [Google Scholar] [CrossRef] [PubMed]
- Vlachos, I.S.; Konstantinos, Z.; Maria, D.; Paraskevopoulou, G.G.; Dimitra, K.; Thanasis, V.; Theodore, D.; Artemis, G.H. DIANA-miRPath v3.0: Deciphering microRNA function with experimental support. Nucleic Acids Res. 2015, 43, W460–W466. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk?/projects/fastqc/ (accessed on 18 April 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Alexa, A.; Rahnenfuehrer, J. topGO: Enrichment Analysis for Gene Ontology. R Package Version 2010, 2, 2010. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Attwood, T.K.; Babbitt, P.C.; Blum, M.; Bork, P.; Bridgem, A.; Brown, S.D.; Chang, H.Y.; El-Gebali, S.; Fraser, M.I.; et al. InterPro in 2019: Improving coverage, classification and access to protein sequence annotations. Nucleic Acids Res. 2019, 47, D351–D360. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2009, 38, D492–D496. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 43, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 43, 1739–1740. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Slenter, D.N.; Kutmon, M.; Hanspers, K.; Riutta, A.; Windsor, J.; Nunes, N.; Mélius, J.; Cirillo, E.; Coort, S.L.; Digles, D.; et al. WikiPathways: A multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2017, 46, D661–D667. [Google Scholar] [CrossRef]
- Saçar-Demirci, M.D.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Cai, S.J.; Wu, L.L.; Chen, D.F.; Li, Y.X.; Liu, Y.J.; Fan, Y.Q.; Du, S.H.; Huang, H.; Liu, N.; Caserta, S.; et al. Circulating Plasma microRNAs can differentiate Human Sepsis and Systemic Inflammatory Response Syndrome (SIRS). Sci. Rep. 2016, 6, 28006. [Google Scholar]
- Pluta, L.; Yousefi, B.; Damania, B.; Khan, A.A. Endosomal TLR-8 Senses microRNA-1294 Resulting in the Production of NFḱB Dependent Cytokines. Front. Immunol. 2019, 10, 2860. [Google Scholar] [CrossRef]
- Liao, B.; Zhou, M.X.; Zhou, F.K.; Luo, X.M.; Zhong, S.X.; Zhou, Y.F.; Qin, Y.S.; Li, P.P.; Qin, C. Exosome-Derived MiRNAs as Biomarkers of the Development and Progression of Intracranial Aneurysms. J. Atheroscler. Thromb. 2019, 27(6), 51102. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Rippo, M.R.; Procopio, A.D.; Fazioli, F. Circulating inflamma-miRs in aging and age-related diseases. Front. Genet. 2013, 4, 121. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [PubMed]
- Vaschetto, L. A putative miRNA in the spike gene of SARS-CoV-2 has perfect sequence identity to both the forward and reverse complementary strands of hsa-mir-8055 involved in T-cell response to antigen. OSF Preprints 2020. [Google Scholar] [CrossRef]
- Zhang, L.; Han, B.; Wang, J.; Liu, Q.; Kong, Y.; Jiang, D.; Jia, H. Differential expression profiles and functional analysis of circular RNAs in children with fulminant myocarditis. Epigenomics 2019, 11, 1129–1141. [Google Scholar] [CrossRef] [PubMed]
- Voss, D.; Pfefferle, S.; Drosten, C.; Stevermann, L.; Traggiai, E.; Lanzavecchia, A.; Becker, S. Studies on membrane topology, N-glycosylation and functionality of SARS-CoV membrane protein. Virol. J. 2009, 6, 79. [Google Scholar] [CrossRef]
- Shajahan, A.; Supekar, N.T.; Gleinich, A.; Azadi, P. Deducing the N- and O-glycosylation profile of the spike protein of novel coronavirus SARS-CoV-2. bioRxiv 2019. [Google Scholar] [CrossRef]
- Zhao, X.; Nicholls, J.M.; Chen, Y.G. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J. Biol. Chem. 2008, 283, 3272–3280. [Google Scholar] [CrossRef]
- Iwuchukwu, I.; Nguyen, D.; Beavers, M.; Tran, V.; Sulaiman, W.; Fannin, E.; Lasseigne, L.; Ramsay, E.; Wilson, J.; Bazan, N.G.; et al. MicroRNA Regulatory Network as Biomarkers of Late Seizure in Patients with Spontaneous Intracerebral Hemorrhage. Mol. Neurobiol. 2020. [Google Scholar] [CrossRef]
- Zhao, Y.; Wei, Q.; Hu, L.; Chen, F.; Hu, Z.; Heist, R.S.; Su, L.; Amos, C.I.; Shen, H.; Christiani, D.C. Polymorphisms in MicroRNAs are associated with survival in non-small cell lung cancer. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2503–2511. [Google Scholar] [CrossRef]
- Schotte, D.; Akbari Moqadam, F.; Lange-Turenhout, E.A.; Chen, C.; van Ijcken, W.F.; Pieters, R.; den Boer, M.L. Discovery of new microRNAs by small RNAome deep sequencing in childhood acute lymphoblastic leukemia. Leukemia 2011, 25, 1389–1399. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Li, X.P.; Gong, W.J.; Wu, N.Y.; Tang, J.; Yin, J.Y.; Li, X.; Zhang, W.; Zhou, H.H.; Liu, Z.Q. Age-related common miRNA polymorphism associated with severe toxicity in lung cancer patients treated with platinum-based chemotherapy. Clin. Exp. Pharmacol. Physiol. 2017, 44 (Suppl. S1), 21–29. [Google Scholar] [CrossRef]
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Mazurov, D.; Ilinskaya, A.; Heidecker, G.; Filatov, A. Role of O-glycosylation and expression of CD43 and CD45 on the surfaces of effector T cells in human T cell leukemia virus type 1 cell-to-cell infection. J. Virol. 2012, 86, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.J.; Linstedt, A.D. Site-specific glycosylation of Ebola virus glycoprotein by human polypeptide GalNAc-transferase 1 induces cell adhesion defects. J. Biol. Chem. 2018, 293, 19866–19873. [Google Scholar] [CrossRef]
- Lantéri, M.; Giordanengo, V.; Hiraoka, N.; Fuzibet, J.G.; Auberger, P.; Fukuda, M.; Baum, L.G.; Lefebvre, J.C. Altered T cell surface glycosylation in HIV-1 infection results in increased susceptibility to galectin-1-induced cell death. Glycobiology 2003, 13, 909–918. [Google Scholar] [CrossRef]
- Gaunitz, S.; Liu, J.; Nilsson, A.; Karlsson, N.; Holgersson, J. Avian influenza H5 hemagglutinin binds with high avidity to sialic acid on different O-linked core structures on mucin-type fusion proteins. Glycoconj. J. 2014, 31, 145–159. [Google Scholar] [CrossRef]
- Nordén, R.; Nyström, K.; Adamiak, B.; Halim, A.; Nilsson, J.; Larson, G.; Trybala, E.; Olofsson, S. Involvement of viral glycoprotein gC-1 in expression of the selectin ligand sialyl-Lewis X induced after infection with herpes simplex virus type 1. Apmis 2013, 121, 280–289. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, L.; Qiu, L.; Pan, R.; Bai, H.; Jiang, Y.; Wang, Z.; Bi, Y.; Chen, G.; Chang, G. Expression patterns of novel circular RNAs in chicken cells after avian leukosis virus subgroup J infection. Gene 2019, 15, 72–81. [Google Scholar] [CrossRef]
- Reid, D.J.; Pham, N.T. Emerging Therapeutic Options for the Management of COPD. Clin. Med. Insights Circ. Respir. Pulm. Med. 2013, 9, 7–15. [Google Scholar] [CrossRef]
- Shen, Z.; Tang, W.; Guo, J.; Sun, S. miR-483-5p plays a protective role in chronic obstructive pulmonary disease. Int. J. Mol. Med. 2017, 40, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Raveh, E.; Matouk, I.J.; Gilon, M.; Hochberg, A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis—A proposed unifying theory. Mol. Cancer 2015, 4, 184. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Martinez, P.; Séron, K.; Luo, G.; Allain, F.; Dubuisson, J.; Belouzard, S. Characterization of hepatitis C virus interaction with heparan sulfate proteoglycans. J. Virol. 2015, 89, 3846–3858. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Jiang, J.; Liang, B.; Wei, F.; Huang, J.; Pan, P.; Su, J.; Zhou, B.; Zang, N.; Ye, L.; et al. Opiate use inhibits TLR9 signaling pathway in vivo: Possible role in pathogenesis of HIV-1 infection. Sci. Rep. 2017, 7, 13071. [Google Scholar] [CrossRef]
- Nyland, S.B.; Cao, C.; Bai, Y.; Loughran, T.P.; Ugen, K.E. Modulation of infection and type 1 cytokine expression parameters by morphine during in vitro coinfection with human T-cell leukemia virus type I and HIV-1. J. Acquir. Immune Defic. Syndr. 2003, 32, 406–416. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Douglas, S.D.; Lai, J.P.; Xiao, W.D.; Pleasure, D.E.; Ho, W.Z. Morphine enhances hepatitis C virus (HCV) replicon expression. Am. J. Pathol. 2003, 163, 1167–1175. [Google Scholar] [CrossRef]
- Li, Y.; Ye, L.; Peng, J.S.; Wang, C.Q.; Luo, G.X.; Zhang, T.; Wan, Q.; Ho, W.Z. Morphine inhibits intrahepatic interferon- alpha expression and enhances complete hepatitis C virus replication. J. Infect. Dis. 2007, 196, 719–730. [Google Scholar] [CrossRef]
- Chuang, R.Y.; Suzuki, S.; Chuang, T.K.; Miyagi, T.; Chuang, L.F.; Doi, R.H. Opioids and the progression of simian AIDS. Front. Biosci. 2005, 10, 1666–1677. [Google Scholar] [CrossRef][Green Version]
- Coussons-Read, M.E.; Daniels, M.; Gilmour, M.I. Morphine alters the immune response to influenza virus infection in Lewis rats. Adv. Exp. Med. Biol. 1998, 437, 73–82. [Google Scholar]
- Chung, K.F. Drugs to suppress cough. Expert Opin. Investig. Drugs 2005, 14, 19–27. [Google Scholar] [CrossRef]
- Hiew, M.S.Y.; Cheng, H.P.; Huang, C.J.; Chong, K.Y.; Cheong, S.K.; Choo, K.B.; Kamarul, T. Incomplete cellular reprogramming of colorectal cancer cells elicits an epithelial/mesenchymal hybrid phenotype. J. Biomed. Sci. 2018, 25, 57. [Google Scholar] [CrossRef] [PubMed]
- Yerukala Sathipati, S.; Ho, S.Y. Identifying the miRNA signature associated with survival time in patients with lung adenocarcinoma using miRNA expression profiles. Sci. Rep. 2017, 7, 7507. [Google Scholar] [CrossRef] [PubMed]
- Ren, A.; Wen, Z.; Zheng, L. Downregulation of miR-3934-5p enhances A549 cell sensitivity to cisplatin by targeting TP53INP1. Exp. Ther. Med. 2019, 18, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Trivellas, A.; Pellatt, A.J.; Mullany, L.E.; Stevens, J.R.; Wolff, R.K.; Herrick, J.S. Genetic variants in the TGF-β-signaling pathway influence expression of miRNAs in colon and rectal normal mucosa and tumor tissue. Oncotarget 2017, 8, 16765–16783. [Google Scholar] [CrossRef]
- Chinnapaiyan, S.; Dutta, R.K.; Nair, M.; Chand, H.S.; Rahman, I.; Unwalla, H.J. TGF-β1 increases viral burden and promotes HIV-1 latency in primary differentiated human bronchial epithelial cells. Sci. Rep. 2019, 9, 12552. [Google Scholar] [CrossRef]
- Lang, J.; Yang, N.; Deng, J.; Liu, K.; Yang, P.; Zhang, G.; Jiang, C. Inhibition of SARS pseudovirus cell entry by lactoferrin binding to heparan sulfate proteoglycans. PLoS ONE 2011, 6, e23710. [Google Scholar] [CrossRef]
- Milewska, A.; Zarebski, M.; Nowak, P.; Stozek, K.; Potempa, J.; Pyrc, K. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 2014, 88, 13221–13230. [Google Scholar] [CrossRef]
- Tamhankar, M.; Gerhardt, D.M.; Bennett, R.S.; Murphy, N.; Jahrling, P.B.; Patterson, J.L. Heparan sulfate is an important mediator of Ebola virus infection in polarized epithelial cells. Virol. J. 2018, 15, 135. [Google Scholar] [CrossRef]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef]
- Robinson-McCarthy, L.R.; McCarthy, K.R.; Raaben, M.; Piccinotti, S.; Nieuwenhuis, J.; Stubbs, S.H.; Bakkers, M.J.G.; Whelan, S.P.J. Reconstruction of the cell entry pathway of an extinct virus. PLoS Pathog. 2018, 14, e1007123. [Google Scholar] [CrossRef]
- Kim, S.Y.; Koetzner, C.A.; Payne, A.F.; Nierode, G.J.; Yu, Y.; Wang, R.; Barr, E.; Dordick, J.S.; Kramer, L.D.; Zhang, F.; et al. Glycosaminoglycan Compositional Analysis of Relevant Tissues in Zika Virus Pathogenesis and in Vitro Evaluation of Heparin as an Antiviral against Zika Virus Infection. Biochemistry 2019, 58, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Huan, C.C.; Wang, Y.; Ni, B.; Wang, R.; Huang, L.; Ren, X.F.; Tong, G.Z.; Ding, C.; Fan, H.J.; Mao, X. Porcine epidemic diarrhea virus uses cell-surface heparan sulfate as an attachment factor. Arch. Virol. 2015, 160, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Anindita, P.D.; Ito, N.; Sugiyama, M.; Carr, M.; Fukuhara, H.; Ose, T.; Maenaka, K.; Takada, A.; Hall, W.W.; et al. The Role of Heparan Sulfate Proteoglycans as an Attachment Factor for Rabies Virus Entry and Infection. J. Infect. Dis. 2018, 217, 1740–1749. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Dhondt, K.P.; Châlons, M.; Mély, S.; Raoul, H.; Negre, D.; Cosset, F.L.; Gerlier, D.; Vivès, R.R.; Horvat, B. Heparan sulfate-dependent enhancement of henipavirus infection. Mbio 2015, 6, e02427. [Google Scholar] [CrossRef]
- Picchianti-Diamanti, A.; Rosado, M.M.; D’Amelio, R. Infectious Agents and Inflammation: The Role of Microbiota in Autoimmune Arthritis. Front. Microbiol. 2018, 8, 2696. [Google Scholar] [CrossRef] [PubMed]
- Demignot, S.; Beilsteinm, F.; Morel, E. Triglyceride-rich lipoproteins and cytosolic lipid droplets in enterocytes: Key players in intestinal physiology and metabolic disorders. Biochimie 2014, 96, 48–55. [Google Scholar] [CrossRef]
- Herzlich, B.C.; Schiano, T.D.; Moussa, Z.; Zimbalist, E.; Panagopoulos, G.; Ast, A.; Nawabi, I. Decreased intrinsic factor secretion in AIDS: Relation to parietal cell acid secretory capacity and vitamin B12 malabsorption. Am. J. Gastroenterol. 1992, 87, 1781–1788. [Google Scholar]
- Ehrenpreis, E.D.; Carlson, S.J.; Boorstein, H.L.; Craig, R.M. Malabsorption and deficiency of vitamin B12 in HIV-infected patients with chronic diarrhea. Dig. Dis. Sci. 1994, 39, 2159–2162. [Google Scholar] [CrossRef]
- Sultana, S.; Li, H.; Puche, A.; Jones, O.; Bryant, J.L.; Royal, W. Quantitation of parvalbumin+ neurons and human immunodeficiency virus type 1 (HIV-1) regulatory gene expression in the HIV-1 transgenic rat: Effects of vitamin A deficiency and morphine. J. Neurovirol. 2010, 16, 33–40. [Google Scholar] [CrossRef][Green Version]
- West, C.E.; Sijtsma, S.R.; Kouwenhoven, B.; Rombout, J.H.; van der Zijpp, A.J. Epithelia-damaging virus infections affect vitamin A status in chickens. J. Nutr. 1992, 122, 333–339. [Google Scholar] [CrossRef]
- Daneshkhah, A.; Eshein, A.; Subramanian, H.; Roy, H.K.; Backman, V. The Role of Vitamin D in Suppressing Cytokine Storm in COVID-19 Patients and Associated Mortality. medRxiv 2020. [Google Scholar] [CrossRef]
- Nagai, A.; Matsumiya, H.; Hayashi, M.; Yasui, S.; Okamoto, H.; Konno, K. Effects of nicotinamide and niacin on bleomycin-induced acute injury and subsequent fibrosis in hamster lungs. Exp. Lung Res. 1994, 20, 263–281. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Rivero, M.; Zhang, R.; Heilmann-Heimbach, S.; Mueller, A.; Bagci, S.; Dresbach, T.; Schröder, L.; Holdenrieder, S.; Reutter, H.M.; Kipfmueller, F. Circulating microRNAs are associated with Pulmonary Hypertension and Development of Chronic Lung Disease in Congenital Diaphragmatic Hernia. Sci. Rep. 2018, 8, 10735. [Google Scholar] [CrossRef] [PubMed]
- Ruan, D.T.; Gao, S.; Shelat, H.; King, B.; Geng, Y.J. Differential expression of microRNA and arachidonic acid metabolism in aspirin-treated human cardiac and peri-cardiac fat-derived mesenchymal stem cells. Vascul. Pharmacol. 2020, 127, 106651. [Google Scholar] [CrossRef] [PubMed]
- Grant, J.S.; White, K.; MacLean, M.R.; Baker, A.H. MicroRNAs in pulmonary arterial remodeling. Cell Mol. Life Sci. 2013, 70, 4479–4494. [Google Scholar] [CrossRef] [PubMed]
- Alejandre-Alcázar, M.A.; Michiels-Corsten, M.; Vicencio, A.G.; Reiss, I.; Ryu, J.; de Krijger, R.R.; Haddad, G.G.; Tibboel, D.; Seeger, W.; Eickelberg, O.; et al. TGF-beta signaling is dynamically regulated during the alveolarization of rodents and human lungs. Dev. Dyn. 2008, 237, 259–269. [Google Scholar] [CrossRef]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim. Biophys. Acta 2008, 1782, 197–228. [Google Scholar] [CrossRef]
- Morty, R.E.; Königshoff, M.; Eickelberg, O. Transforming growth factor-beta signaling across ages: From distorted lung development to chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2009, 6, 607–613. [Google Scholar] [CrossRef]
- Lal, C.V.; Olave, N.; Travers, C.; Rezonzew, G.; Dolma, K.; Simpson, A.; Halloran, B.; Aghai, Z.; Das, P.; Sharma, N.; et al. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 2018, 3, 93994. [Google Scholar] [CrossRef]
- Lin, R.; Rahtu-Korpela, L.; Magga, J.; Ulvila, J.; Swan, J.; Kemppi, A.; Pakanen, L.; Porvari, K.; Huikuri, H.; Junttila, J.; et al. miR-1468-3p promotes aging-related cardiac fibrosis. Mol. Ther. Nucl. Acids 2020. [Google Scholar] [CrossRef] [PubMed]
- Erener, S.; Marwaha, A.; Tan, R.; Panagiotopoulos, C.; Kieffer, T.J. Profiling of circulating microRNAs in children with recent onset of type 1 diabetes. JCI Insight 2017, 2, e89656. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Zhi, T.; Xu, W.; Xu, X.; Wu, W.; Yu, T.; Nie, E.; Zhou, X.; Bao, Z.; Jin, X.; et al. MicroRNA-1468-5p inhibits glioma cell proliferation and induces cell cycle arrest by targeting RRM1. Am. J. Cancer Res. 2017, 7, 784–800. [Google Scholar] [PubMed]
- Liu, F.; Zhao, H.; Gong, L.; Yao, L.; Li, Y.; Zhang, W. MicroRNA-129-3p functions as a tumor suppressor in serous ovarian cancer by targeting BZW1. Int. J. Clin. Exp. Pathol. 2018, 11, 5901–5908. [Google Scholar]
- Ludwig, N.; Fehlmann, T.; Kern, F.; Gogol, M.; Maetzler, W.; Deutscher, S.; Gurlit, S.; Schulte, C.; von Thaler, A.K.; Deuschle, C.; et al. Machine Learning to Detect Alzheimer’s Disease from Circulating Non-coding RNAs. Genom. Proteom. Bioinform. 2019, 17, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Feng, B.; Han, S.; Lu, L.; Chen, Y.; Chu, X.; Wang, R.; Chen, L. MicroRNA-129 in Human Cancers: From Tumorigenesis to Clinical Treatment. Cell Physiol. Biochem. 2016, 39, 2186–2202. [Google Scholar] [CrossRef]
- Tang, X.; Tang, J.; Liu, X.; Zeng, L.; Cheng, C.; Luo, Y.; Li, L.; Qin, S.L.; Sang, Y.; Deng, L.M.; et al. Downregulation of miR-129-2 by promoter hypermethylation regulates breast cancer cell proliferation and apoptosis. Oncol. Rep. 2016, 35, 2963–2969. [Google Scholar] [CrossRef]
- Chen, X.; Ruan, A.; Wang, X.; Han, W.; Wang, R.; Lou, N.; Ruan, H.; Qiu, B.; Yang, H.; Zhang, X. miR-129-3p, as a diagnostic and prognostic biomarker for renal cell carcinoma, attenuates cell migration and invasion via downregulating multiple metastasis-related genes. J. Cancer Res. Clin. Oncol. 2014, 140, 1295–1304. [Google Scholar] [CrossRef]
- Wong, K.Y.; Yim, R.L.; Kwong, Y.L.; Leung, C.Y.; Hui, P.K.; Cheung, F.; Liang, R.; Jin, D.Y.; Chim, C.S. Epigenetic inactivation of the MIR129-2 in hematological malignancies. J. Hematol. Oncol. 2013, 6, 16. [Google Scholar] [CrossRef]
- Wu, J.; Qian, J.; Li, C.; Kwok, L.; Cheng, F.; Liu, P.; Perdomo, C.; Kotton, D.; Vaziri, C.; Anderlind, C.; et al. miR-129 regulates cell proliferation by downregulating Cdk6 expression. Cell Cycle 2010, 9, 1809–1818. [Google Scholar] [CrossRef]
- Katayama, Y.; Maeda, M.; Miyaguchi, K.; Nemoto, S.; Yasen, M.; Tanaka, S.; Mizushima, H.; Fukuoka, Y.; Arii, S.; Tanaka, H. Identification of pathogenesis-related microRNAs in hepatocellular carcinoma by expression profiling. Oncol. Lett. 2012, 4, 817–823. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sannigrahi, M.K.; Sharma, R.; Singh, V.; Panda, N.K.; Rattan, V.; Khullar, M. DNA methylation regulated microRNAs in HPV-16-induced head and neck squamous cell carcinoma (HNSCC). Mol. Cell Biochem. 2018, 448, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Dreher, A.; Rossing, M.; Kaczkowski, B.; Andersen, D.K.; Larsen, T.J.; Christophersen, M.K.; Nielsen, F.C.; Norrild, B. Differential expression of cellular microRNAs in HPV 11, -16, and -45 transfected cells. Biochem. Biophys. Res. Commun. 2011, 412, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–106. [Google Scholar] [PubMed]
- Liu, Z.; Wang, J.; Xu, Y.; Guo, M.; Mi, K.; Xu, R.; Pei, Y.; Zhang, Q.; Luan, X.; Hu, Z.; et al. Implications of the virus-encoded miRNA and host miRNA in the pathogenicity of SARS-CoV-2. arXiv 2020, arXiv:2004.04874. [Google Scholar]
- Rakhmetullina, A.; Ivashchenko, A.; Akimniyazova, A.; Aisina, D.; Pyrkova, A. The miRNA Complexes Against Coronaviruses COVID-19, SARS-CoV, And MERS-CoV. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Chen, L.; Zhong, L. Genomics functional analysis and drug screening of SARS-CoV-2. Genes Dis. 2020. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRs | Score | E-Value | Alignment | Wuhan | Italy | UK | Valencia | Turkey | Vero E6 |
|---|---|---|---|---|---|---|---|---|---|
| NC_045512.2 | MT066156.1 | hCoV-19/England/20136087804/2020|EPI_ISL_420910 | MT198652.2 | hCoV-19/Turkey/GLAB-CoV008/2020 | hCoV-19/Turkey/ERAGEM-001/2020 | ||||
| hsa-miR-8066 | 80 | 1.6–2.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-5197-3p | 79 | 1.6–2.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-3611 | 77 | 2.8–3.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-3934-3p | 76 | 3.4–5.0 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-1468-5p | 71 | 4.7–8.8 |  | √ | √ | √ | √ | √ | √ |
| hsa-miR-1307-3p | 72 | 4.3–6.3 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3691-3p | 74 | 5.0–9.5 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3120-5p | 73 | 6.0–7.2 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3914 | 73 | 6.0–8.5 |  | √ | √ | √ | √ | √ | |
| hsa-miR-3672 | 72 | 7.3–9.8 |  | X | X | X | X | ||
| hsa-miR-7107-3p | 73 | 6.0–6.2 |  | √ | √ | √ | √ | √ | |
| hsa-miR-1287-5p | 73 | 6.0–8.3 |  | √ | √ | √ | √ | √ | |
| hsa-miR-129-2-3p | 73 | 6.0–7.7 |  | √ | √ | √ | |||
| hsa-miR-378c | 71 | 8.8–9.3 |  | √ | √ | √ | |||
| hsa-miR-10397-5p | 72 | 6.9–10.0 |  | √ | √ | √ | |||
| hsa-miR-584-3p | 72 | 7.3–9.8 |  | √ | √ | ||||
| hsa-miR-3085-3p | 71 | 8.8–9.9 |  | √ | √ | √ | |||
| hsa-miR-3191-3p | 70 | 7.4–8.5 |  | √ | √ | ||||
| hsa-miR-148b-3p | 72 | 8.2–9.8 |  | √ | √ | ||||
| hsa-miR-3529-3p | 69 | 9.0 |  | √ | |||||
| hsa-miR-3682-5p | 68 | 9.0 |  | √ | √ |
| KEGG Pathway (A) | p-Value | #genes | #miRNAs |
| Mucin type O-Glycan biosynthesis | 2.52 × 10−2 | 7 | 3 |
| TGF-beta signaling pathway | 4.96 × 10−1 | 12 | 4 |
| Morphine addiction | 0.0001128919 | 14 | 5 |
| Metabolism of xenobiotics by cytochrome P450 | 0.0002215491 | 5 | 2 |
| Other types of O-glycan biosynthesis | 0.0003646344 | 1 | 1 |
| Vitamin digestion and absorption | 0.001008222 | 2 | 1 |
| Glycosaminoglycan biosynthesis—heparan sulfate/heparin | 0.00385809 | 1 | 1 |
| GABAergic synapse | 0.01342039 | 13 | 4 |
| Cytokine-cytokine receptor interaction | 0.02096334 | 9 | 1 |
| Signaling pathways regulating pluripotency of stem cells | 0.180299 | 9 | 1 |
| Amphetamine addiction | 0.2150865 | 7 | 1 |
| Axon guidance | 0.2239648 | 22 | 3 |
| Hippo signaling pathway | 0.2278356 | 7 | 1 |
| Prolactin signaling pathway | 0.2284669 | 5 | 1 |
| mRNA surveillance pathway | 0.2795597 | 1 | 1 |
| Glycosphingolipid biosynthesis—lacto and neolacto series | 0.3157068 | 1 | 1 |
| Bile secretion | 0.4120997 | 1 | 1 |
| Circadian entrainment | 0.4608082 | 9 | 1 |
| N-Glycan biosynthesis | 0.488078 | 2 | 1 |
| Mismatch repair | 0.6174557 | 1 | 1 |
| Drug metabolism—cytochrome P450 | 0.7063987 | 6 | 1 |
| Glutamatergic synapse | 0.7319762 | 6 | 1 |
| Glycosaminoglycan degradation | 0.7395672 | 2 | 1 |
| Antigen processing and presentation | 0.7591685 | 1 | 1 |
| GO Category (B) | p-Value | #genes | #miRNAs |
| organelle | 1 × 10−38 | 848 | 6 |
| cellular nitrogen compound metabolic process | 1 × 10−12 | 414 | 7 |
| ion binding | 8 × 10−8 | 495 | 7 |
| biosynthetic process | 3 × 10−7 | 351 | 7 |
| nucleic acid binding transcription factor activity | 4 × 10−2 | 115 | 6 |
| cellular protein modification process | 2 × 101 | 205 | 7 |
| molecular_function | 5 × 103 | 1303 | 7 |
| cellular_component | 1 × 105 | 1312 | 7 |
| enzyme binding | 2 × 105 | 119 | 5 |
| gene expression | 3 × 105 | 54 | 6 |
| protein binding transcription factor activity | 1 × 106 | 52 | 5 |
| blood coagulation | 0.000176061599974 | 44 | 6 |
| protein complex | 0.00115693944276 | 290 | 5 |
| post-translational protein modification | 0.00185490003064 | 19 | 5 |
| neurotrophin TRK receptor signaling pathway | 0.00197464302174 | 24 | 5 |
| synaptic transmission | 0.00204631087649 | 42 | 5 |
| cellular protein metabolic process | 0.00275618650845 | 39 | 5 |
| small molecule metabolic process | 0.00275618650845 | 170 | 7 |
| cytoskeletal protein binding | 0.00396272124679 | 68 | 4 |
| cell-cell signaling | 0.00396272124679 | 60 | 5 |
| transcription, DNA-templated | 0.00450420995446 | 208 | 6 |
| symbiosis, encompassing mutualism through parasitism | 0.0140041886634 | 41 | 5 |
| catabolic process | 0.0141620388146 | 142 | 6 |
| Fc-epsilon receptor signaling pathway | 0.0222360628043 | 15 | 6 |
| cellular component assembly | 0.02375306792 | 99 | 5 |
| transcription initiation from RNA polymerase II promoter | 0.0250016205995 | 24 | 5 |
| nucleoplasm | 0.0335128910566 | 92 | 6 |
| platelet activation | 0.0350801245107 | 20 | 5 |
| positive regulation of telomere maintenance via telomerase | 0.0370638891992 | 3 | 3 |
| RNA polymerase II core promoter proximal region sequence-specific DNA binding transcription factor activity involved in positive regulation of transcription | 0.0448871926331 | 32 | 5 |
| O-glycan processing | 0.0449415771561 | 8 | 5 |
| miRs | Alignment | Wuhan/China | Italy | Spain | France | England | USA | India |
|---|---|---|---|---|---|---|---|---|
| n = 28 | n = 44 | n = 133 | n = 104 | n = 104 | n = 104 | n = 34 | ||
| hsa-miR-8066 | ccaaaagaucacauug | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-5197-3p | auucgaagacccagucccuacuu | 0 | 0 | 0 | 0 | 0 | 0.9% | 0 |
| hsa-miR-3611 | ugagaagcaagaaauucuu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3934-3p | ucagguuggacagcugg | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-1307-3p | accgaggccacgcggagu | 3.5% | 2.2% | 8.27% | 1.92% | 2.88% | 2.88% | 38.23% |
| hsa-miR-3691-3p | gagauguugacacagacuuugu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-1468-5p | cucaguuugccuguuu | 0 | 0 | 2.25% | 0.96% | 0 | 0 | 8.83% |
| hsa-miR-3120-5p | uguagaggaggcaaagacag | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3914 | caucucacuugcugguuccu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3672 | ugagucucauggaaaaca | 0 | 0 | 0.75% | 0.96% | 0 | 0 | 0 |
| hsa-miR-378c | acugggcauugauuuagaugagugg | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-7107-3p | ccaaaaagagaaagucaaca | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-1287-5p | acucaaaccacugaaacagc | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-10397-5p | uucuucaccugaugcugu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-584-3p | gccugguuugccuggcac | 0 | 0 | 0.75% | 0 | 0 | 0 | 0 |
| hsa-miR-3085-3p | ucuggcuguuauggcc | 0 | 0 | 0 | 0 | 0 | 0.96% | 0 |
| hsa-miR-3191-3p | cugucuauccaguugcgucacca | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-3529-3p | uggcagacgggcgauuuuguu | 0 | 0 | 0 | 0 | 0 | 0 | 2.94% |
| hsa-miR-3682-5p | auagcacaaguagauguag | 0 | 0 | 0 | 0 | 0 | 0.96% | 0 |
| hsa-miR-148b-3p | aaguucuaugaugcacag | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| hsa-miR-129-2-3p | ugauuuuuguggaaagggcu | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| WikiPathways | p-Adj |
| Photodynamic therapy-induced NF-kB survival signaling | 0 |
| IL-18 signaling pathway | 8.6 × 10−9 |
| miRNAs involvement in the immune response in sepsis | 2.4 × 10−8 |
| Cytokines and Inflammatory Response | 9.9 × 10−7 |
| Lung fibrosis | 2.5 × 10−6 |
| BioPlanet | p-Adj |
| Oncostatin M | 0 |
| Interleukin-1 regulation of extracellular matrix | 0 |
| Interleukin-5 regulation of apoptosis | 0 |
| TNF-alpha effects on cytokine activity, cell motility, and apoptosis | 0 |
| Immune system signaling by interferons, interleukins, prolactin, and growth hormones | 0 |
| KEGG | p-Adj |
| IL-17 signaling pathway | 1.3 × 10−9 |
| TNF signaling pathway | 1.6 × 10−9 |
| Legionellosis | 3.5 × 10−9 |
| Rheumatoid arthritis | 5.4 × 10−9 |
| Cytokine-cytokine receptor interaction | 6.6 × 10−9 |
| PANTHER | p-Adj |
| Plasminogen activating cascade | 0.00156 |
| Toll receptor signaling pathway | 0.00911 |
| CCKR signaling map ST | 0.02550 |
| Apoptosis signaling pathway | 0.10282 |
| Blood coagulation | 0.10433 |
| REACTOME | p-Adj |
| Interferon alpha/beta signaling | 1.6 × 10−9 |
| Interleukin-10 signaling | 2.5 × 10−9 |
| Interleukin-4 and Interleukin-13 signaling | 2.4 × 10−7 |
| Formation of the cornified envelope | 1.3 × 10−5 |
| Chemokine receptors bind chemokines | 0.00047 |
| Small Molecule Pathway DB | p-Adj |
| CD40L Signalling Pathway | 0.25268 |
| NF-kB Signaling Pathway | 0.25268 |
| Toll-Like Receptor Pathway 2 | 0.25268 |
| Capecitabine Metabolism Pathway | 0.25268 |
| Capecitabine Action Pathway | 0.25268 |
| BIOCYC | p-Adj |
| vitamin D3 biosynthesis | 0.03597 |
| guanosine nucleotides degradation | 0.03597 |
| retinoate biosynthesis II | 0.03597 |
| guanosine nucleotides degradation III | 0.03597 |
| adenosine nucleotides degradation II | 0.03597 |
| Pathway Interaction DB | p-Adj |
| Validated transcriptional targets of AP1 family members Fra1 and Fra2 | 3.8 × 10−5 |
| IL23-mediated signaling events | 0.00050 |
| CD40/CD40L signaling | 0.02539 |
| Glucocorticoid receptor regulatory network | 0.02603 |
| LPA receptor mediated events | 0.04171 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arisan, E.D.; Dart, A.; Grant, G.H.; Arisan, S.; Cuhadaroglu, S.; Lange, S.; Uysal-Onganer, P. The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities. Viruses 2020, 12, 614. https://doi.org/10.3390/v12060614
Arisan ED, Dart A, Grant GH, Arisan S, Cuhadaroglu S, Lange S, Uysal-Onganer P. The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities. Viruses. 2020; 12(6):614. https://doi.org/10.3390/v12060614
Chicago/Turabian StyleArisan, Elif Damla, Alwyn Dart, Guy H. Grant, Serdar Arisan, Songul Cuhadaroglu, Sigrun Lange, and Pinar Uysal-Onganer. 2020. "The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities" Viruses 12, no. 6: 614. https://doi.org/10.3390/v12060614
APA StyleArisan, E. D., Dart, A., Grant, G. H., Arisan, S., Cuhadaroglu, S., Lange, S., & Uysal-Onganer, P. (2020). The Prediction of miRNAs in SARS-CoV-2 Genomes: hsa-miR Databases Identify 7 Key miRs Linked to Host Responses and Virus Pathogenicity-Related KEGG Pathways Significant for Comorbidities. Viruses, 12(6), 614. https://doi.org/10.3390/v12060614