Genome Analysis of a Novel Tembusu Virus in Taiwan


Abstract
1. Introduction
2. Materials and Methods
2.1. Collections of Mosquitoes
2.2. RNA Extraction and Real Time RT-PCR
2.3. Virus Isolation and Immunofluorescence Assay (IFA)
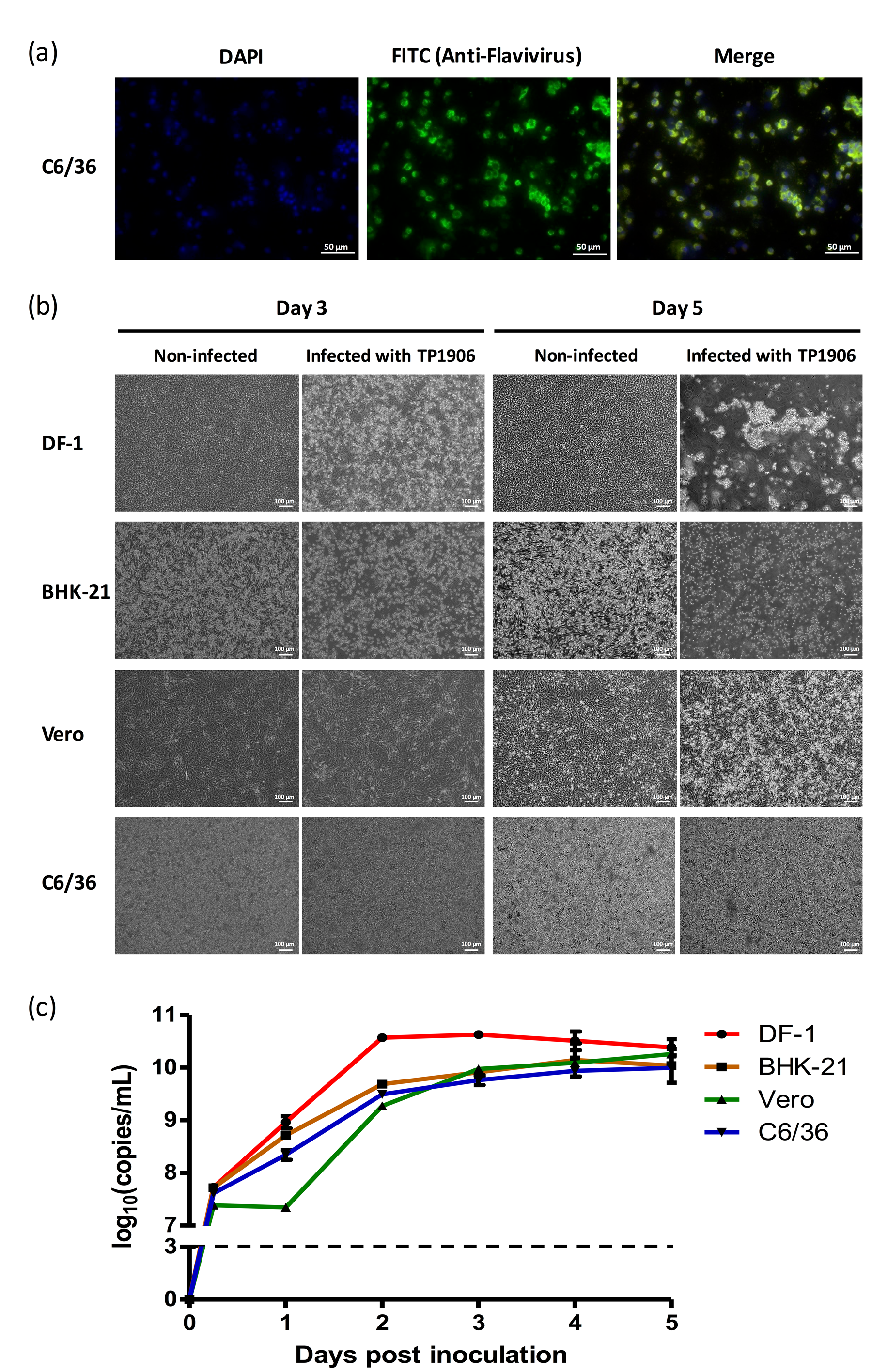
2.4. Viral Growth Kinetics
2.5. Complete Genome Sequencing
2.6. Phylogenetic Analysis
3. Results
3.1. Identification of Novel TMUV Strains from Culex Mosquitoes in Taiwan
3.2. Isolation of a Novel TMUV from the Cx. Annulus Mosquito Pool
3.3. Different Cell Lines Were Permissive for TMUV-TP1906 Infection
3.4. Characteristics of the Genome Sequence of TMUV Strain TP1906
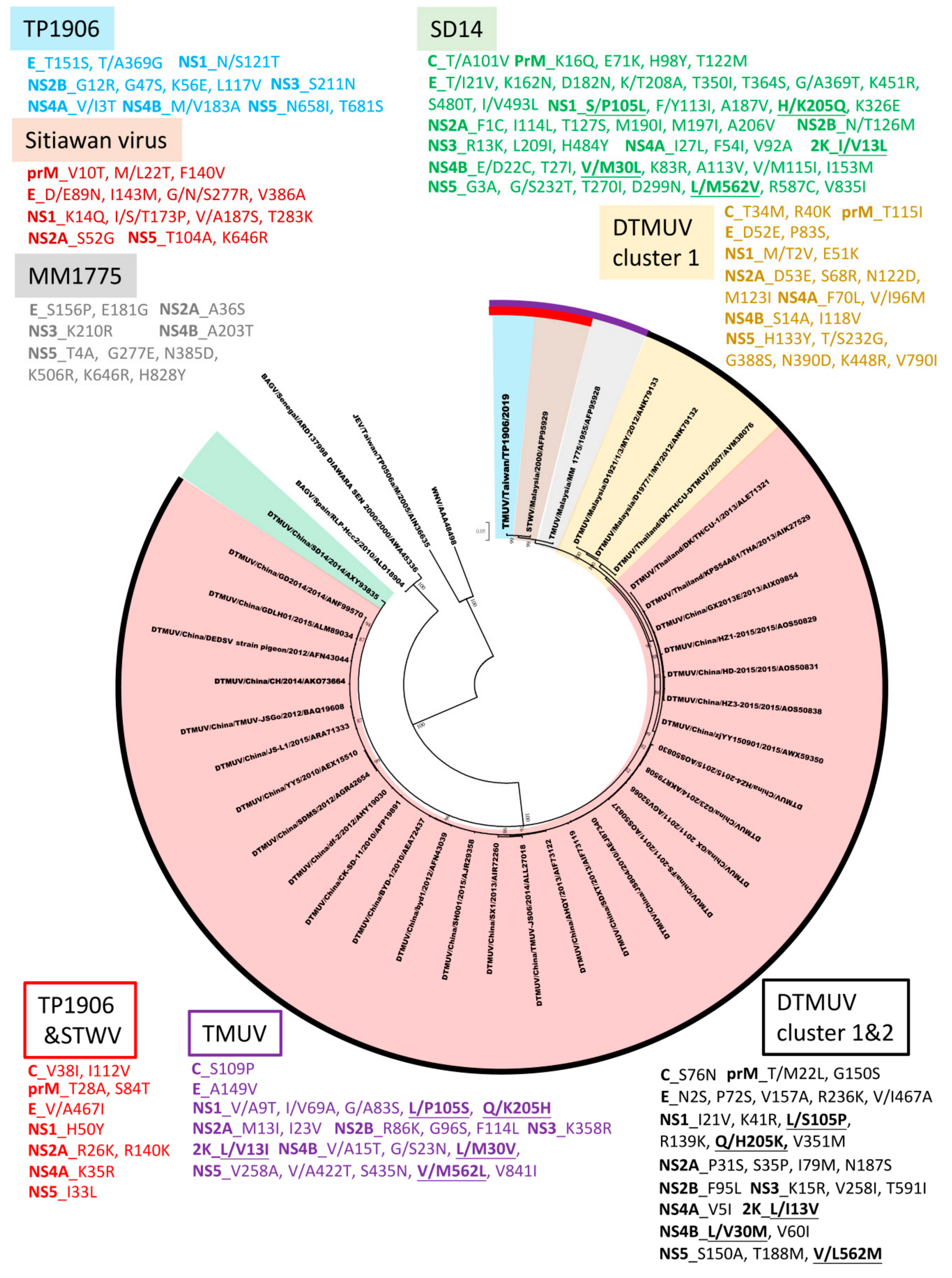
3.5. Phylogenetic Analysis of TMUV-TP1906 and Other Flaviviruses Based on ORF Sequences
3.6. Amino Acid Sequence Comparison between TMUV-TP1906 and Other TMUVs
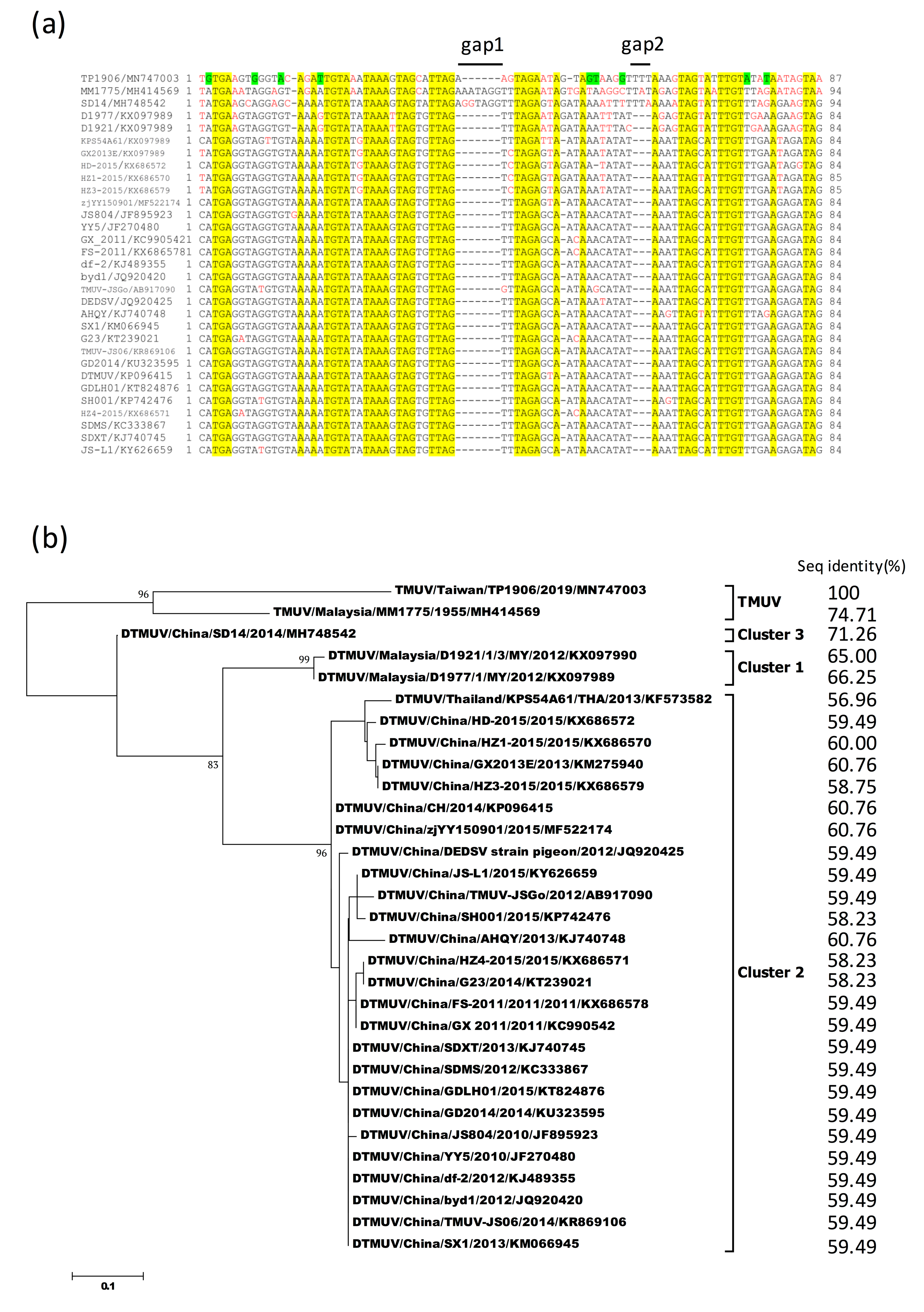
3.7. Sequence Alignment and Phylogenetic Analysis of 3′-UTR Variable Region of TMUV-TP1906 and Other TMUVs
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Benzarti, E.; Linden, A.; Desmecht, D.; Garigliany, M. Mosquito-borne epornitic flaviviruses: An update and review. J. Gen. Virol. 2019, 100, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kono, Y.; Tsukamoto, K.; Abd Hamid, M.; Darus, A.; Lian, T.C.; Sam, L.S.; Yok, C.N.; Di, K.B.; Lim, K.T.; Yamaguchi, S.; et al. Encephalitis and retarded growth of chicks caused by Sitiawan virus, a new isolate belonging to the genus Flavivirus. Am. J. Trop. Med. Hyg. 2000, 63, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, S.; Hu, X.; Yu, X.; Wang, Y.; Liu, P.; Lu, X.; Zhang, G.; Hu, X.; Liu, D.; et al. Duck egg-drop syndrome caused by BYD virus, a new Tembusu-related flavivirus. PLoS ONE 2011, 24, e18106. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Zhang, C.; Liu, Y.; Liu, Y.; Ye, W.; Han, J.; Ma, G.; Zhang, D.; Xu, F.; Gao, X.; et al. Tembusu Virus in Ducks, China. Emerg. Infect. Dis. 2011, 17, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Homonnay, Z.G.; Kovács, E.W.; Bányai, K.; Albert, M.; Fehér, E.; Mató, T.; Tatár-Kis, T.; Palya, V. Tembusu-like flavivirus (Perak virus) as the cause of neurological disease outbreaks in young Pekin ducks. Avian Pathol. 2014, 43, 552–560. [Google Scholar] [CrossRef]
- Thontiravong, A.; Ninvilai, P.; Tunterak, W.; Nonthabenjawan, N.; Chaiyavong, S.; Angkabkingkaew, K.; Mungkundar, C.; Phuengpho, W.; Oraveerakul, K.; Amonsin, A. Tembusu-Related Flavivirus in Ducks, Thailand. Emerg. Infect. Dis. 2015, 21, 2164–2167. [Google Scholar] [CrossRef]
- Liu, P.; Lu, H.; Li, S.; Moureau, G.; Deng, Y.Q.; Wang, Y.; Zhang, L.; Jiang, T.; de Lamballerie, X.; Qin, C.F.; et al. Genomic and antigenic characterization of the newly emerging Chinese duck egg-drop syndrome flavivirus: Genomic comparison with Tembusu and Sitiawan viruses. J. Gen. Virol. 2012, 93 Pt 10, 2158–2170. [Google Scholar] [CrossRef]
- Lei, W.; Guo, X.; Fu, S.; Feng, Y.; Tao, X.; Gao, X.; Song, J.; Yang, Z.; Zhou, H.; Liang, G. The genetic characteristics and evolution of Tembusu virus. Vet. Microbiol. 2017, 201, 32–41. [Google Scholar] [CrossRef]
- Pandey, B.D.; Karabatsos, N.; Cropp, B.; Tagaki, M.; Tsuda, Y.; Ichinose, A.; Igarashi, A. Identification of a flavivirus isolated from mosquitos in Chiang Mai Thailand. Southeast Asian J. Trop. Med. Public Health 1999, 30, 161–165. [Google Scholar]
- Tang, Y.; Diao, Y.; Chen, H.; Ou, Q.; Liu, X.; Gao, X.; Yu, C.; Wang, L. Isolation and genetic characterization of a tembusu virus strain isolated from mosquitoes in Shandong, China. Transbound. Emerg. Dis. 2015, 62, 209–216. [Google Scholar] [CrossRef]
- O’Guinn, M.L.; Turell, M.J.; Kengluecha, A.; Jaichapor, B.; Kankaew, P.; Miller, R.S.; Endy, T.P.; Jones, J.W.; Coleman, R.E.; Lee, J.S. Field detection of Tembusu virus in western Thailand by RT-PCR and vector competence determination of select Culex mosquitoes for transmission of the virus. Am. J. Trop. Med. Hyg. 2013, 89, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Shi, Y.; Liu, Q.; Wang, Y.; Li, G.; Teng, Q.; Zhang, Y.; Liu, S.; Li, Z. Airborne transmission of a novel Tembusu virus in ducks. J. Clin. Microbiol. 2015, 53, 2734–2736. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Zhao, Y.; Zhang, X.; Xu, D.; Dai, X.; Teng, Q.; Yan, L.; Zhou, J.; Ji, X.; Zhang, S.; et al. An infectious disease of ducks caused by a newly emerged Tembusu virus strain in mainland China. Virology 2011, 417, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Birding Taiwan. Available online: https://taiwantoday.tw/news.php?unit=14&post=23956&unitname=Environment-Taiwan-Review&postname=Birding-Taiwan (accessed on 1 August 2013).
- Su, C.L.; Yang, C.F.; Teng, H.J.; Lu, L.C.; Lin, C.; Tsai, K.H.; Chen, Y.Y.; Chen, L.Y.; Chang, S.F.; Shu, P.Y. Molecular epidemiology of Japanese encephalitis virus in mosquitoes in Taiwan during 2005–2012. PLoS Negl. Trop. Dis. 2014, 8, e3122. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Lin, T.H.; Teng, H.J.; Su, C.L.; Tsai, K.H.; Lu, L.C.; Lin, C.; Yang, C.F.; Chang, S.F.; Liao, T.L.; et al. Molecular epidemiology of Japanese encephalitis virus, Taiwan. Emerg. Infect. Dis. 2010, 16, 876–878. [Google Scholar] [CrossRef]
- Shu, P.Y.; Chang, S.F.; Kuo, Y.C.; Yueh, Y.Y.; Chien, L.J.; Sue, C.L.; Lin, T.H.; Huang, J.H. Development of group- and serotype-specific one-step SYBR green I-based real-time reverse transcription-PCR assay for dengue virus. J. Clin. Microbiol. 2003, 41, 2408–2416. [Google Scholar] [CrossRef]
- Huang, X.; Han, K.; Zhao, D.; Liu, Y.; Zhang, J.; Niu, H.; Zhang, K.; Zhu, J.; Wu, D.; Gao, L.; et al. Identification and molecular characterization of a novel flavivirus isolated from geese in China. Res. Vet. Sci. 2013, 94, 774–780. [Google Scholar] [CrossRef]
- Yu, G.; Lin, Y.; Tang, Y.; Diao, Y. Evolution of Tembusu virus in ducks, chikens, geeses, sparrows, and mosquitoes in northern China. Viruses 2018, 10, 485. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Shi, Y.; Wang, H.; Li, G.; Li, X.; Wang, B.; Su, X.; Wang, J.; Teng, Q.; Yang, J.; et al. A single mutation at position 156 in the envelope protein of Tembusu virus is responsible for virus tissue tropism and transmissibility in ducks. J. Virol. 2018, 16, e00427-18. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, L.; Cao, Y.; Wang, J.; Yu, Z.; Sun, X.; Liu, F.; Li, Z.; Liu, P.; Su, J. Basic amino acid substitution at residue 367 of Tembusu virus plays critical role in pathogenesis. J. Virol. 2020, 31, e02011-19. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.C.; Soto-Acosta, R.; Bradrick, S.S.; Garcia-Blanco, M.A.; Ooi, E.E. The 5′ and 3′ untranslated regions of the flaviviral genome. Viruses 2017, 6, 137. [Google Scholar] [CrossRef]
- Alvarez, D.E.; De Lella Ezcurra, A.L.; Fucito, S.; Gamarnik, A.V. Role of RNA structures present at the 3′-UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339, 200–212. [Google Scholar] [CrossRef]
- Villordo, S.M.; Filomatori, C.V.; Sanchez-Vargas, I.; Blair, C.D.; Gamarnik, A.V. Dengue virus RNA structure specialization facilities host adaption. PLoS Pathog. 2015, 30, e1004604. [Google Scholar] [CrossRef]
- Wang, H.J.; Li, X.F.; Liu, L.; Xu, Y.P.; Ye, Q.; Deng, Y.Q.; Huang, X.Y.; Zhao, H.; Qin, E.D.; Shi, P.Y.; et al. The emerging duck flavivirus is not pathogenic for primates and is highly sensitive to mammalian interferon antiviral signaling. J. Virol. 2016, 90, 6538–6548. [Google Scholar] [CrossRef]
- Cheng, M.C.; Lee, M.S.; Ho, Y.H.; Chyi, W.L.; Wang, C.H. Avian influenza monitoring in migrating in Taiwan during 1998–2007. Avian Dis. 2010, 54, 109–114. [Google Scholar] [CrossRef]
- Gao, X.; Liu, H.; Wang, H.; Fu, S.; Guo, Z.; Liang, G. Southernmost Asia is the source of Japanese encephalitis virus (genotype 1) diversity from which the viruses disperse and evolve throughout Asia. PLoS Negl. Trop. Dis. 2013, 19, e2459. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′ to 3′) | Amplicon (bp) | Annealing Temp (°C) | References |
|---|---|---|---|---|
| P1f | AGAAGTTCRYCTGTGTGA | 638 | 50 | [6,18] |
| DF_R638 | CAGCAGTCTATGTCTTCAGG | |||
| DF_F441 | CGATAGTTGCTGGGCTGAAGC | 675 | 50 | [6] |
| DF_R1115 | GCAGTAAGATCTCACAACCGC | |||
| DF_F954 | GCTTCAGCTGTCTGGGGATGC | 696 | 50 | |
| DF_R1650 | CAATGACTCTTTGTTTTGCCACG | |||
| DF_F2353 | GGCACTGCTATTGTGGATGGG | 987 | 50 | |
| DF_R3339 | GGTGGGGTGGTGCAAGACC | |||
| DF_F3302 | GGAACAACTGTCACAGTAACG | 1393 | 50 | |
| DF_R4694 | GCATGACTCCCACTCCAGCC | |||
| DF_F4406 | GCATCACAGAGATTTGATGTGG | 757 | 54 | |
| DF_R5162 | CCTGAACCTGGATGTAGGTCC | |||
| DF_F4874 | GCAAGTCATCGTCGTGCAACC | 709 | 50 | |
| DF_R5582 | GCTCTTCAATGTCTGTTATTGGC | |||
| DF_F5399 | GCTCACACCTCAGCGAGTGC | 851 | 50 | |
| DF_R6249 | GGTCATTGTAACTTATCCCAGC | |||
| DF_F6494 | CGCTCACAGAATGACAGAATCC | 855 | 50 | |
| DF_R7348 | GGAACATCTGTAGCCACTATGC | |||
| DF_F6807 | GAACCAGAGAGACAGAGATCGC | 1352 | 50 | |
| DF_R8158 | CCCTAGCTAGCCATTCCTCGG | |||
| DF_F7940 | GCAGGTTCAGGAAGTGAGAGG | 597 | 50 | |
| DF_R8536 | GGATTGTCTTGGTCATAATGCC | |||
| DF_F8383 | GGATGCACAAAACCAACCGC | 833 | 50 | |
| DF_R9215 | GGCCGAGATGTCACGCAGC | |||
| DF_F9274 | GGGACACTAGAATAACCAAGGC | 1212 | 50 | |
| DF_R10485 | CCAACATCCGGTGGCAGGG | |||
| TMUV-E_F | TTCAGCTGTCTGGGGATGCA | 1503 | 50 | [19] |
| TMUV-E_R | GGCATTGACATTTACTGCCA | |||
| TMUV-NS1_F | GACACGGGGTGCTCAATCGACTT | 1056 | 50 | |
| TMUV-NS1_R | AGCCATGACCTTTGATTTGAT | |||
| TMUV-NS3_F | GGAGGAGTCATCTGGGATGTG | 1857 | 50 | |
| TMUV-NS3_R | TCTCTTTCCACTCGCAAAATC | |||
| TMUV-NS5_F | GAACTGGCAGAACTTTGGGGGAG | 2711 | 50 | |
| TMUV-NS5_R | TTACAAGACACCTTCACTCCAGC | |||
| 1_F | AAATGACTTCAGGACACCTC | 723 | 50 | This study * |
| 1_R | ACATACCTTGTCCACACTTC | |||
| 2_F | ATGTCATGGATCACTCAAGG | 400 | 50 | |
| 2_R | CAGTCAAGTCAATGCTGTTG | |||
| 3_F | AAAAGAAAGGAGGCATGCTA | 523 | 50 | |
| 3_R | GGAACATCCCATATGACTCC | |||
| 5_F | CAGTCGGAAGTGCATTAAAC | 683 | 54 | |
| 5_R | CAGCTGTAGTCAGCATGTAT | |||
| 7_F | TTAGAATCCTGTCAAAGCCC | 767 | 50 | |
| 7_R | CACCTTCACCACCTTATTCA | |||
| 8_F | CTGGAATCTCGTTGATAGGG | 572 | 50 | |
| 8_R | TAATGAGTTGAACGCACAGA | |||
| 3′UTR-2_F | CCAACCCTCAATAGGTTCAA | 612 | 50 | This study † |
| 3′UTR-2_R | GAGGGTCTCCTAGTCTATCC | |||
| 3′UTR-5_F | ATACATGGAAGACAAGACCC | 465 | 50 | |
| 3′UTR-5_R | GAGACGGTATTGAACGCTTA | |||
| 3′UTR-10_F | AGGAGCTAAGCGTTCAATAC | 427 | 50 | |
| 3′UTR-10_R | GACTCTGTGTTCTACCACC | |||
| GSP-5′ end | GGTCGCCTCACTGACCCCAACTAGC | 331 | 55 | For RACE |
| 3′UTR-10_F | AGGAGCTAAGCGTTCAATAC | 424 | 55 | |
| oligo d(T)-anchor | GACCACGCGTATCGATGTCGACT(16)V | 55 |
| TMUV /DTMUV Strains | Host | Location | Year/ Month | GenBank Accession Number /ORF Sequence Identity (%)/ Length (Nucleotides) | GenBank Accession Number /Protein Sequence Identity (%)/ Length (Amino Acids) |
|---|---|---|---|---|---|
| TP1906 | Cx. annulus | Taipei/ Taiwan | 2019/ Jun | MN747003 100 (10,990) | 100 (3425) |
| TC1906 † | Cx. tritaeniorhynchus | Taichung/ Taiwan | 2019/ Jun | MN958524 NS1:99.79 (964) | NS1:99.69 (964) |
| Sitiawan virus | Broiler Chicks | Perak state/ Malaysia | 2000 | JX477686 93.71 (10,278) | AFP95929 98.92 (3425) |
| MM1775 | Cx. tritaeniorhynchus | Kuala Lumpar/ Malaysia | 1955 | JX477685 91.27 (10,278) | AFP95928 98.57 (3425) |
| YY5 | Duck | Zhejiang Province/ China | 2010 | JF270480 87.05 (10,990) | AEX15510 96.50 (3425) |
| BYD-1 | Duck | China | 2010 | JF312912 87.15 (10,278) | AEA72437 96.55 (3425) |
| JS804 | Goose | Jiangsu Province/ China | 2010 | JF895923 87.12 (10,990) | AEJ87340 96.38 (3425) |
| CK-SD-11 | Chicken | China | 2010 | JQ627862 87.01 (10,278) | AFP19891 96.41 (3425) |
| GX_2011 | Duck | Guangxi Province/ China | 2011 | KC990542 87.05 (10,990) | AGV52066 96.47 (3425) |
| FS-2011 | Duck | China | 2011 | KX686578 87.00 (10,991) | AOS50837 96.44 (3425) |
| df-2 | Duck | China | 2012 | KJ489355 87.16 (10,990) | AHY19030 96.53 (3425) |
| byd1 | Duck | Hebei Province/ China | 2012 | JQ920420 87.15 (10,990) | AFN43039 96.55 (3425) |
| JSGo | Goose | China | 2012 | AB917090 87.21 (10,959) | BAQ19608 96.53 (3425) |
| DEDSV strain pigeon | Pigeon | Beijing Autonomous City/ China | 2012 | JQ920425 87.09 (10,990) | AFN43044 96.61 (3425) |
| AHQY | Layer Duck | China | 2013 | KJ740748 86.65 (10,990) | AIF73122 96.35 (3425) |
| SX1 | Chicken | China | 2013 | KM066945 86.80 (10,990) | AIR72260 95.97 (3425) |
| G23 | Goose | China | 2014 | KT239021 86.92 (10,881) | AKR79508 96.61 (3425) |
| JS06 | Chicken | China | 2014 | KR869106 86.69 (10,990) | ALL27018 95.7 (3425) |
| GD2014 | Duck | China | 2014 | KU323595 87.06 (10,990) | ANF99570 96.50 (3425) |
| DTMUV/CH/2014 | Duck | China | 2014 | KP096415 87.05 (10,990) | AKO73664 96.44 (3425) |
| GDLH01 | Duck | China | 2015 | KT824876 87.07 (10,990) | ALM89034 96.47 (3425) |
| SH001 | Duck | Shanghai Province /China | 2015 | KP742476 87.10 (10,990) | AJR29358 96.50 (3425) |
| HZ4-2015 | Broiler Duck | China | 2015 | KX686571 86.63 (10,991) | AOS50830 95.94 (3425) |
| SD14 | Anas platyrhynchos | China | 2014 | MH748542 88.00 (11,001) | AXY93835 96.64 (3425) |
| DK/TH/CU-DTMUV | Duck | Thailand | 2007 | MF621927 87.21 (10,278) | AVM38076 96.41 (3425) |
| D1977/1/MY | Pekin Duck | Malaysia | 2012 | KX097989 87.09 (10,988) | ANK79132 96.20 (3424) |
| D1921/1/3/MY | Pekin Duck | Malaysia | 2012 | KX097990 87.10 (10,988) | ANK79133 96.18 (3424) |
| DK/TH/CU-1 | Duck | Thailand | 2013 | KR061333 86.91 (10,278) | ALE71321 96.55 (3425) |
| KPS54A61 | Duck | Thailand | 2013 | KF573582 86.84 (10,990) | AIK27529 96.35 (3425) |
| GX2013E | Duck | China | 2013 | KM275940 86.97 (10,990) | AIX09854 96.23 (3425) |
| HD-2015 | Layer Duck | China | 2015 | KX686572 86.75 (10,991) | AOS50831 96.15 (3425) |
| HZ1-2015 | Layer Duck | China | 2015 | KX686570 86.52 (10,990) | AOS50829 96.12 (3425) |
| HZ3-2015 | Duck | China | 2015 | KX686579 86.94 (10,992) | AOS50838 96.18 (3425) |
| zjYY150901 | Duck | China | 2015 | MF522174 86.82 (10,990) | AWX59350 96.29 (3425) |
| SDMS | Culex Mosquito | Shangdong Province/China | 2012 | KC333867 87.04 (10,990) | AGR42654 96.47 (3425) |
| SDXT | Layer Duck | China | 2013 | KJ740745 87.08 (10,990) | AIF73119 96.58 (3425) |
| JS-L1 | Duck | China | 2015 | KY626659 87.14 (10,890) | ARA71333 96.53 (3425) |
| Species | Taipei | Taichung | Tainan | Yilan | Hualien | Num Individuals | Num Pools | Num TMUV Pos Pools | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Park | Wetland | Park | Pig Farm | Park | Pig Farm | Park | Pig Farm | Pig Farm | ||||
| Aedes aegypti | 0 | 0 | 0 | 0 | 7 | 0 | 0 | 0 | 0 | 7 | 3 | 0 |
| Aedes albopictus | 86 | 110 | 4 | 0 | 33 | 0 | 43 | 0 | 33 | 309 | 23 | 0 |
| Aedes malikuli | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 3 | 0 |
| Aedes penghuensis | 0 | 9 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 9 | 4 | 0 |
| Aedes vexans | 0 | 75 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 75 | 5 | 0 |
| Anopheles sinensis | 0 | 0 | 0 | 106 | 0 | 3 | 0 | 12 | 35 | 156 | 8 | 0 |
| Anopheles tessellatus | 0 | 179 | 2 | 0 | 0 | 0 | 0 | 0 | 10 | 191 | 7 | 0 |
| Armigeres subalbatus | 5 | 40 | 0 | 0 | 105 | 0 | 0 | 0 | 66 | 216 | 14 | 0 |
| Coquillettidia crassipes | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
| Culex annulus | 4 | 606 | 22 | 7 | 52 | 2 | 645 | 15 | 472 | 1825 | 50 | 1 * |
| Culex fuscocephala | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 12 | 14 | 3 | 0 |
| Culex pipiens form molestus | 63 | 19 | 53 | 0 | 87 | 0 | 17 | 0 | 21 | 260 | 21 | 0 |
| Culex quinquefasciatus | 15 | 11 | 29 | 0 | 174 | 7 | 116 | 0 | 133 | 485 | 25 | 0 |
| Culex sitiens | 0 | 0 | 0 | 0 | 62 | 0 | 0 | 0 | 4 | 66 | 5 | 0 |
| Culex tritaeniorhynchus | 2 | 2470 | 71 | 1301 | 914 | 2280 | 327 | 1473 | 2062 | 10,900 | 231 | 1* |
| Heizmannia taiwanensis | 4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 4 | 2 | 0 |
| Mansonia uniformis | 0 | 809 | 0 | 0 | 0 | 0 | 0 | 0 | 29 | 838 | 19 | 0 |
| Tripteroides bambusa | 0 | 11 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 11 | 4 | 0 |
| Uranotaenia novobscura | 0 | 2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 1 | 0 |
| Total | 183 | 4343 | 181 | 1414 | 1434 | 2294 | 1148 | 1500 | 2877 | 15,374 | 429 | 2 |
| Genomic Region (% Sequence Identity) | TMUV-TP1906 | Sitiawan Virus (JX477686) | TMUV-MM1775 (MH414569) | DTMUV-SD14 (MH748542) | DTMUVs (see Table 2) |
|---|---|---|---|---|---|
| Complete genome sequence | 10,990 nt (100) | NA # | 11,001 (91.54) | 11,001 (88.31) | 10,890–10,992 * (86.93–87.49) |
| 5′-UTR | 94 nt (100) | NA # | 94 nt (94.68) | 94 nt (92.55) | 93–95 (90.43–93.68) |
| 3′-UTR (total length) | 618 nt (100) | NA # | 629 nt (95.15) | 629 nt (92.72) | 618 nt (89.28–92.14) |
| 3′-UTR (variable region) | 87 nt(100) | NA # | 94 nt (74.71) | 94 nt (71.26) | 84–85 nt (56.96–66.25) |
| ORF | 10,278 nt (100) | 10,278 nt (93.71) | 10,278 nt (91.27) | 10,278 nt (88.00) | 10,275–10,278 nt † (86.52–87.21) |
| polyprotein | 3425 aa (100) | 3425 aa (98.92) | 3425 aa (98.57) | 3425 aa (96.64) | 3424–3425 aa (95.71–96.61) |
| C | 360 nt (100) 120 aa (100) | 360 nt (96.67) 120 aa (99.17) | 360 nt (94.71) 120 aa (96.67) | 360 nt (90.53) 120 aa (95.00) | 360 nt (88.89–90.28) 120 aa (93.33–95.00) |
| prM | 501 nt (100) 167 aa (100) | 501 nt (91.20) 167 aa (98.20) | 501 nt (90.80) 167 aa (98.80) | 501 nt (87.00) 167 aa (95.21) | 501 nt (84.40–85.40) 167 aa (94.61–96.41) |
| E | 1503 nt (100) 501 aa (100) | 1503 nt (93.68) 501 aa( 98.40) | 1503 nt (90.21) 501 aa (98.60) | 1503 nt (87.62) 501 aa (96.41) | 1503 nt (86.09–86.96) 501 aa (96.41–97.8) |
| NS1 | 1056 nt (100) 352 aa (100) | 1056 nt (93.93) 352 aa (97.73) | 1056 nt (91.86) 352 aa (98.86) | 1056 nt (89.67) 352 aa (96.31) | 1056 nt (85.13–88.35) 352 aa (90.62–94.89) |
| NS2A | 681 nt (100) 227 aa (100) | 681 nt (92.22) 227 aa (99.12) | 681 nt (89.88) 227 aa (97.8) | 681 nt (86.43) 227 aa (93.81) | 681 nt (84.14–85.61) 227 aa (90.75–92.51) |
| NS2B | 393 nt (100) 131 aa (100) | 393 nt (91.09) 131 aa (96.95) | 393 nt (89.57) 131 aa (96.95) | 393 nt (87.28) 131 aa (93.13) | 393 nt (85.24–86.77) 131 aa (90.84–93.89) |
| NS3 | 1857 nt (100) 619 aa (100) | 1857 nt (94.18) 619 aa (99.52) | 1857 nt (91.76) 619 aa (99.03) | 1857 nt (89.01) 619 aa (98.71) | 1857 nt (87.51–88.26) 619 aa (97.58–98.71) |
| NS4A | 378 nt (100) 126 aa (100) | 378 nt (93.39) 126 aa (99.21) | 378 nt (90.74) 126 aa (98.41) | 378 nt (86.21) 126 aa (96.03) | 378 nt (84.62–87.04) 126 aa (92.86–96.03) |
| 2K | 69 nt (100) 23 aa (100) | 69 nt (95.65) 23 aa (100) | 69 nt (94.12) 23 aa (100) | 69 nt (94.12) 23 aa (95.65) | 69 nt (85.51–91.18) 23 aa (95.65) |
| NS4B | 762 nt (100) 254 aa (100) | 762 nt (93.83) 254 aa (99.61) | 762 nt (90.94) 254 aa (99.21) | 762 nt (85.9) 254 aa (95.28) | 762 nt (84.83–88.52) 254 aa (91.34–97.64) |
| NS5 | 2715 nt (100) 905 aa (100) | 2715 nt (94.18) 905 aa (99.34) | 2715 nt (91.71) 905 aa (98.56) | 2715 nt (88.32) 905 aa (97.79) | 2712–2715 nt † (87.18–87.81) 904–905 aa † (96.91–98.34) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, S.-H.; Su, C.-L.; Chang, M.-C.; Hu, H.-C.; Yang, S.-L.; Shu, P.-Y. Genome Analysis of a Novel Tembusu Virus in Taiwan. Viruses 2020, 12, 567. https://doi.org/10.3390/v12050567
Peng S-H, Su C-L, Chang M-C, Hu H-C, Yang S-L, Shu P-Y. Genome Analysis of a Novel Tembusu Virus in Taiwan. Viruses. 2020; 12(5):567. https://doi.org/10.3390/v12050567
Chicago/Turabian StylePeng, Shih-Huan, Chien-Ling Su, Mei-Chun Chang, Huai-Chin Hu, Su-Lin Yang, and Pei-Yun Shu. 2020. "Genome Analysis of a Novel Tembusu Virus in Taiwan" Viruses 12, no. 5: 567. https://doi.org/10.3390/v12050567
APA StylePeng, S.-H., Su, C.-L., Chang, M.-C., Hu, H.-C., Yang, S.-L., & Shu, P.-Y. (2020). Genome Analysis of a Novel Tembusu Virus in Taiwan. Viruses, 12(5), 567. https://doi.org/10.3390/v12050567