Substitution of the CD81 Binding Site and β-Sandwich Area in E2 of HCV in Cambodia

Abstract
1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Ethical Issues
2.3. Serological Tests
2.4. Genotype Analysis
2.5. HCV Full-Length Genome Sequence Analysis
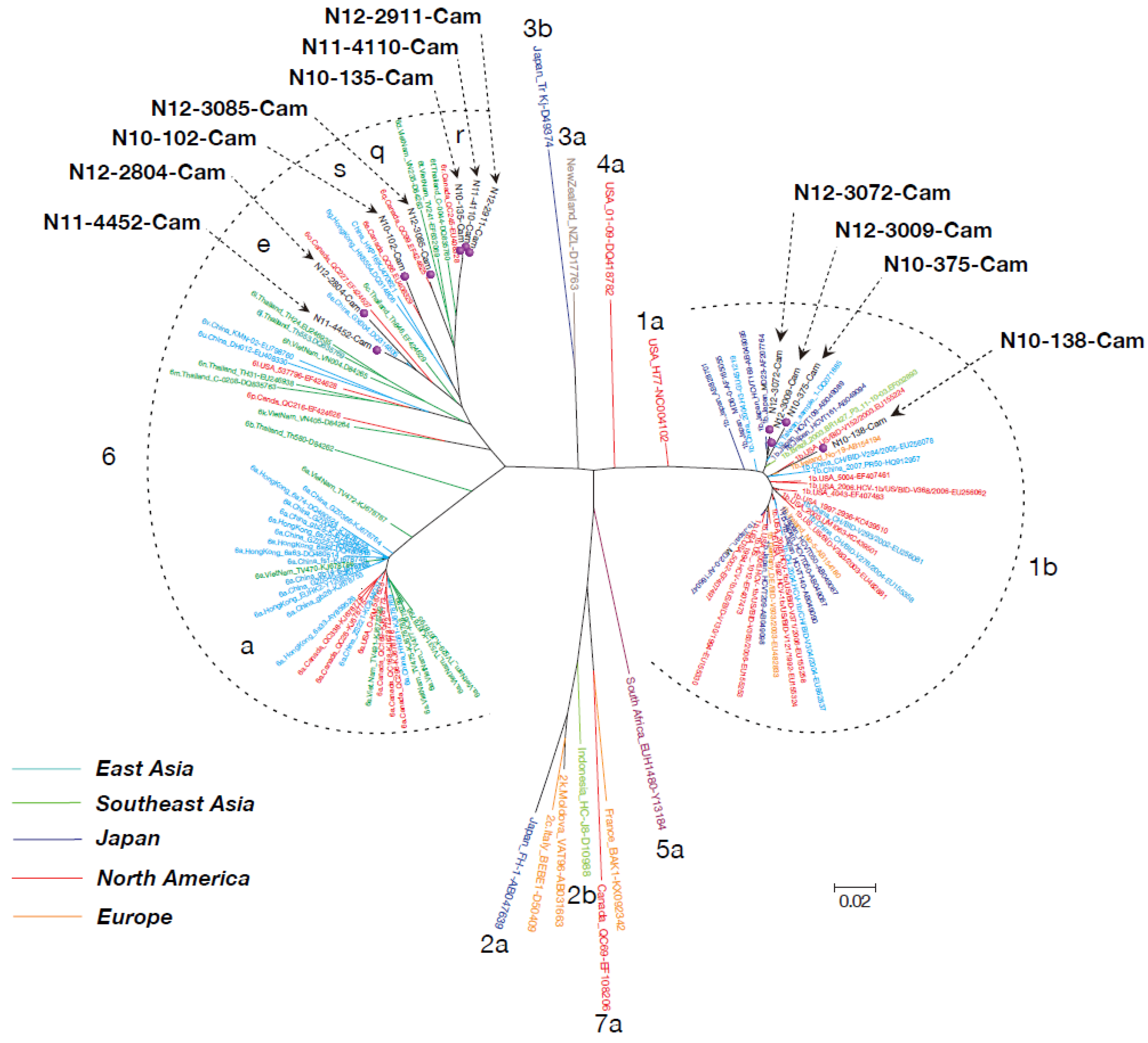
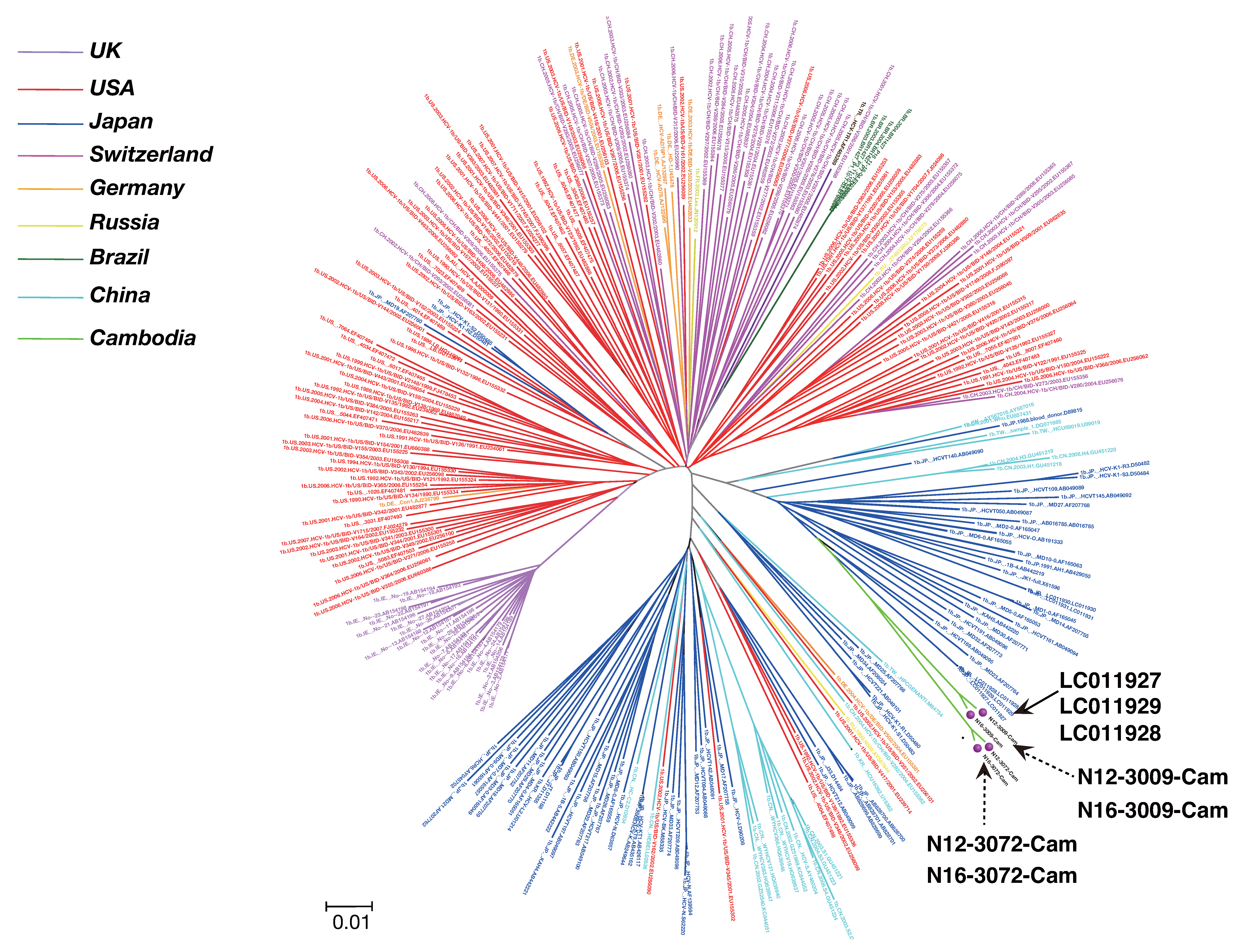
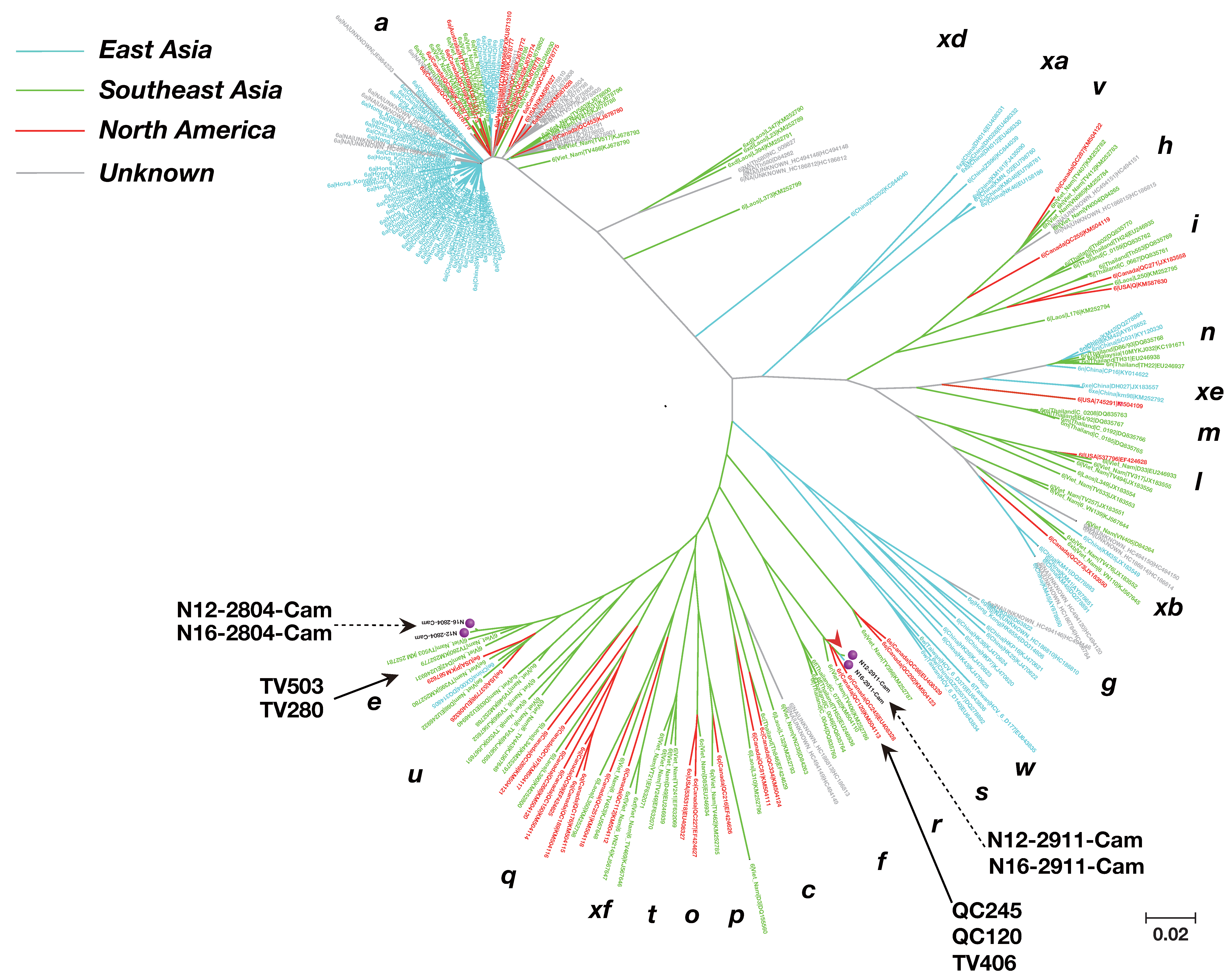
2.6. Phylogenetic Tree Analysis
2.7. Calculation of HCV Mutation Rate in the Same Individuals of Cambodia
2.8. Estimation of Mutation in Each Genetic Region of HCV Sampled from the Same Cambodian Subjects
2.9. Nonsynonymous Substitution at E2 in the Same Cambodian Subject
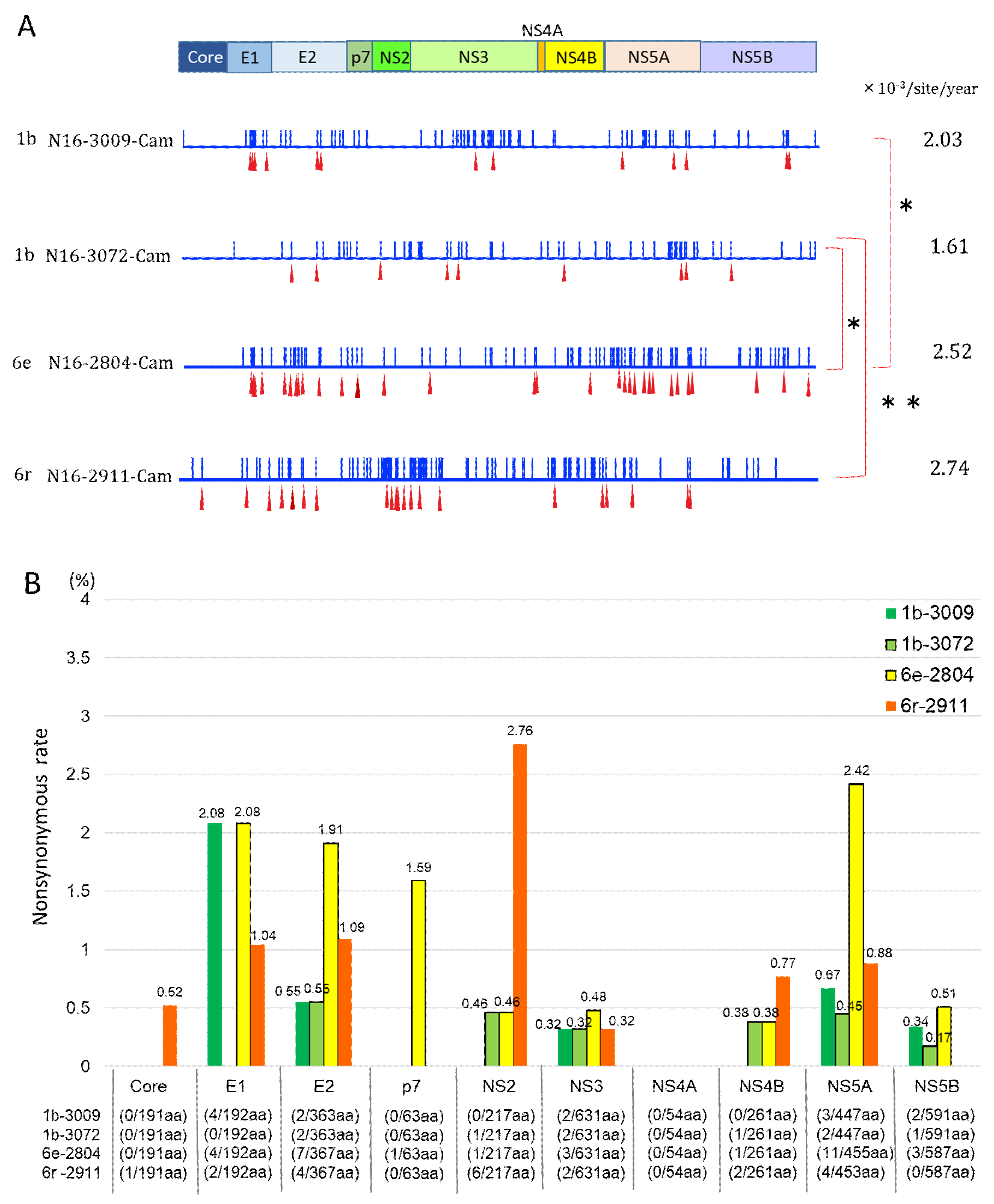
3. Results
3.1. Serologic Analysis
3.2. HCV Genotype
3.3. Phylogenetic Tree Analysis of HCV Near Full-Length Genome Sequence
3.4. Mutation Rate of HCV Genotypes 1b and 6
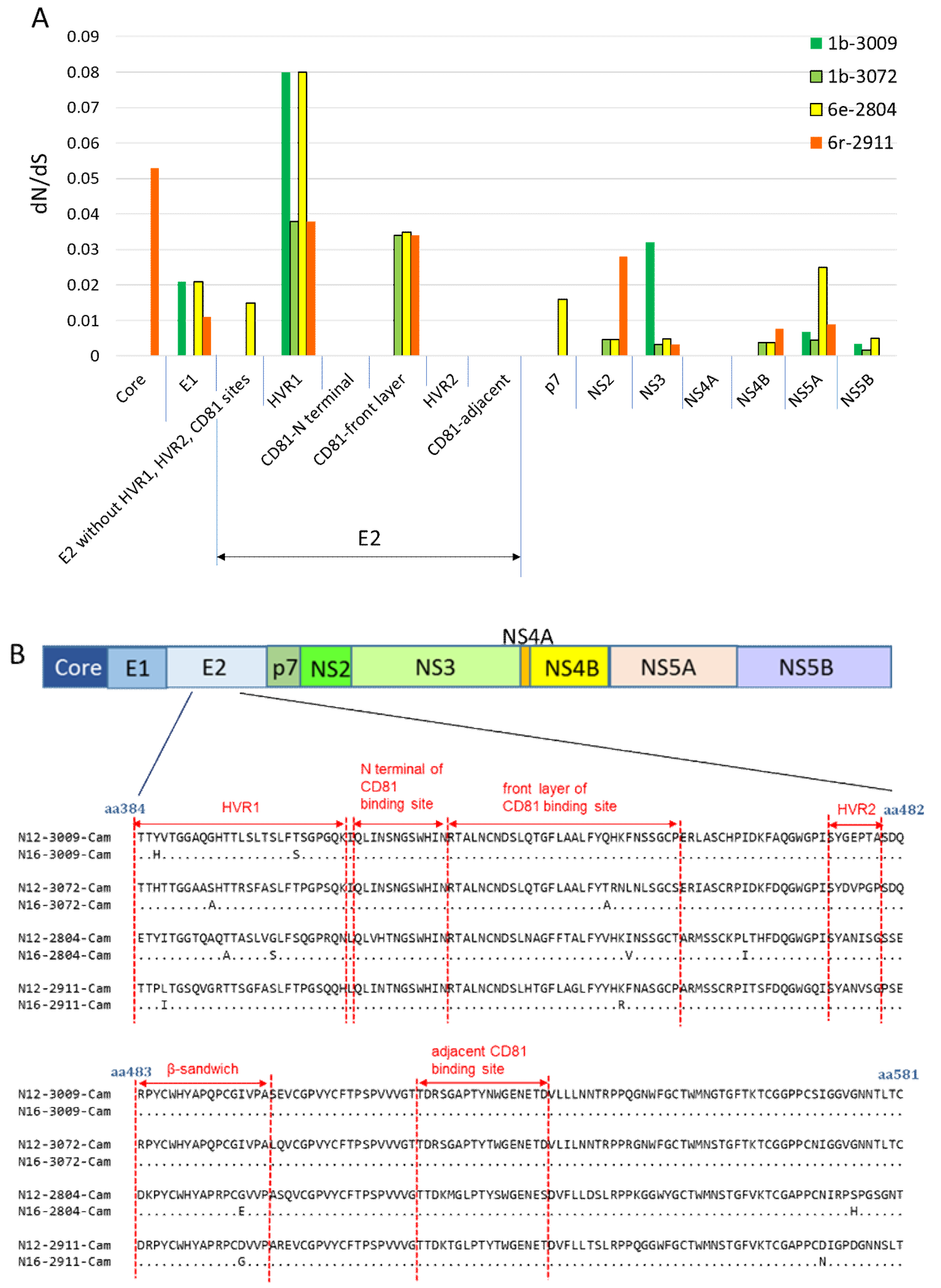
3.5. Nonsynonymous Substitution Rate and Immune Pressure in the HCV RNA
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- WHO. WHO HCV Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-c (accessed on 20 July 2019).
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Von Schaewen, M.; Hrebikova, G.; Heller, B.; Sandmann, L.; Plaas, M.; Ploss, A. Mice Expressing Minimally Humanized CD81 and Occludin Genes Support Hepatitis C Virus Uptake In Vivo. J. Virol. 2016, 91, e01799-16. [Google Scholar] [CrossRef] [PubMed]
- Netski, D.M.; Mao, Q.; Ray, S.C.; Klein, R.S. Genetic divergence of HCV: The role of HIV-related immunosuppression. J. Acquir. Immune. Defic. Syndr. 2005, 49, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.; Pybus, O.G.; Barnes, E. Global Distribution and Prevalence of Hepatitis C Virus Genotypes. Hepatology 2014, 61, 77–87. [Google Scholar] [CrossRef]
- Simmonds, P. The origin and evolution of hepatitis viruses in humans. J. Gen. Virol. 2001, 82, 693–712. [Google Scholar] [CrossRef]
- Adhikari, V.P.; Lu, L.-J.; Kong, L.-Q. Does hepatitis B virus infection cause breast cancer? Chin. Clin. Oncol. 2016, 5, 804. [Google Scholar] [CrossRef]
- Lu, L.; Wu, T.; Xiong, L.; Li, C.; Nguyen, M.H.; Murphy, D.G. Analysis of HCV-6 isolates among Asian-born immigrants in North America reveals their high genetic diversity and a new subtype. Virology 2016, 492, 25–31. [Google Scholar] [CrossRef]
- Cheng, X.-D.; Xu, H.-F.; Wei, X.-M.; Zhou, H.-Z. Variation analysis of E1 and E2 in HCV subtypes. Arch. Virol. 2015, 160, 2479–2482. [Google Scholar] [CrossRef]
- Stamataki, Z.; Coates, S.; Evans, M.; Wininger, M.; Crawford, K.; Dong, C.; Fong, Y.-L.; Chien, D.; Abrignani, S.; Balfe, P.; et al. Hepatitis C virus envelope glycoprotein immunization of rodents elicits cross-reactive neutralizing antibodies. Vaccine 2007, 25, 7773–7784. [Google Scholar] [CrossRef]
- Stamataki, Z.; Coates, S.; Abrignani, S.; Houghton, M.; McKeating, J.A. Immunization of human volunteers with hepatitis C virus envelope glycoproteins elicits antibodies that cross-neutralize heterologous virus strains. J. Infect. Dis. 2011, 204, 811–813. [Google Scholar] [CrossRef]
- Frey, S.E.; Houghton, M.; Coates, S.; Abrignani, S.; Chien, D.; Rosa, M.; Pileri, P.; Ray, R.; Di Bisceglie, A.M.; Rinella, P.; et al. Safety and immunogenicity of HCV E1E2 vaccine adjuvanted with MF59 administered to healthy adults. Vaccine 2010, 28, 6367–6373. [Google Scholar] [CrossRef] [PubMed]
- Krapchev, V.B.; Rychłowska, M.; Chmielewska, A.; Zimmer, K.; Patel, A.H.; Bieńkowska-Szewczyk, K. Recombinant Flag-tagged E1E2 glycoproteins from three hepatitis C virus genotypes are biologically functional and elicit cross-reactive neutralizing antibodies in mice. Virology 2018, 519, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Ströh, L.J.; Nagarathinam, K.; Krey, T. Conformational Flexibility in the CD81-Binding Site of the Hepatitis C Virus Glycoprotein E2. Front. Immunol. 2018, 9, 9. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Baumert, T.F.; Bukh, J.; Houghton, M.; Lemon, S.M.; Lindenbach, B.D.; Lohmann, V.; Moradpour, D.; Pietschmann, T.; Rice, C.M.; et al. Critical challenges and emerging opportunities in hepatitis C virus research in an era of potent antiviral therapy: Considerations for scientists and funding agencies. Virus Res. 2018, 248, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Olbrich, A.; Wardemann, H.; Böhm, S.; Rother, K.; Colpitts, C.C.; Wrensch, F.; Baumert, T.F.; Berg, T.; Benckert, J. Repertoire and Neutralizing Activity of Antibodies Against Hepatitis C Virus E2 Peptide in Patients With Spontaneous Resolution of Hepatitis C. J. Infect. Dis. 2019, 220, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization; International Agency for Research on Cancer. Globocan. 2018. Available online: https://gco.iarc.fr/today/data/factsheets/populations/116-cambodia-fact-sheets.pdf#search=%27cambodia+liver+cancer+globocan%27 (accessed on 25 June 2019).
- Yamada, H.; Fujimoto, M.; Svay, S.; Lim, O.; Hok, S.; Goto, N.; Ohisa, M.; Akita, T.; Matsuo, J.; Do, S.H.; et al. Seroprevalence, genotypic distribution and potential risk factors of hepatitis B and C virus infections among adults in Siem Reap, Cambodia. Hepatol. Res. 2014, 45, 480–487. [Google Scholar] [CrossRef]
- Yamada, H.; Takahashi, K.; Lim, O.; Svay, S.; Chuon, C.; Hok, S.; Do, S.H.; Fujimoto, M.; Akita, T.; Goto, N.; et al. Hepatitis E Virus in Cambodia: Prevalence among the General Population and Complete Genome Sequence of Genotype 4. PLoS ONE 2015, 10, e0136903. [Google Scholar] [CrossRef]
- Fujimoto, M.; Chuon, C.; Nagashima, S.; Yamamoto, C.; Ko, K.; Svay, S.; Hok, S.; Lim, O.; Ohisa, M.; Akita, T.; et al. A seroepidemiological survey of the effect of hepatitis B vaccine and hepatitis B and C virus infections among elementary school students in Siem Reap province, Cambodia. Hepatol. Res. 2017, 48, E172–E182. [Google Scholar] [CrossRef]
- Do, S.H.; Yamada, H.; Fujimoto, M.; Ohisa, M.; Matsuo, J.; Akita, T.; Katayama, K.; Van Nguyen, N.; Miyakawa, Y.; Tanaka, J. High prevalences of hepatitis B and C virus infections among adults living in Binh Thuan province, Vietnam. Hepatol. Res. 2014, 45, 259–268. [Google Scholar] [CrossRef]
- Matsuo, J.; Do, S.H.; Yamamoto, C.; Nagashima, S.; Chuon, C.; Katayama, K.; Takahashi, K.; Tanaka, J. Clustering infection of hepatitis B virus genotype B4 among residents in Vietnam, and its genomic characters both intra- and extra-family. PLoS ONE 2017, 12, e0177248. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Rzhetsky, A.; Nei, M. A simple method for estimating and testing minimum evolution trees. Mol. Biol. Evol. 1992, 9, 945–967. [Google Scholar]
- Major, M.E.; Mihalik, K.; Fernandez, J.; Seidman, J.; Kleiner, D.E.; Kolykhalov, A.A.; Rice, C.M.; Feinstone, S.M. Long-Term Follow-Up of Chimpanzees Inoculated with the First Infectious Clone for Hepatitis C Virus. J. Virol. 1999, 73, 3317–3325. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.C.; Wang, Y.-M.; Laeyendecker, O.; Ticehurst, J.R.; Villano, S.A.; Thomas, D.L. Acute Hepatitis C Virus Structural Gene Sequences as Predictors of Persistent Viremia: Hypervariable Region 1 as a Decoy. J. Virol. 1999, 73, 2938–2946. [Google Scholar] [CrossRef]
- Le Guillou-Guillemette, H.; Vallet, S.; Gaudy-Graffin, C.; Payan, C.; Pivert, A.; Goudeau, A.; Lunel-Fabiani, F. Genetic diversity of the hepatitis C virus: Impact and issues in the antiviral therapy. World J. Gastroenterol. 2007, 13, 2416–2426. [Google Scholar] [CrossRef]
- Kong, L.; Lee, D.E.; Kadam, R.U.; Liu, T.; Giang, E.; Nieusma, T.; Garcés, F.; Tzarum, N.; Woods, V.L.; Ward, A.B.; et al. Structural flexibility at a major conserved antibody target on hepatitis C virus E2 antigen. Proc. Natl. Acad. Sci. USA 2016, 113, 12768–12773. [Google Scholar] [CrossRef]
- Caruana, S.R.; Kelly, H.A.; De Silva, S.L.; Chea, L.; Nuon, S.; Saykao, P.; Bak, N.; Biggs, B.-A. Knowledge about hepatitis and previous exposure to hepatitis viruses in immigrants and refugees from the Mekong Region. Aust. N. Z. J. Public Health 2005, 29, 64–68. [Google Scholar] [CrossRef]
- Thüring, E.G.; Joller-Jemelka, H.I.; Sareth, H.; Sokhan, U.; Reth, C.; Grob, P. Prevalence of markers of hepatitis viruses A, B, C and of HIV in healthy individuals and patients of a Cambodian province. Southeast Asian J. Trop. Med. Public Health 1993, 24, 239–249. [Google Scholar]
- Pybus, O.G.; Barnes, E.; Taggart, R.; Lemey, P.; Markov, P.V.; Rasachak, B.; Syhavong, B.; Phetsouvanah, R.; Sheridan, I.; Humphreys, I.S.; et al. Genetic History of Hepatitis C Virus in East Asia. J. Virol. 2008, 83, 1071–1082. [Google Scholar] [CrossRef]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, A.S.; De Almeida-Neto, C.; Romano, C.M.; Alencar, C.S.; Ferreira, S.C.; Di-Lorenzo-Oliveira, C.; Levi, J.E.; Salles, N.A.; Mendrone-Junior, A.; Sabino, E.C. Phylogenetic analysis of the emergence of main hepatitis C virus subtypes in São Paulo, Brazil. Braz. J. Infect. Dis. 2015, 19, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, I.A.; Chen, J.J.; Lane, D.S.; Allmer, J.; Jimenez-Lucho, V.E. Analysis of a hepatitis C screening programme for US veterans. Epidemiol. Infect. 2005, 134, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Roselle, G.A.; Danko, L.H.; Kralovic, S.M.; Simbartl, L.A.; Kizer, K.W. National Hepatitis C Surveillance Day in the Veterans Health Administration of the Department of Veterans Affairs. Mil. Med. 2002, 167, 756–759. [Google Scholar] [CrossRef]
- Bernier, L.; Willems, B.; Delage, G.; Murphy, D.G. Identification of numerous hepatitis C virus genotypes in Montreal, Canada. J. Clin. Microbiol. 1996, 34, 2815–2818. [Google Scholar] [CrossRef]
- Murphy, D.G.; Willems, B.; Deschênes, M.; Hilzenrat, N.; Mousseau, R.; Sabbah, S. Use of Sequence Analysis of the NS5B Region for Routine Genotyping of Hepatitis C Virus with Reference to C/E1 and 5′ Untranslated Region Sequences. J. Clin. Microbiol. 2007, 45, 1102–1112. [Google Scholar] [CrossRef]
- Bull, R.A.; Eltahla, A.A.; Rodrigo, C.; Koekkoek, S.M.; Walker, M.R.; Pirozyan, M.R.; Betz-Stablein, B.; Toepfer, A.; Smith, M.L.; Oh, S.; et al. A method for near full-length amplification and sequencing for six hepatitis C virus genotypes. BMC Genom. 2016, 17, 247. [Google Scholar] [CrossRef]
- Ogata, N.; Alter, H.J.; Miller, R.H.; Purcell, R.H. Nucleotide sequence and mutation rate of the H strain of Hepatitis C virus. Proc. Natl. Acad. Sci. USA 1991, 88, 3392–3396. [Google Scholar] [CrossRef]
- Salemi, M.; Vandamme, A.-M. Hepatitis C Virus Evolutionary Patterns Studied Through Analysis of Full-Genome Sequences. J. Mol. Evol. 2002, 54, 62–70. [Google Scholar] [CrossRef]
- Gojobori, T.; Yamaguchi, Y.; Ikeo, K.; Mizokami, M. Evolution of pathogenic viruses with special reference to the rates of synonymous and nonsynonymous substitutions. Jpn. J. Genet. 1994, 69, 481–488. [Google Scholar] [CrossRef][Green Version]
- Caraballo, C.K.; Laskus, T.; Bukowska-Ośko, I.; Pawełczyk, A.; Berak, H.; Horban, A.; Fic, M.; Radkowski, M. Variability of hepatitis C virus hypervariable resion 1 (HVR1) during the early phase of pegylated interferon and rebavirin therapy. Adv. Med Sci. 2012, 57, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Wang, W.; Liu, X.; Tong, Y.; Liu, Y.; Ren, H.; Zhu, S.; Dubuisson, J.D.; Baumert, T.F.; Zhu, Y.; et al. Three Different Functional Microdomains in the Hepatitis C Virus Hypervariable Region 1 (HVR1) Mediate Entry and Immune Evasion. J. Boil. Chem. 2012, 287, 35631–35645. [Google Scholar] [CrossRef]
- Khera, T.; Behrendt, P.; Bankwitz, D.; Brown, R.J.P.; Todt, D.; Doepke, M.; Khan, A.G.; Schulze, K.; Law, J.; Logan, M.; et al. Functional and immunogenic characterization of diverse HCV glycoprotein E2 variants. J. Hepatol. 2019, 70, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Vasiliauskaite, I.; Owsianka, A.; England, P.; Khan, A.G.; Cole, S.; Bankwitz, D.; Foung, S.K.H.; Pietschmann, T.; Marcotrigiano, J.; Rey, F.A.; et al. Conformational Flexibility in the Immunoglobulin-Like Domain of the Hepatitis C Virus Glycoprotein E2. mBio 2017, 8, e00382-17. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Okamoto, H.; Sakamoto, M.; Kojima, M.; Tsuda, F.; Tanaka, T.; Munekata, E.; Muchmore, E.E.; Peterson, D.A.; Mishiro, S. A structurally frexible and antigenically variable N-terminal domain of hepatitis C virus E2/NS1protein: Implication for an escape from antibody. Virology 1993, 195, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Rispeter, K.; Lu, M.; Behrens, S.-E.; Fumiko, C.; Yoshida, T.; Roggendorf, M. Hepatitis C virus variability: Sequence analysis of an isolate after 10 years of chronic infection. Virus Genes 2000, 21, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Lavillette, D.; Tarr, A.W.; Donot, P.; Bartosch, B.; Bain, C.; Patel, A.H.; Dubuisson, J.D.; Ball, J.K. Characterization of host-range and cell entry properties of the major genotypes and subtypes of hepatitis C virus. Hepatology 2005, 41, 265–274. [Google Scholar] [CrossRef]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; et al. Hepatitis C Virus E2 Envelope Glycoprotein Core Structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef]
- Tseng, C.-T.K.; Klimpel, G.R. Binding of the Hepatitis C Virus Envelope Protein E2 to CD81 Inhibits Natural Killer Cell Functions. J. Exp. Med. 2001, 195, 43–50. [Google Scholar] [CrossRef]
- Sabahi, A.; Uprichard, S.L.; Wimley, W.C.; Dash, S.; Garry, R.F. Unexpected Structural Features of the Hepatitis C Virus Envelope Protein 2 Ectodomain. J. Virol. 2014, 88, 10280–10288. [Google Scholar] [CrossRef][Green Version]
- Li, W.; Krishnadas, D.K.; Li, J.; Tyrrell, D.L.J.; Agrawal, B. Induction of Primary Human T Cell Responses against Hepatitis C Virus-Derived Antigens NS3 or Core by Autologous Dendritic Cells Expressing Hepatitis C Virus Antigens: Potential for Vaccine and Immunotherapy. J. Immunol. 2006, 176, 6065–6075. [Google Scholar] [CrossRef] [PubMed]
- Dawood, R.M.; Moustafa, R.I.; Abdelhafez, T.H.; El-Shenawy, R.; El-Abd, Y.; El Din, N.G.B.; Dubuisson, J.; El Awady, M.K. A multiepitope peptide vaccine against HCV stimulates neutralizing humoral and persistent cellular responses in mice. BMC Infect. Dis. 2019, 19, 932. [Google Scholar] [CrossRef] [PubMed]
- Ikram, A.; Zaheer, T.; Awan, F.M.; Obaid, A.; Naz, A.; Hanif, R.; Paracha, R.Z.; Ali, A.; Naveed, A.K.; Janjua, H.A. Exploring NS3/4A, NS5A and NS5B proteins to design conserved subunit multi-epitope vaccine against HCV utilizing immunoinformatics approaches. Sci. Rep. 2018, 8, 16107. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Donato, G.; Amador-Cañizares, Y.; Alvarez-Lajonchere, L.; Guerra, I.; Perez, A.; Dubuisson, J.D.; Wychowsk, C.; Musacchio, A.; Aguilar, D.; Dueñas-Carrera, S. Neutralizing antibodies and broad, functional T cell immune response following immunization with hepatitis C virus proteins-based vaccine formulation. Vaccine 2014, 32, 1720–1726. [Google Scholar] [CrossRef] [PubMed]
- Keck, Z.-Y.; Pierce, B.G.; Lau, P.; Lu, J.; Wang, Y.; Underwood, A.; Bull, R.A.; Prentoe, J.; Velázquez-Moctezuma, R.; Walker, M.R.; et al. Broadly neutralizing antibodies from an individual that naturally cleared multiple hepatitis C virus infections uncover molecular determinants for E2 targeting and vaccine design. PLoS Pathog. 2019, 15, e1007772. [Google Scholar] [CrossRef] [PubMed]
- Masavuli, M.G.; Wijesundara, D.K.; Torresi, J.; Gowans, E.J.; Grubor-Bauk, B. Preclinical Development and Production of Virus-Like Particles as Vaccine Candidates for Hepatitis C. Front. Microbiol. 2017, 8, 2413. [Google Scholar] [CrossRef]
- Torresi, J. The Rationale for a Preventative HCV Virus-Like Particle (VLP) Vaccine. Front. Microbiol. 2017, 8, 2163. [Google Scholar] [CrossRef]
- Sheridan, D.; Hajarizadeh, B.; Fenwick, F.I.; Matthews, G.V.; Applegate, T.L.; Douglas, M.; Neely, D.; Askew, B.; Dore, G.J.; Lloyd, A.; et al. Maximum levels of hepatitis C virus lipoviral particles are associated with early and persistent infection. Liver Int. 2016, 36, 1774–1782. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Isolate Name | Nucleotide Length | Polyprotein Length | Blood Sampling Date | Country | Province | Village | Sex | Age | Occupation |
|---|---|---|---|---|---|---|---|---|---|---|
| 1b | N12-3009-Cam | 9323 | 3010 | 2012.08.22 | Cambodia | Siem Reap | Krabei Riel | male | 60s | teacher |
| N16-3009-Cam | 9326 | 3010 | 2016.09.03 | |||||||
| N12-3072-Cam | 9378 | 3010 | 2012.08.22 | Cambodia | Siem Reap | Chrey | female | 70s | housewife | |
| N16-3072-Cam | 9321 | 3010 | 2016.09.03 | |||||||
| 6e | N12-2804-Cam | 9341 | 3018 | 2012.08.22 | Cambodia | Siem Reap | Krabei Riel | male | 20s | office worker |
| N16-2804-Cam | 9326 | 3018 | 2016.09.03 | |||||||
| 6r | N12-2911-Cam | 9374 | 3016 | 2012.08.22 | Cambodia | Siem Reap | Krabei Riel | female | 60s | housewife |
| N16-2911-Cam | 9374 | 3016 | 2016.09.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, C.; Nagashima, S.; Chuon, C.; Ko, K.; Huy Do, S.; Lim, O.; Hok, S.; Svay, S.; Matsuo, J.; Katayama, K.; et al. Substitution of the CD81 Binding Site and β-Sandwich Area in E2 of HCV in Cambodia. Viruses 2020, 12, 551. https://doi.org/10.3390/v12050551
Yamamoto C, Nagashima S, Chuon C, Ko K, Huy Do S, Lim O, Hok S, Svay S, Matsuo J, Katayama K, et al. Substitution of the CD81 Binding Site and β-Sandwich Area in E2 of HCV in Cambodia. Viruses. 2020; 12(5):551. https://doi.org/10.3390/v12050551
Chicago/Turabian StyleYamamoto, Chikako, Shintaro Nagashima, Channarena Chuon, Ko Ko, Son Huy Do, Oline Lim, Sirany Hok, Somana Svay, Junko Matsuo, Keiko Katayama, and et al. 2020. "Substitution of the CD81 Binding Site and β-Sandwich Area in E2 of HCV in Cambodia" Viruses 12, no. 5: 551. https://doi.org/10.3390/v12050551
APA StyleYamamoto, C., Nagashima, S., Chuon, C., Ko, K., Huy Do, S., Lim, O., Hok, S., Svay, S., Matsuo, J., Katayama, K., Takahashi, K., & Tanaka, J. (2020). Substitution of the CD81 Binding Site and β-Sandwich Area in E2 of HCV in Cambodia. Viruses, 12(5), 551. https://doi.org/10.3390/v12050551