Infectious Bronchitis Virus Regulates Cellular Stress Granule Signaling

 , , , and
, , , and 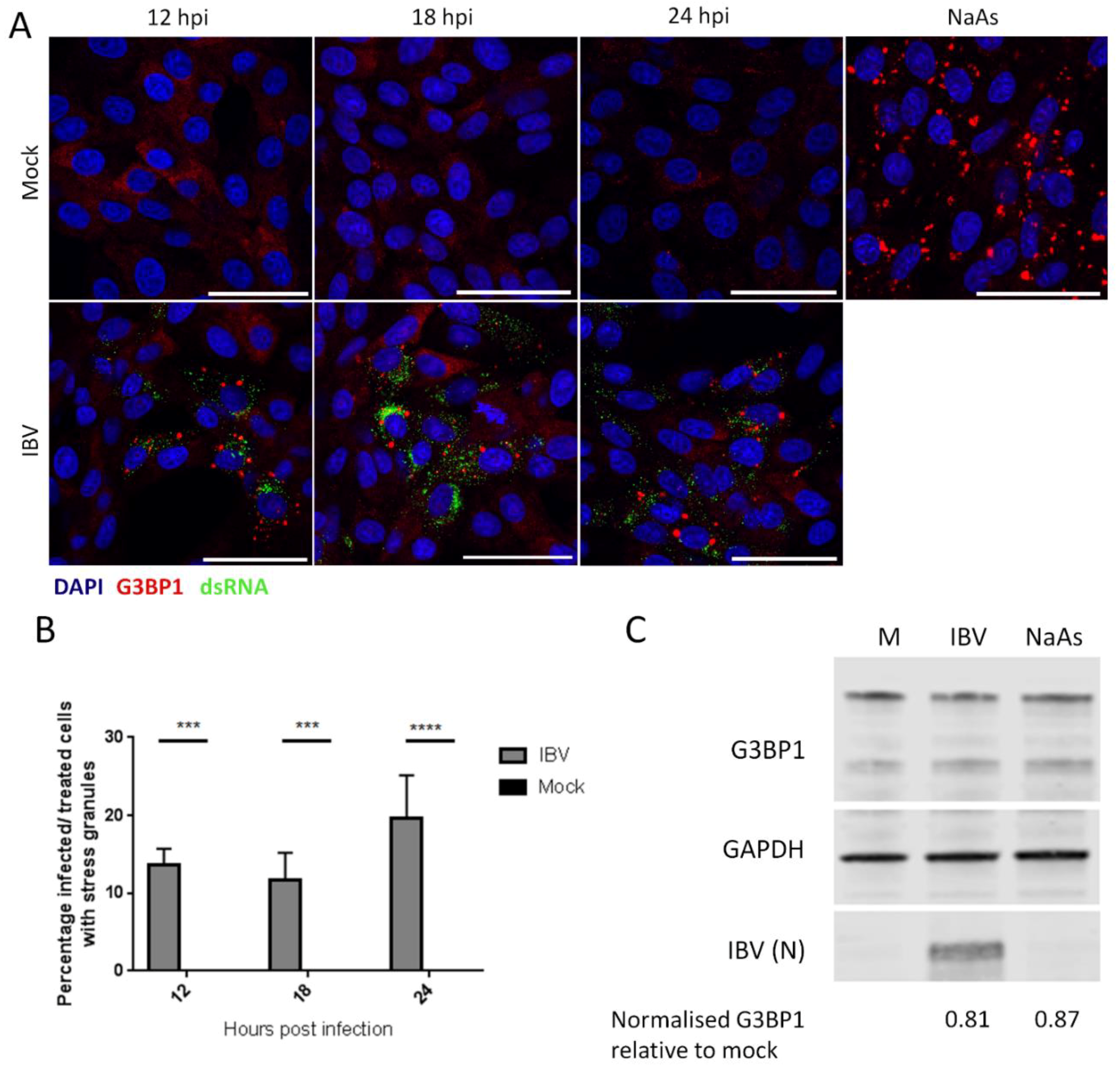{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells, Viruses and Reagents
2.2. Immunofluorescence
2.3. Fluorescent in situ Hybridization (FISH)
2.4. Cell Lysis and Western Blot
2.5. Ribopuromycylation (RPM)
3. Results
3.1. IBV Replication Induces Stress Granules in a Proportion of Infected Cells
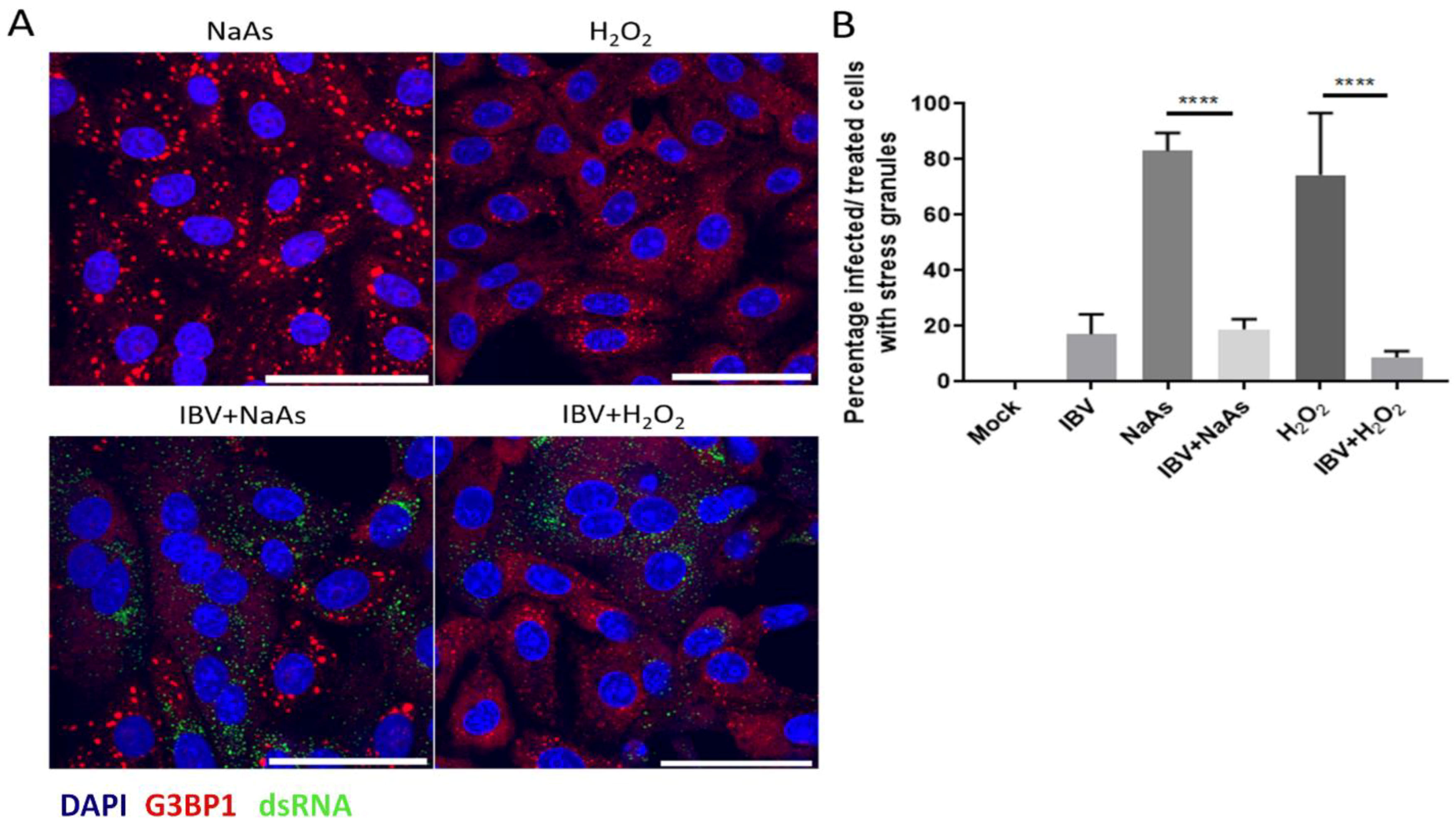
3.2. IBV Replication Inhibits Chemical Induction of Stress Granules
3.3. Stress Granules in IBV Infected Cells Are Canonical
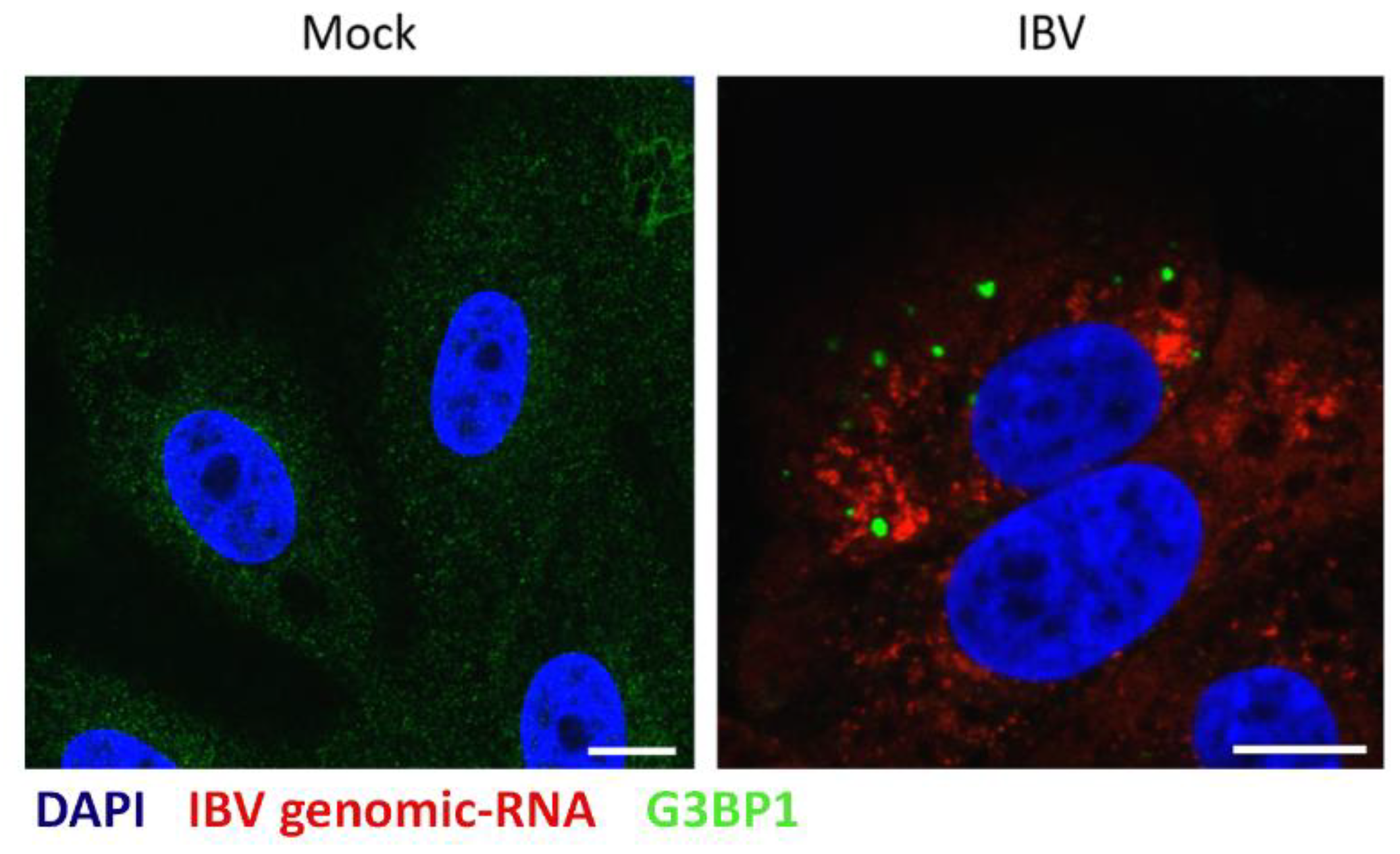
3.4. IBV Genomic RNA Is not Diverted to Stress Granules during Infection
3.5. Stress Granules Are not Diverted to Sites of Virus Replication
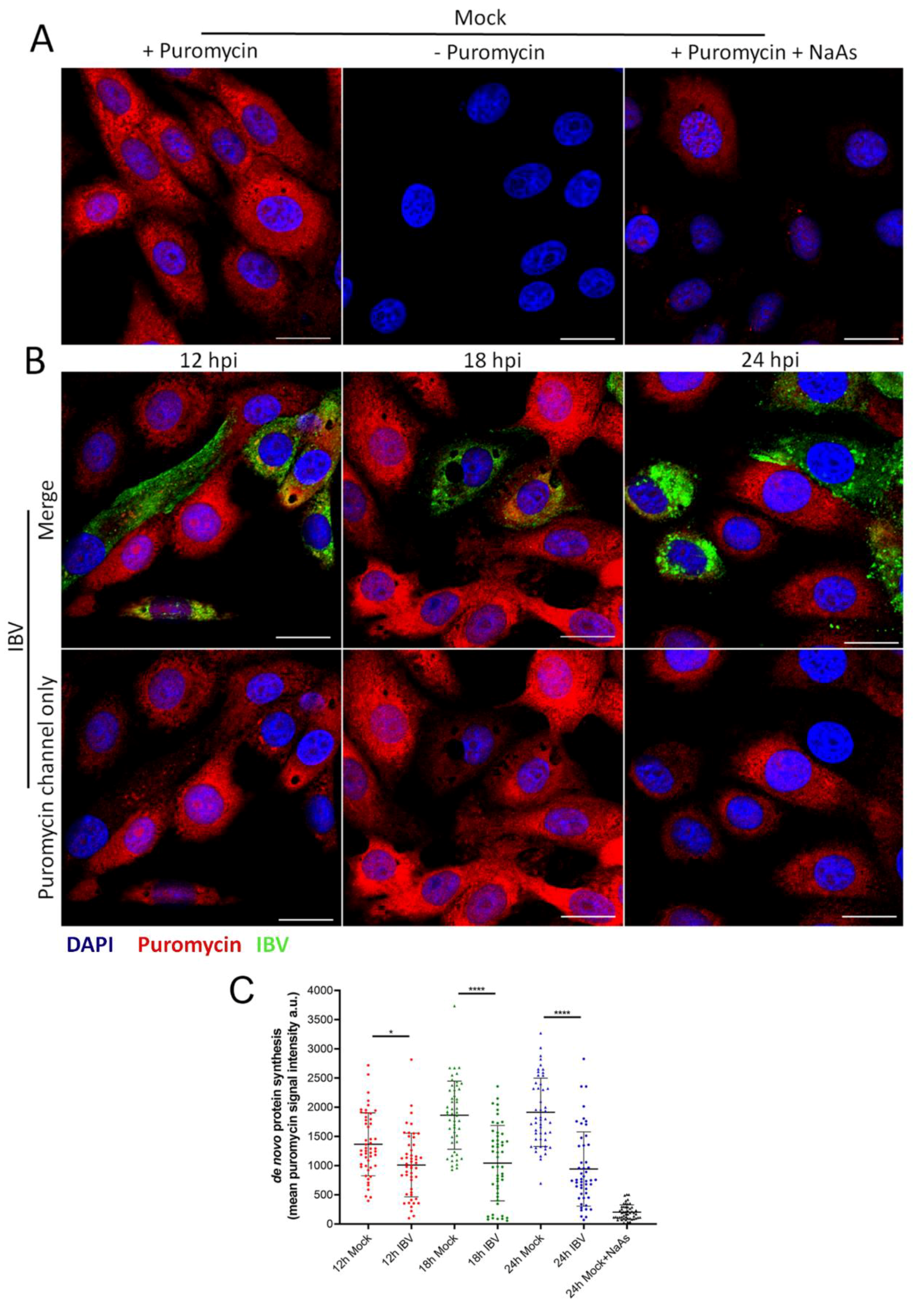
3.6. IBV Infection Induces A Translational Shut-Off that Increases with the Duration of Infection
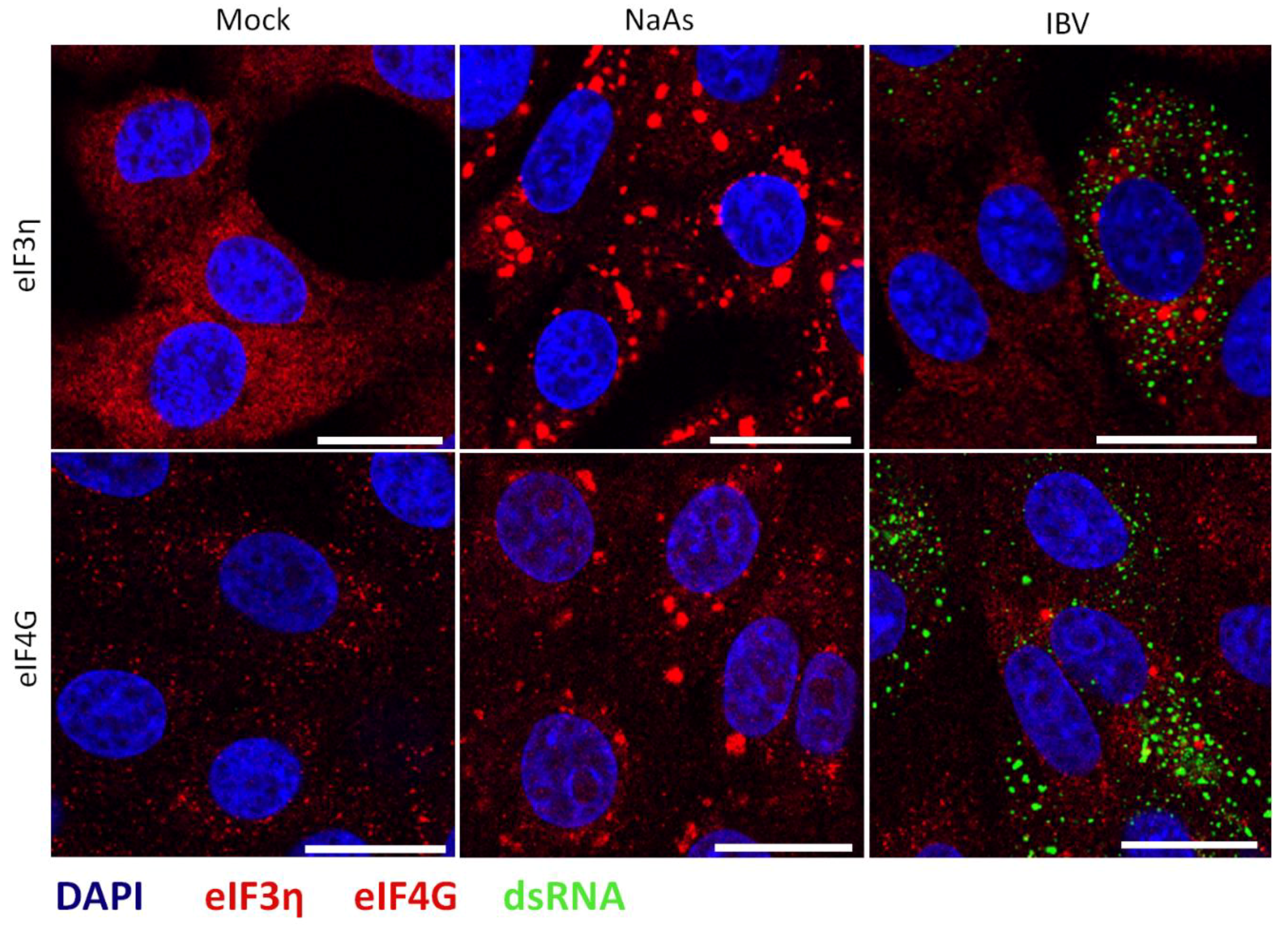
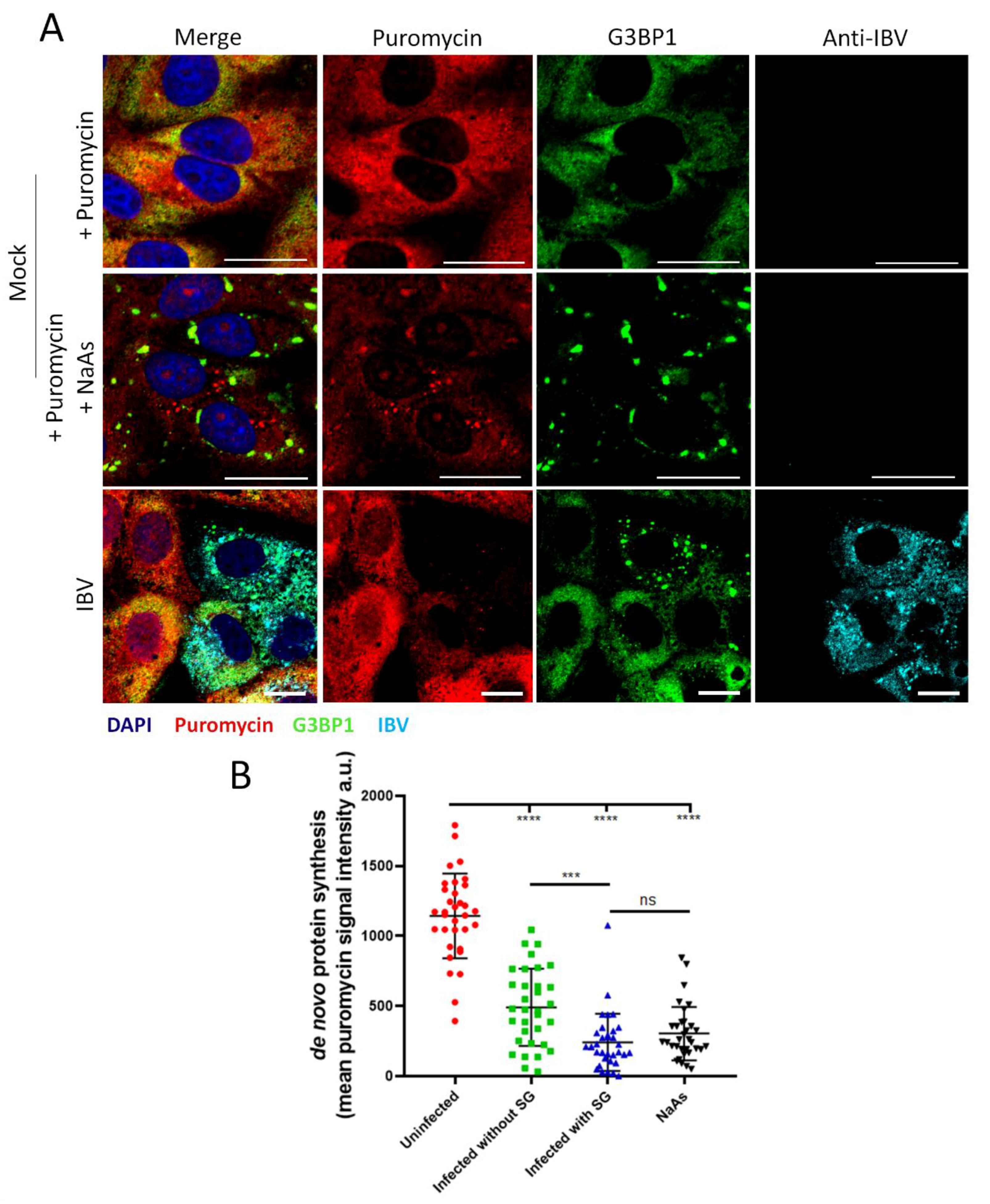
3.7. Stress Granule Formation in IBV Infected Cells Is Correlated with Translational Shut-Off
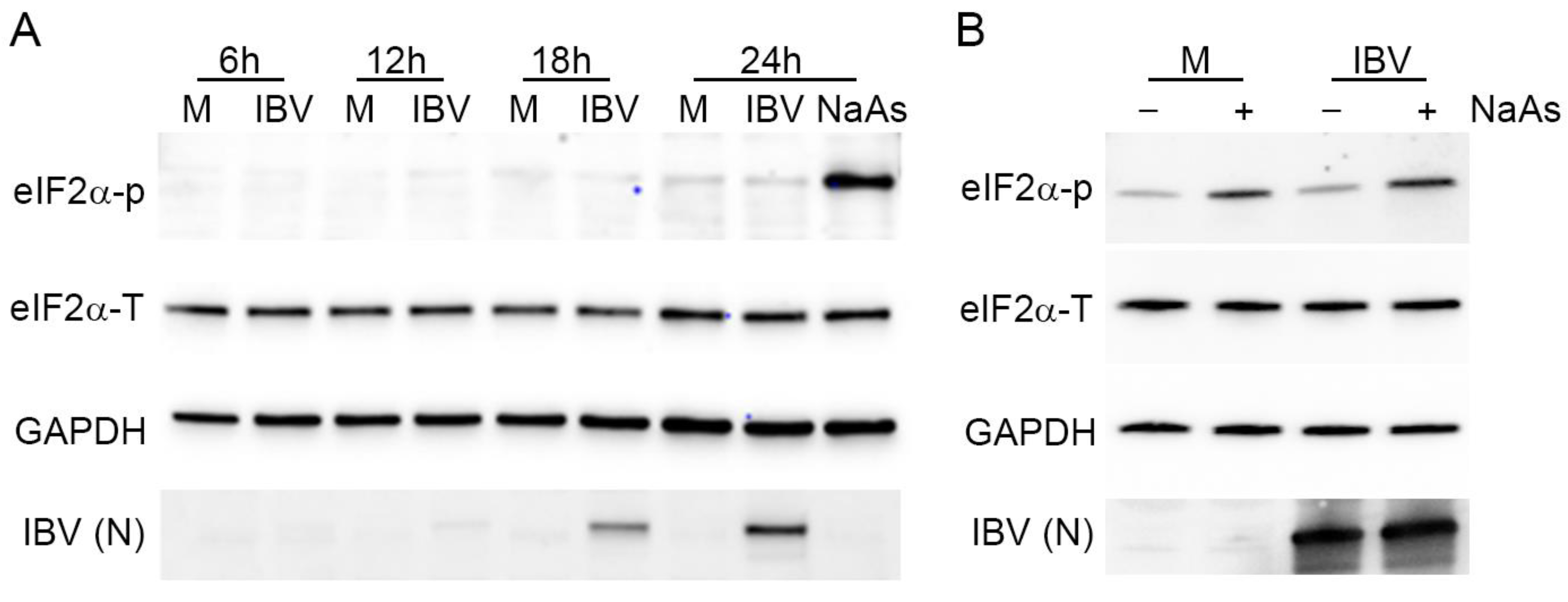
3.8. Stress Granule Formation and Translational Shut-Off during IBV Replication Are Independent of eIF2α Phosphorylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- McCormick, C.; Khaperskyy, D.A. Translation inhibition and stress granules in the antiviral immune response. Nat. Rev. Immunol. 2017, 17, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.; Mathews, M.B.; Mohr, I. Tinkering with translation: Protein synthesis in virus-infected cells. Cold Spring Harb. Perspect. Biol. 2013, 5, a012351. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.; Anderson, P. Stress granules: Sites of mRNA triage that regulate mRNA stability and translatability. Biochem. Soc. Trans. 2002, 30, 963–969. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef]
- Deng, J.; Harding, H.P.; Raught, B.; Gingras, A.C.; Berlanga, J.J.; Scheuner, D.; Kaufman, R.J.; Ron, D.; Sonenberg, N. Activation of GCN2 in UV-irradiated cells inhibits translation. Curr. Biol. 2002, 12, 1279–1286. [Google Scholar] [CrossRef]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef]
- Berlanga, J.J.; Ventoso, I.; Harding, H.P.; Deng, J.; Ron, D.; Sonenberg, N.; Carrasco, L.; de Haro, C. Antiviral effect of the mammalian translation initiation factor 2alpha kinase GCN2 against RNA viruses. EMBO J. 2006, 25, 1730–1740. [Google Scholar] [CrossRef]
- Mazroui, R.; Sukarieh, R.; Bordeleau, M.E.; Kaufman, R.J.; Northcote, P.; Tanaka, J.; Gallouzi, I.; Pelletier, J. Inhibition of ribosome recruitment induces stress granule formation independently of eukaryotic initiation factor 2alpha phosphorylation. Mol. Biol. Cell 2006, 17, 4212–4219. [Google Scholar] [CrossRef]
- Low, W.K.; Dang, Y.; Schneider-Poetsch, T.; Shi, Z.; Choi, N.S.; Merrick, W.C.; Romo, D.; Liu, J.O. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol. Cell 2005, 20, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.E.; Matthews, J.; Wojnar, J.M.; Lindqvist, L.; Novac, O.; Jankowsky, E.; Sonenberg, N.; Northcote, P.; Teesdale-Spittle, P.; Pelletier, J. Stimulation of mammalian translation initiation factor eIF4A activity by a small molecule inhibitor of eukaryotic translation. Proc. Natl. Acad. Sci. USA 2005, 102, 10460–10465. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, M.E.; Mori, A.; Oberer, M.; Lindqvist, L.; Chard, L.S.; Higa, T.; Belsham, G.J.; Wagner, G.; Tanaka, J.; Pelletier, J. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat. Chem. Biol. 2006, 2, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Fujimura, K.; Sciaranghella, D.; Ivanova, V.; Ivanov, P.; Anderson, P. Hydrogen peroxide induces stress granule formation independent of eIF2alpha phosphorylation. Biochem. Biophys. Res. Commun. 2012, 423, 763–769. [Google Scholar] [CrossRef]
- Protter, D.S.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Wheeler, J.R.; Matheny, T.; Jain, S.; Abrisch, R.; Parker, R. Distinct stages in stress granule assembly and disassembly. Elife 2016, 5, e18413. [Google Scholar] [CrossRef]
- Contu, L.; Steiner, S.; Thiel, V.; Muhlemann, O. The Role of Stress Granules and the Nonsense-mediated mRNA Decay Pathway in Antiviral Defence. Chim. Int. J. Chem. 2019, 73, 374–379. [Google Scholar] [CrossRef]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and Maturation of Phase-Separated Liquid Droplets by RNA-Binding Proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef]
- Van Treeck, B.; Protter, D.S.W.; Matheny, T.; Khong, A.; Link, C.D.; Parker, R. RNA self-assembly contributes to stress granule formation and defining the stress granule transcriptome. Proc. Natl. Acad. Sci. USA 2018, 115, 2734–2739. [Google Scholar] [CrossRef]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; van Kuppeveld, F.J.; Lloyd, R.E. Stress granules regulate double-stranded RNA-dependent protein kinase activation through a complex containing G3BP1 and Caprin1. MBIO 2015, 6, e02486. [Google Scholar] [CrossRef] [PubMed]
- Poblete-Duran, N.; Prades-Perez, Y.; Vera-Otarola, J.; Soto-Rifo, R.; Valiente-Echeverria, F. Who Regulates Whom? An Overview of RNA Granules and Viral Infections. Viruses 2016, 8, 180. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Cardenas, A.M.; Marissen, W.E.; Lloyd, R.E. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2007, 2, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Domeradzka, N.E.; Baggen, J.; Geertsema, C.; Flipse, J.; Vlak, J.M.; Pijlman, G.P. Chikungunya virus nsP3 blocks stress granule assembly by recruitment of G3BP into cytoplasmic foci. J. Virol. 2012, 86, 10873–10879. [Google Scholar] [CrossRef]
- Matthews, J.D.; Frey, T.K. Analysis of subcellular G3BP redistribution during rubella virus infection. J. Gen. Virol. 2012, 93, 267–274. [Google Scholar] [CrossRef]
- Sola, I.; Galan, C.; Mateos-Gomez, P.A.; Palacio, L.; Zuniga, S.; Cruz, J.L.; Almazan, F.; Enjuanes, L. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J. Virol. 2011, 85, 5136–5149. [Google Scholar] [CrossRef]
- Raaben, M.; Groot Koerkamp, M.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell. Microbiol. 2007, 9, 2218–2229. [Google Scholar] [CrossRef]
- Galan, A.; Lozano, G.; Pineiro, D.; Martinez-Salas, E. G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J. 2017, 284, 3202–3217. [Google Scholar] [CrossRef]
- Bidet, K.; Dadlani, D.; Garcia-Blanco, M.A. Correction: G3BP1, G3BP2 and CAPRIN1 Are Required for Translation of Interferon Stimulated mRNAs and Are Targeted by a Dengue Virus Non-coding RNA. PLoS Pathog. 2017, 13, e1006295. [Google Scholar] [CrossRef]
- Nakagawa, K.; Narayanan, K.; Wada, M.; Makino, S. Inhibition of Stress Granule Formation by Middle East Respiratory Syndrome Coronavirus 4a Accessory Protein Facilitates Viral Translation, Leading to Efficient Virus Replication. J. Virol. 2018, 92, e00902-18. [Google Scholar] [CrossRef]
- Rabouw, H.H.; Langereis, M.A.; Knaap, R.C.; Dalebout, T.J.; Canton, J.; Sola, I.; Enjuanes, L.; Bredenbeek, P.J.; Kikkert, M.; De Groot, R.J.; et al. Middle East Respiratory Coronavirus Accessory Protein 4a Inhibits PKR-Mediated Antiviral Stress Responses. PLoS Pathog. 2016, 12, e1005982. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liao, Y.; Yap, P.L.; Png, K.J.; Tam, J.P.; Liu, D.X. Inhibition of protein kinase R activation and upregulation of GADD34 expression play a synergistic role in facilitating coronavirus replication by maintaining de novo protein synthesis in virus-infected cells. J. Virol. 2009, 83, 12462–12472. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J. Virol. 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Kint, J.; Langereis, M.A.; Maier, H.J.; Britton, P.; van Kuppeveld, F.J.; Koumans, J.; Wiegertjes, G.F.; Forlenza, M. Infectious Bronchitis Coronavirus Limits Interferon Production by Inducing a Host Shutoff That Requires Accessory Protein 5b. J. Virol. 2016, 90, 7519–7528. [Google Scholar] [CrossRef]
- Britton, P.; Evans, S.; Dove, B.; Davies, M.; Casais, R.; Cavanagh, D. Generation of a recombinant avian coronavirus infectious bronchitis virus using transient dominant selection. J. Virol. Methods 2005, 123, 203–211. [Google Scholar] [CrossRef]
- Maier, H.J.; Hawes, P.C.; Cottam, E.M.; Mantell, J.; Verkade, P.; Monaghan, P.; Wileman, T.; Britton, P. Infectious bronchitis virus generates spherules from zippered endoplasmic reticulum membranes. MBio 2013, 4, e00801-13. [Google Scholar] [CrossRef]
- David, A.; Dolan, B.P.; Hickman, H.D.; Knowlton, J.J.; Clavarino, G.; Pierre, P.; Bennink, J.R.; Yewdell, J.W. Nuclear translation visualized by ribosome-bound nascent chain puromycylation. J. Cell Biol. 2012, 197, 45–57. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Taniuchi, S.; Miyake, M.; Tsugawa, K.; Oyadomari, M.; Oyadomari, S. Integrated stress response of vertebrates is regulated by four eIF2α kinases. Sci. Rep. 2016, 6, 32886. [Google Scholar] [CrossRef]
- McInerney, G.M.; Kedersha, N.L.; Kaufman, R.J.; Anderson, P.; Liljestrom, P. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 2005, 16, 3753–3763. [Google Scholar] [CrossRef]
- Nelson, E.V.; Schmidt, K.M.; Deflube, L.R.; Doganay, S.; Banadyga, L.; Olejnik, J.; Hume, A.J.; Ryabchikova, E.; Ebihara, H.; Kedersha, N.; et al. Ebola Virus Does Not Induce Stress Granule Formation during Infection and Sequesters Stress Granule Proteins within Viral Inclusions. J. Virol. 2016, 90, 7268–7284. [Google Scholar] [CrossRef] [PubMed]
- Albornoz, A.; Carletti, T.; Corazza, G.; Marcello, A. The stress granule component TIA-1 binds tick-borne encephalitis virus RNA and is recruited to perinuclear sites of viral replication to inhibit viral translation. J. Virol. 2014, 88, 6611–6622. [Google Scholar] [CrossRef] [PubMed]
- Emara, M.M.; Brinton, M.A. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 9041–9046. [Google Scholar] [CrossRef] [PubMed]
- Amorim, R.; Temzi, A.; Griffin, B.D.; Mouland, A.J. Zika virus inhibits eIF2alpha-dependent stress granule assembly. PLoS Negl. Trop. Dis. 2017, 11, e0005775. [Google Scholar] [CrossRef]
- Sharma, N.R.; Majerciak, V.; Kruhlak, M.J.; Zheng, Z.M. KSHV inhibits stress granule formation by viral ORF57 blocking PKR activation. PLoS Pathog. 2017, 13, e1006677. [Google Scholar] [CrossRef]
- Basu, M.; Courtney, S.C.; Brinton, M.A. Arsenite-induced stress granule formation is inhibited by elevated levels of reduced glutathione in West Nile virus-infected cells. PLoS Pathog. 2017, 13, e1006240. [Google Scholar] [CrossRef]
- Linero, F.N.; Thomas, M.G.; Boccaccio, G.L.; Scolaro, L.A. Junin virus infection impairs stress-granule formation in Vero cells treated with arsenite via inhibition of eIF2alpha phosphorylation. J. Gen. Virol. 2011, 92, 2889–2899. [Google Scholar] [CrossRef]
- Le Sage, V.; Cinti, A.; McCarthy, S.; Amorim, R.; Rao, S.; Daino, G.L.; Tramontano, E.; Branch, D.R.; Mouland, A.J. Ebola virus VP35 blocks stress granule assembly. Virology 2017, 502, 73–83. [Google Scholar] [CrossRef]
- Roth, H.; Magg, V.; Uch, F.; Mutz, P.; Klein, P.; Haneke, K.; Lohmann, V.; Bartenschlager, R.; Fackler, O.T.; Locker, N.; et al. Flavivirus Infection Uncouples Translation Suppression from Cellular Stress Responses. MBio 2017, 8, e02150-16. [Google Scholar] [CrossRef]
- Arimoto-Matsuzaki, K.; Saito, H.; Takekawa, M. TIA1 oxidation inhibits stress granule assembly and sensitizes cells to stress-induced apoptosis. Nat. Commun. 2016, 7, 10252. [Google Scholar] [CrossRef]
- Fung, T.S.; Liao, Y.; Liu, D.X. The endoplasmic reticulum stress sensor IRE1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J. Virol. 2014, 88, 12752–12764. [Google Scholar] [CrossRef]
- Katsafanas, G.C.; Moss, B. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2007, 2, 221–228. [Google Scholar] [CrossRef]
- Desmet, E.A.; Anguish, L.J.; Parker, J.S. Virus-mediated compartmentalization of the host translational machinery. MBio 2014, 5, e01463-14. [Google Scholar] [CrossRef]
- Panas, M.D.; Varjak, M.; Lulla, A.; Eng, K.E.; Merits, A.; Karlsson Hedestam, G.B.; McInerney, G.M. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 2012, 23, 4701–4712. [Google Scholar] [CrossRef]
- Ruggieri, A.; Dazert, E.; Metz, P.; Hofmann, S.; Bergeest, J.-P.; Mazur, J.; Bankhead, P.; Hiet, M.-S.; Kallis, S.; Alvisi, G.; et al. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 2012, 12, 71–85. [Google Scholar] [CrossRef]
- Fritzlar, S.; Aktepe, T.E.; Chao, Y.W.; Kenney, N.D.; McAllaster, M.R.; Wilen, C.B.; White, P.A.; Mackenzie, J.M. Mouse Norovirus Infection Arrests Host Cell Translation Uncoupled from the Stress Granule-PKR-eIF2alpha Axis. MBio 2019, 10. [Google Scholar] [CrossRef]
- Brocard, M.; Iadevaia, V.; Klein, P.; Hall, B.; Lewis, G.; Lu, J.; Burke, J.; Willcocks, M.; Parker, R.; Ruggieri, A.; et al. Norovirus infection results in eIF2α independent host translation shut-off and remodels the G3BP1 interactome evading stress granule formation. PLoS Pathog. 2020, 16, e1008250. [Google Scholar] [CrossRef]
- Waskiewicz, A.J.; Flynn, A.; Proud, C.G.; Cooper, J.A. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J. 1997, 16, 1909–1920. [Google Scholar] [CrossRef]
- Royall, E.; Doyle, N.; Abdul-Wahab, A.; Emmott, E.; Morley, S.J.; Goodfellow, I.; Roberts, L.O.; Locker, N. Murine norovirus 1 (MNV1) replication induces translational control of the host by regulating eIF4E activity during infection. J. Biol. Chem. 2015, 290, 4748–4758. [Google Scholar] [CrossRef]
- Thornbrough, J.M.; Jha, B.K.; Yount, B.; Goldstein, S.A.; Li, Y.; Elliott, R.; Sims, A.C.; Baric, R.S.; Silverman, R.H.; Weiss, S.R. Middle East Respiratory Syndrome Coronavirus NS4b Protein Inhibits Host RNase L Activation. MBio 2016, 7, e00258. [Google Scholar] [CrossRef]
- Birdwell, L.D.; Zalinger, Z.B.; Li, Y.; Wright, P.W.; Elliott, R.; Rose, K.M.; Silverman, R.H.; Weiss, S.R. Activation of RNase L by Murine Coronavirus in Myeloid Cells Is Dependent on Basal Oas Gene Expression and Independent of Virus-Induced Interferon. J. Virol. 2016, 90, 3160–3172. [Google Scholar] [CrossRef]
- Silverman, R.H. A scientific journey through the 2-5A/RNase L system. Cytokine Growth Factor Rev. 2007, 18, 381–388. [Google Scholar] [CrossRef]
- Burke, J.M.; Lester, E.T.; Tauber, D.; Parker, R. RNase L promotes the formation of unique ribonucleoprotein granules distinct from stress granules. J. Biol. Chem. 2020, 295, 1426–1438. [Google Scholar] [CrossRef]
- Manivannan, P.; Siddiqui, M.A.; Malathi, K. RNase L amplifies interferon signaling by inducing PKR-mediated antiviral stress granules. J. Virol. 2020. [Google Scholar] [CrossRef]
- Narayanan, K.; Ramirez, S.I.; Lokugamage, K.G.; Makino, S. Coronavirus nonstructural protein 1: Common and distinct functions in the regulation of host and viral gene expression. Virus Res. 2015, 202, 89–100. [Google Scholar] [CrossRef]
- Dalet, A.; Argüello, R.J.; Combes, A.; Spinelli, L.; Jaeger, S.; Fallet, M.; Vu Manh, T.-P.; Mendes, A.; Perego, J.; Reverendo, M.; et al. Protein synthesis inhibition and GADD34 control IFN-β heterogeneous expression in response to dsRNA. EMBO J. 2017, 36, 761–782. [Google Scholar] [CrossRef]
- Knoops, K.; Kikkert, M.; van den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; Van der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 2008, 6, 1957–1974. [Google Scholar] [CrossRef]
- Kint, J.; Fernandez-Gutierrez, M.; Maier, H.J.; Britton, P.; Langereis, M.A.; Koumans, J.; Wiegertjes, G.F.; Forlenza, M. Activation of the Chicken Type I Interferon Response by Infectious Bronchitis Coronavirus. J. Virol. 2015, 89, 1156–1167. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brownsword, M.J.; Doyle, N.; Brocard, M.; Locker, N.; Maier, H.J. Infectious Bronchitis Virus Regulates Cellular Stress Granule Signaling. Viruses 2020, 12, 536. https://doi.org/10.3390/v12050536
Brownsword MJ, Doyle N, Brocard M, Locker N, Maier HJ. Infectious Bronchitis Virus Regulates Cellular Stress Granule Signaling. Viruses. 2020; 12(5):536. https://doi.org/10.3390/v12050536
Chicago/Turabian StyleBrownsword, Matthew J., Nicole Doyle, Michèle Brocard, Nicolas Locker, and Helena J. Maier. 2020. "Infectious Bronchitis Virus Regulates Cellular Stress Granule Signaling" Viruses 12, no. 5: 536. https://doi.org/10.3390/v12050536
APA StyleBrownsword, M. J., Doyle, N., Brocard, M., Locker, N., & Maier, H. J. (2020). Infectious Bronchitis Virus Regulates Cellular Stress Granule Signaling. Viruses, 12(5), 536. https://doi.org/10.3390/v12050536