ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel?



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
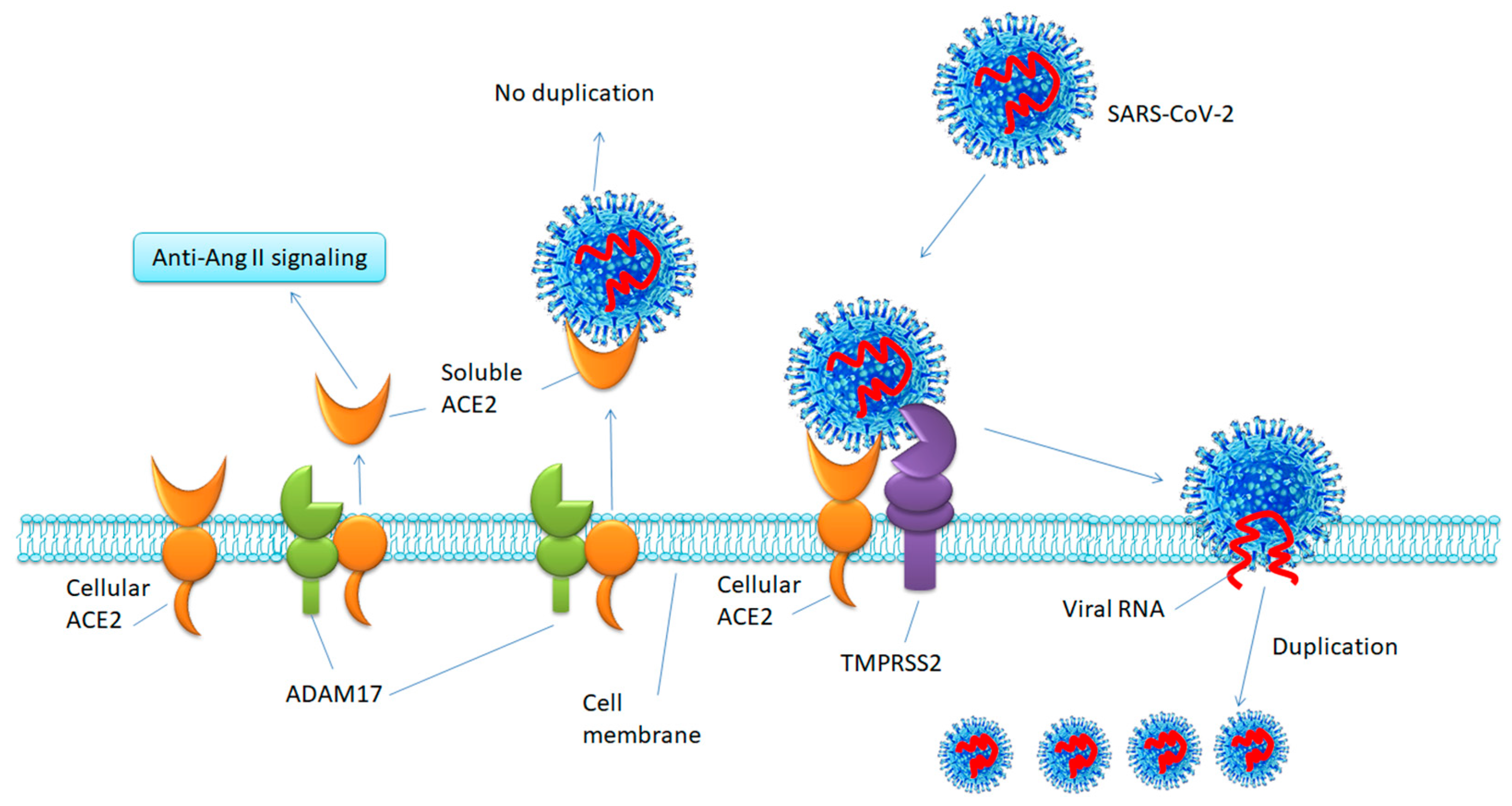
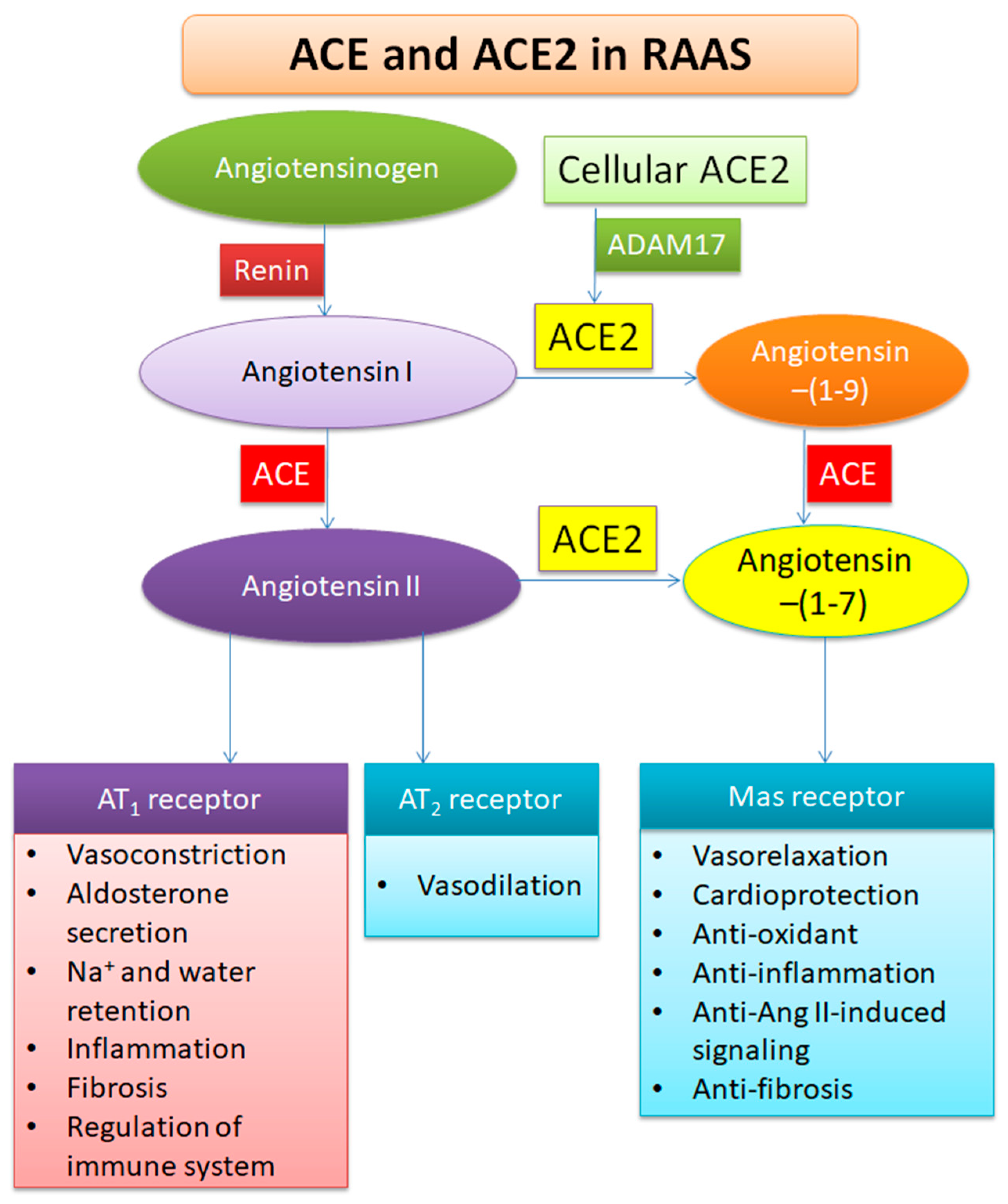
2. Profile of ACE2
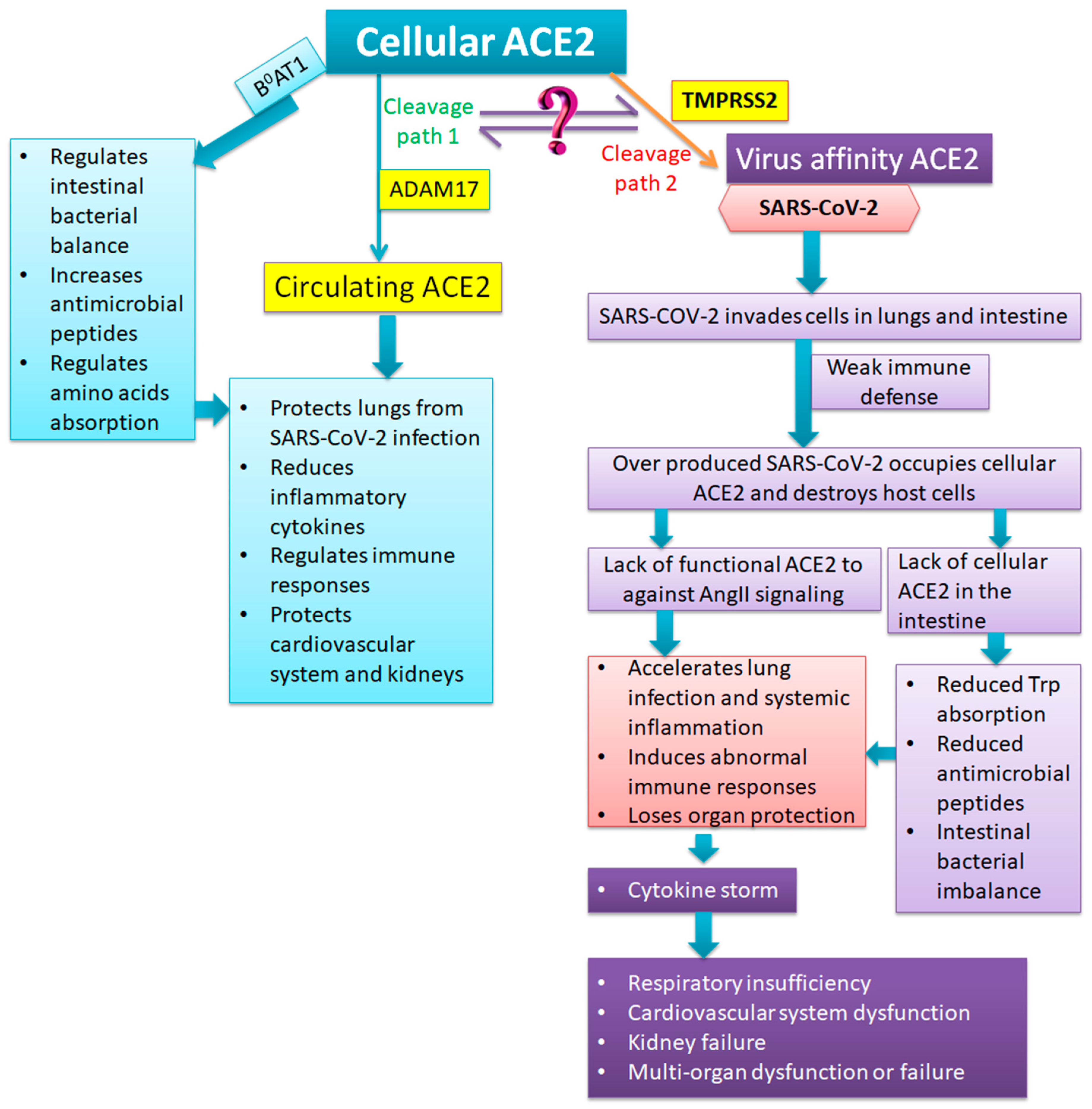
3. Functions of ACE2
4. Anatomic and Pathological Findings of Severe and Critical COVID-19: What is the Role of ACE2?
5. Strategies for Preventing and Reversing Severe and Critical Conditions of COVID-19
5.1. Inhibiting TMPRSS2 to Block SARS-CoV-2 Cell Entry
5.2. Increasing ACE2 or Ang-(1-7)
5.3. Supplementation of Essential Amino Acids
Funding
Acknowledgments
Conflicts of Interest
References
- COVID-19 Dashboard by the Center for Systems Science and Engineering (CSSE) at Johns Hopkins University (JHU). Available online: https://coronavirus.jhu.edu/map.html (accessed on 24 April 2020).
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.; Lely, A.T.; Navis, G.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Crackower, M.A.; Sarao, R.; Oudit, G.Y.; Yagil, C.; Kozieradzki, I.; Scanga, S.E.; Oliveira-dos-Santos, A.J.; da Costa, J.; Zhang, L.; Pei, Y.; et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002, 417, 822–828. [Google Scholar] [CrossRef]
- Feng, Y.; Yue, X.; Xia, H.; Bindom, S.M.; Hickman, P.J.; Filipeanu, C.M.; Wu, G.; Lazartigues, E. Angiotensin-converting enzyme 2 overexpression in the subfornical organ prevents the angiotensin II-mediated pressor and drinking responses and is associated with angiotensin II type 1 receptor downregulation. Circ. Res. 2008, 102, 729–736. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential downregulation of ACE2 by the spike proteins of severe acute respiratory syndrome coronavirus and human coronavirus NL63. J. Virol. 2010, 84, 1198–1205. [Google Scholar] [CrossRef]
- Matsuyama, S.; Ujike, M.; Morikawa, S.; Tashiro, M.; Taguchi, F. Protease-mediated enhancement of severe acute respiratory syndrome coronavirus infection. Proc. Natl. Acad. Sci. USA 2005, 102, 12543–12547. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, e01815-18. [Google Scholar] [CrossRef]
- Jacquinet, E.; Rao, N.V.; Rao, G.V.; Zhengming, W.; Albertine, K.H.; Hoidal, J.R. Cloning and characterization of the cDNA and gene for human epitheliasin. Eur. J. Biochem. 2001, 268, 2687–2699. [Google Scholar] [CrossRef]
- Lin, B.; Ferguson, C.; White, J.T.; Wang, S.; Vessella, R.; True, L.D.; Hood, L.; Nelson, P.S. Prostate-localized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999, 59, 4180–4184. [Google Scholar]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef]
- Turner, A.J.; Tipnis, S.R.; Guy, J.L.; Rice, G.; Hooper, N.M. ACEH/ACE2 is a novel mammalian metallocarboxypeptidase and a homologue of angiotensin-converting enzyme insensitive to ACE inhibitors. Can. J. Physiol. Pharmacol. 2002, 80, 346–353. [Google Scholar] [CrossRef]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F.; et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- Benter, I.F.; Yousif, M.H.; Dhaunsi, G.S.; Kaur, J.; Chappell, M.C.; Diz, D.I. Angiotensin-(1-7) prevents activation of NADPH oxidase and renal vascular dysfunction in diabetic hypertensive rats. Am. J. Nephrol. 2008, 28, 25–33. [Google Scholar] [CrossRef]
- El-Hashim, A.Z.; Renno, W.M.; Raghupathy, R.; Abduo, H.T.; Akhtar, S.; Benter, I.F. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-κB-dependent pathways. Br. J. Pharmacol. 2012, 166, 1964–1976. [Google Scholar] [CrossRef]
- Santos, R.A. Angiotensin-(1-7). Hypertension 2014, 63, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.; Simoes e Silva, A.C.; Maric, C.; Silva, D.M.; Machado, R.P.; de Buhr, I.; Heringer-Walther, S.; Pinheiro, S.V.; Lopes, M.T.; Bader, M.; et al. Angiotensin-(1-7) is an endogenous ligand for the G protein-coupled receptor Mas. Proc. Natl. Acad. Sci. USA 2003, 100, 8258–8263. [Google Scholar] [CrossRef] [PubMed]
- Chamsi-Pasha, M.A.; Shao, Z.; Tang, W.H. Angiotensin-converting enzyme 2 as a therapeutic target for heart failure. Curr. Heart Fail. Rep. 2014, 11, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Rice, G.I.; Jones, A.L.; Grant, P.J.; Carter, A.M.; Turner, A.J.; Hooper, N.M. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension 2006, 48, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, L.; Riera, M.; Pascual, J.; Valdivielso, J.M.; Barrios, C.; Betriu, A.; Mojal, S.; Fernández, E.; Soler, M.J.; NEFRONA Study. Circulating angiotensin-converting enzyme 2 activity in patients with chronic kidney disease without previous history of cardiovascular disease. Nephrol. Dial. Transplant. 2015, 30, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Lai, F.M.; Kwan, B.C.; Lai, K.B.; Chow, K.M.; Li, P.K.; Szeto, C.C. Expression of ACE and ACE2 in patients with hypertensive nephrosclerosis. Kidney Blood Press. Res. 2011, 34, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.; Liu, J.; Cheng, G.; An, C.; Deo, K.; Gorret, A.M.; Qin, X. A review of urinary angiotensin converting enzyme 2 in diabetes and diabetic nephropathy. Biochem. Med. 2019, 29, 010501. [Google Scholar] [CrossRef] [PubMed]
- Benigni, A.; Cassis, P.; Remuzzi, G. Angiotensin II revisited: New roles in inflammation, immunology and aging. EMBO Mol. Med. 2010, 2, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef]
- Thomas, M.C.; Pickering, R.J.; Tsorotes, D.; Koitka, A.; Sheehy, K.; Bernardi, S.; Toffoli, B.; Nguyen-Huu, T.P.; Head, G.A.; Fu, Y.; et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ. Res. 2010, 107, 888–897. [Google Scholar] [CrossRef]
- Alghamri, M.S.; Weir, N.M.; Anstadt, M.P.; Elased, K.M.; Gurley, S.B.; Morris, M. Enhanced angiotensin II-induced cardiac and aortic remodeling in ACE2 knockout mice. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 138–151. [Google Scholar] [CrossRef]
- Rodrigues Prestes, T.R.; Rocha, N.P.; Miranda, A.S.; Teixeira, A.L.; Simoes-E-Silva, A.C. The Anti-Inflammatory Potential of ACE2/Angiotensin-(1-7)/Mas Receptor Axis: Evidence from Basic and Clinical Research. Curr. Drug Targets 2017, 18, 1301–1313. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Jando, J.; Camargo, S.M.R.; Herzog, B.; Verrey, F. Expression and regulation of the neutral amino acid transporter B0AT1 in rat small intestine. PLoS ONE 2017, 12, e0184845. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Zhou, Q. Structure of dimeric full-length human ACE2 in complex with B0AT1. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, R.S.; Qu, G.Q.; Wang, Y.Y.; Liu, P.; Zhu, Y.Z.; Fei, G.; Ren, L.; Zhou, Y.W.; Liu, L. Gross examination report of a COVID-19 death autopsy. Fa Yi Xue Za Zhi 2020, 36, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Ding, Y.; He, L.; Zhang, Q.; Huang, Z.; Che, X.; Hou, J.; Wang, H.; Shen, H.; Qiu, L.; Li, Z.; et al. Organ distribution of severe acute respiratory syndrome (SARS) associated coronavirus (SARS-CoV) in SARS patients: Implications for pathogenesis and virus transmission pathways. J. Pathol. 2004, 203, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, W.A.; Wyne, K. Renin-Angiotensin-aldosterone system in diabetes and hypertension. J. Clin. Hypertens. 2011, 13, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Munger, M.A. Use of Angiotensin receptor blockers in cardiovascular protection: Current evidence and future directions. Pharm. Ther. 2011, 36, 22–40. [Google Scholar]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, NEJMoa2002032. [Google Scholar] [CrossRef]
- Identification of an Existing Japanese Pancreatitis Drug, Nafamostat, Which is Expected to Prevent the Transmission of New Coronavirus Infection (COVID-19), IMSUT Website. Available online: https://www.ims.u-tokyo.ac.jp/imsut/jp/about/press/page_00060.html (accessed on 28 April 2020).
- Gao, Y.; Li, T.; Han, M.; Li, X.; Wu, D.; Xu, Y.; Zhu, Y.; Liu, Y.; Wang, X.; Wang, L. Diagnostic Utility of Clinical Laboratory Data Determinations for Patients with the Severe COVID-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Wan, S.; Xiang, Y.; Fang, W.; Zheng, Y.; Li, B.; Hu, Y.; Lang, C.; Huang, D.; Sun, Q.; Xiong, Y.; et al. Clinical Features and Treatment of COVID-19 Patients in Northeast Chongqing. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.C.; Wang, J.; Jeffers, S.; Edeen, K.E.; Wang, S.; Cosgrove, G.P.; Funk, C.J.; Manzer, R.; Miura, T.A.; Pearson, L.D.; et al. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology 2008, 372, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, P.; Wei, Y.; Yue, H.; Wang, Y.; Hu, M.; Zhang, S.; Cao, T.; Yang, C.; Li, M.; et al. Histopathologic Changes and SARS–CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, M20-0533. [Google Scholar] [CrossRef] [PubMed]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020, 181, 1–9. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, L.; Sakagami, H.; Miwa, N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses 2020, 12, 491. https://doi.org/10.3390/v12050491
Xiao L, Sakagami H, Miwa N. ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses. 2020; 12(5):491. https://doi.org/10.3390/v12050491
Chicago/Turabian StyleXiao, Li, Hiroshi Sakagami, and Nobuhiko Miwa. 2020. "ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel?" Viruses 12, no. 5: 491. https://doi.org/10.3390/v12050491
APA StyleXiao, L., Sakagami, H., & Miwa, N. (2020). ACE2: The key Molecule for Understanding the Pathophysiology of Severe and Critical Conditions of COVID-19: Demon or Angel? Viruses, 12(5), 491. https://doi.org/10.3390/v12050491